KALETRA
ABBVIE
Kaletra es un inhibidor de la proteasa del HIV con actividad contra el virus de la inmunodeficiencia humana.
LISTAS N°: 3956 / 6799 / 0522.
Composición.
Kaletra Solución Oral (Elixir): Cada ml contiene: 80 mg de Lopinavir; 20 mg de Ritonavir; Alcohol deshidratado USP; 200 Proof, Jarabe de maíz con alto contenido de fructosa, Propilenglicol USP; Agua purificada USP; Glicerina USP; Povidona USP; Saborizante Magnasweet-110; Sabor natural y artificial de vainilla; Aceite de castor hidrogenado de polioxilo 40 NF; Sabor artificial de algodón de azúcar; Acesulfame de potasio; Sacarina sódica USP; Cloruro de sodio USP; Aceite de menta NF; Citrato de sodio dihidrato USP; Ácido cítrico anhidro USP, Mentol USP. La solución oral de Kaletra contiene 42,4% de alcohol (v/v) y 15,3% de propilenglicol (p/v). Kaletra Comprimidos Recubiertos: Cada comprimido recubierto de Kaletra contiene 200 mg de Lopinavir, coformulado con 50 mg de Ritonavir. Excipientes: Copovidona, Monolaurato de Sorbitán, Dióxido de Silicio Coloidal y Estearil Fumarato de Sodio. Para la cubierta del comprimido se utilizan los siguientes ingredientes: Hipromelosa, Dióxido de Titanio, Polietilenglicol 400, Hidroxipropilcelulosa, Talco, Dióxido de Silicio Coloidal, Polietileglicol 3350, Oxido Férrico Amarillo, E172 y Polisorbato 80. Kaletra Comprimidos Recubiertos 100/25: Cada comprimido recubierto contiene 100 mg de Lopinavir, coformulado con 25 mg de Ritonavir. Excipientes: Copovidona, Monolaurato de Sorbitán, Dióxido de Silicio Coloidal y Estearil Fumarato de Sodio. Para la cubierta del comprimido se utilizan los siguientes ingredientes: Alcohol polivinílico, Dióxido de titanio, Talco, Polietilenglicol 3350 y Oxido Férrico Amarillo, E172.
Farmacología.
Descripción: Kaletra (Lopinavir/Ritonavir) es una coformulación de Lopinavir y Ritonavir. Lopinavir es un inhibidor de las proteasas del HIV-1 y HIV-2. En esta coformulación, Ritonavir inhibe el metabolismo de Lopinavir mediado por la CYP3A y de esta forma proporciona mayores niveles plasmáticos de Lopinavir. Lopinavir es químicamente designado como [1S-[1R*,(R*), 3R*, 4R*]]-N-[4-[[2,6-(dimetilfenoxi) acetil]amino]-3-hidroxi-5-fenil-1-(fenilmetil)pentil]tetrahidro-alfa-(1-metiletil)-2-oxo-1(2H)-pirimidineacetamida. Su fórmula molecular es C37 H48 N4 O5 y su peso molecular es de 628.80. Lopinavir posee la siguiente fórmula estructural: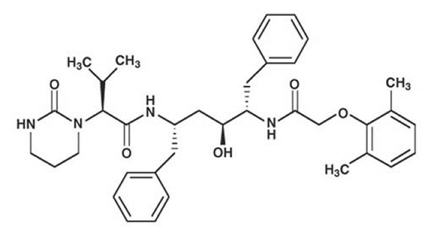
Ritonavir es químicamente designado como 10-hidroxi-2-metil-5-(1-metiletil)- 1-[2-(1-metiletil) -4-tiazolil]-3,6-dioxo-8,11-bis(fenilmetil)-2,4,7,12-tetraaza-tridecan-13-oico, éster 5-tiazolilmetil, [5S-(5R*,8R*,10R*,11R*)]. Su fórmula molecular es C37 H48 N6 O5 S2 y su peso molecular es de 720.95. Ritonavir posee la siguiente fórmula estructural: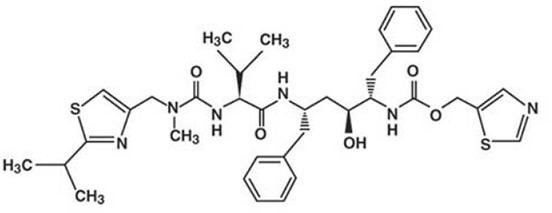
Farmacología: Microbiología: Mecanismo de acción: El Lopinavir, inhibidor de las proteasas del HIV-1 y HIV-2, impide el clivaje de la poliproteína gag-pol, dando como resultado la producción de partículas virales inmaduras no infecciosas. Actividad antiviral in vitro: Se evaluó la actividad antiviral in vitro del Lopinavir contra cepas de laboratorio y aislados clínicos del HIV en líneas celulares linfoblásticas agudamente infectadas y en linfocitos de sangre periférica, respectivamente. En ausencia de suero humano, la concentración efectiva media del 50% (CE50) de Lopinavir contra cinco diferentes cepas de laboratorio del HIV-1 osciló entre 10 y 27 nM (0,006 a 0,017 mcg/ml; 1 mcg/ml = 1,6 microM) y entre 4-11 nM (0,003 a 0,007 mcg/ml) contra distintos aislados clínicos de HIV-1 (N=6). En presencia del 50% de suero humano, la CE50 media de Lopinavir contra estas cinco cepas de laboratorio osciló entre 65 y 289 nM (0,04 a 0,18 mcg/ml), o sea 7 a 11 veces de atenuación. No han finalizado los estudios sobre la actividad de Lopinavir en combinación con otros inhibidores de la proteasa o inhibidores de la transcriptasa inversa. Resistencia: Se seleccionaron aislados de HIV-1 in vitro con susceptibilidad reducida al Lopinavir. La presencia de Ritonavir no parece afectar la selección de virus in vitro resistentes al Lopinavir. No se ha tipificado la selección de resistencia a Kaletra en pacientes vírgenes de tratamiento antirretroviral. En un estudio fase III en 653 pacientes vírgenes de tratamiento antirretroviral (Estudio 863), se analizaron los aislados virales plasmáticos de cada paciente en tratamiento con más de 400 copias/ml de HIV en las semanas 24, 32, 40 y/o 48. No se observó evidencia de resistencia a Kaletra en 37 pacientes evaluables tratados con Kaletra (0%). Se observó evidencia de resistencia genotípica al Nelfinavir definida como la presencia de mutaciones D3ON y/o L9OM en la proteasa de HIV en 25/76 (33%) de los pacientes evaluables tratados con Nelfinavir. La selección de la resistencia a Kaletra en pacientes pediátricos vírgenes de tratamiento antirretroviral (Estudio 940) parece coincidir con lo observado en pacientes adultos (Estudio 863). Se ha observado resistencia a Kaletra en pacientes tratados con otros inhibidores de la proteasa antes de Kaletra. En estudios de fase II en 227 pacientes vírgenes de tratamiento antirretroviral y tratados con inhibidores de la proteasa, los aislados de 4 de 23 pacientes con ARN viral cuantificable ( > 400 copias/ml) después del tratamiento con Kaletra durante 12 a 100 semanas exhibieron susceptibilidad significativamente reducida al Lopinavir en comparación con los correspondientes aislados virales basales. Tres de estos pacientes habían sido tratados con un solo inhibidor de la proteasa (Nelfinavir, Indinavir o Saquinavir) y uno había sido tratado con varios inhibidores de la proteasa (Indinavir, Saquinavir y Ritonavir). Los cuatro pacientes presentaron por lo menos 4 mutaciones asociadas con resistencia a los inhibidores de la proteasa inmediatamente antes del tratamiento con Kaletra. Luego del rebote viral, todos los aislados de estos pacientes contenían más mutaciones, algunas asociadas con resistencia a los inhibidores de la proteasa. Sin embargo, los datos disponibles son insuficientes para identificar patrones de mutación asociados con Lopinavir en pacientes tratados con Kaletra. La evaluación de estos patrones de mutación se encuentra actualmente en estudio. Resistencia cruzada - Estudios preclínicos: Se han observado distintos grados de resistencia cruzada entre los inhibidores de la proteasa. Hay escasa información disponible sobre la resistencia cruzada de virus que desarrollaron susceptibilidad disminuida al Lopinavir durante el tratamiento con Kaletra. Se determinó la actividad in vitro de Lopinavir contra aislados clínicos obtenidos de pacientes previamente tratados con un solo inhibidor de la proteasa. Los aislados que exhibieron susceptibilidad reducida > 4 veces a Nelfinavir (N=13) y Saquinavir (N=4), exhibieron susceptibilidad reducida < 4 veces al Lopinavir. Los aislados con susceptibilidad reducida > 4 veces a Indinavir (N=16) y Ritonavir (N=3) exhibieron una susceptibilidad reducida media al Lopinavir de 5,7 y 8,3 veces respectivamente. Los aislados de pacientes previamente tratados con dos o más inhibidores de la proteasa demostraron mayores reducciones en la susceptibilidad al Lopinavir, según se describe más adelante. (Estudios clínicos/Actividad antiviral de Kaletra en pacientes ya tratados con inhibidores de la proteasa). Resistencia cruzada durante el tratamiento con Kaletra: Es escasa la información sobre la resistencia cruzada de virus seleccionados durante el tratamiento con Kaletra. Los aislados de 4 pacientes previamente tratados con uno o más inhibidores de la proteasa que exhibieron mayor resistencia fenotípica al Lopinavir durante el tratamiento con Kaletra, se mantuvieron resistentes o desarrollaron resistencia cruzada a Ritonavir, Indinavir y Nelfinavir. Todos los virus de rebote permanecieron totalmente sensibles o presentaron una susceptibilidad ligeramente reducida al Amprenavir (hasta 8.5 veces junto con 99 veces de resistencia a Lopinavir). Los aislados de rebote de los 2 sujetos sin tratamiento previo con Saquinavir permanecieron completamente sensibles al Saquinavir. Correlatos genotípicos de respuesta virológica reducida en pacientes con experiencia antirretroviral, iniciando un régimen combinado basado en Kaletra: La respuesta virológica a Kaletra ha demostrado ser afectada por la presencia de tres o más de las siguientes sustituciones de aminoácidos en la proteasa basal: L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T e I84V. La siguiente tabla muestra la respuesta virológica a las 48 semanas (ARN del HIV < 400 copias/ml) de acuerdo al número de las mutaciones de resistencia del inhibidor de la proteasa basal mencionadas, en los estudios 888 y 765, y en el estudio 957.
La Tabla 2 muestra la respuesta virológica a las 48 semanas (ARN de HIV-1 < 50 copias/ml) en el Estudio 802 de acuerdo con el número de mutaciones de resistencia asociadas con Lopinavir señaladas en la Tabla 1 presentes en la evaluación basal. No se dispone de suficientes datos para avalar la administración una vez al día de Kaletra en pacientes adultos con tres o más mutaciones asociadas con Lopinavir.
Estudios Clínicos: Actividad antiviral de Kaletra en pacientes ya tratados con inhibidores de la proteasa. Se evaluó la relevancia clínica de la susceptibilidad in vitro reducida al Lopinavir mediante la respuesta virológica al tratamiento con Kaletra, respecto del genotipo y fenotipo viral basal, en 56 pacientes no tratados con Inhibidores no nucleótidos de la transcriptasa reversa (INNTR) con ARN HIV > 1000 copias/ml a pesar del tratamiento previo con por lo menos dos de los inhibidores de la proteasa Nelfinavir, Indinavir, Saquinavir y Ritonavir. La CE50 de Lopinavir contra los 56 aislados virales basales fue de 0.5 a 96 veces superior que la CE50 frente al HIV de tipo salvaje. El 55% (31/56) de estos aislados basales exhibió una susceptibilidad reducida > 4 veces al Lopinavir. Estos aislados presentaron una reducción media de 27,9 veces en la susceptibilidad a Lopinavir. Después de 48 semanas de tratamiento con Kaletra, Efavirenz e inhibidores nucleósidos de la transcriptasa reversa, se observó ARN HIV plasmático ≤ 400 copias/ml en el 93% (25/27), 73 % (11/15) y 25% (2/8) de los pacientes con menos de 10 veces, 10 a 40 veces y ≥ 40 veces la susceptibilidad reducida al Lopinavir al nivel basal, respectivamente, y ≤ 50 copias/ml en el 81% (22/27), 60% (9/15) y 25% (2/8) respectivamente. Los datos disponibles son insuficientes para identificar patrones de mutación asociados con Lopinavir en pacientes tratados con Kaletra. Es necesaria la realización de estudios para evaluar la asociación entre patrones específicos de mutación e índices de respuesta virológica. La Sociedad Europea Clínica de SIDA (EACS) define que Lopinavir/r es no sólo una droga antirretroviral con clara y demostrada penetración en fluido cefalorraquídeo cuando se la estudia en poblaciones saludables infectadas con HIV (concentración por encima de IC90 en > 90 % de pacientes examinados) pero también con eficacia probada a corto plazo (3-6 meses) sobre la función cognitiva o sobre la disminución de la carga viral en líquido cefalorraquídeo cuando se la evalúa como agente único o en estudios controlados (publicaciones revisadas por pares). Farmacocinética: En la evaluación de las propiedades farmacocinéticas de Lopinavir coadministrado con Ritonavir en voluntarios adultos sanos y en pacientes con HIV no se observaron diferencias significativas entre ambos grupos. El Lopinavir es fundamentalmente metabolizado por la CYP3A. El Ritonavir inhibe el metabolismo del Lopinavir y, por lo tanto, aumenta los niveles plasmáticos de Lopinavir. En los estudios clínicos, la administración de 400/100 mg de Kaletra dos veces por día produjo concentraciones plasmáticas medias de Lopinavir en estado de equilibrio 15 a 20 veces superiores a las de Ritonavir en pacientes con HIV. Las concentraciones plasmáticas de Ritonavir son inferiores al 7% de las obtenidas con dosis de 600 mg de Ritonavir dos veces por día. La CE50 antiviral in vitro de Lopinavir es aproximadamente 10 veces menor que la de Ritonavir. Por lo tanto, la actividad antiviral de Kaletra se debe al Lopinavir. La Figura 1 presenta las concentraciones plasmáticas medias de Lopinavir y Ritonavir en estado de equilibrio después de la administración de 400/100 mg de Kaletra dos veces al día con las comidas, durante 3-4 semanas extraídas de un estudio farmacocinético en pacientes adultos infectados con HIV (N=19).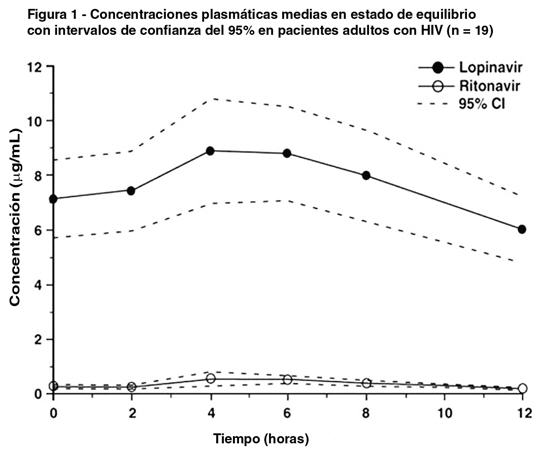
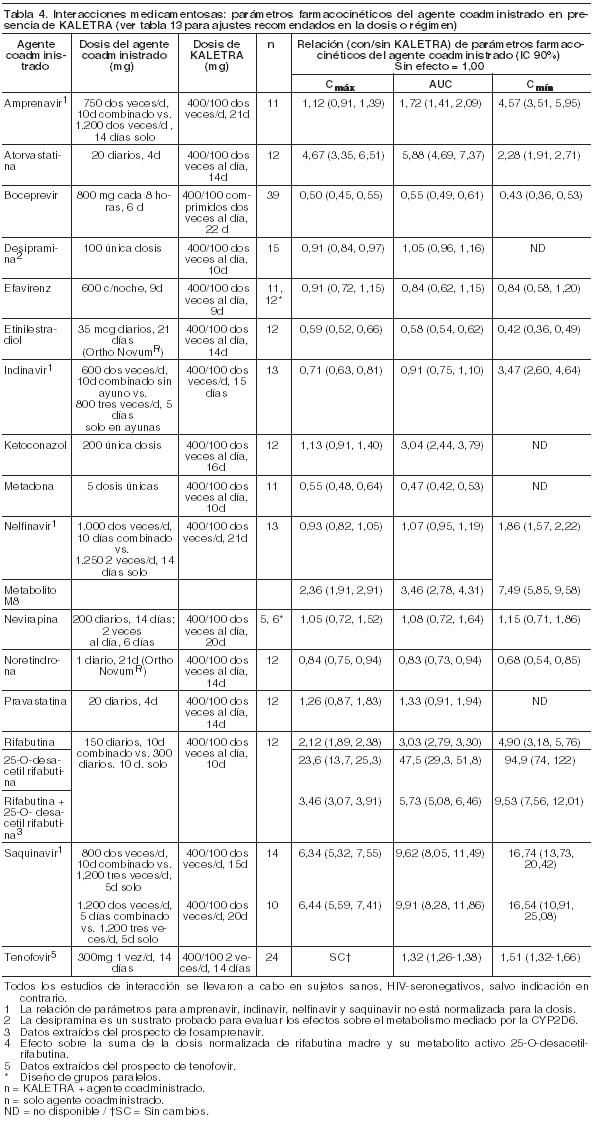
Absorción: En un estudio farmacocinético en sujetos HIV-seropositivos (N=18), el tratamiento con dosis múltiples de 400/100 mg de Kaletra dos veces por día con las comidas, durante 3 semanas produjo una Cmax media de Lopinavir ± DS de 12,3 ± 5,4 mcg/ml en aproximadamente 4 horas después de la administración. La concentración mínima media en estado de equilibrio antes de la dosis de la mañana fue de 8,1 ± 5,7 mcg/ml y la concentración mínima dentro del intervalo de dosis fue de 5,6 ± 4,5 mcg/ml. El AUC de Lopinavir durante un intervalo posológico de 12 horas promedió 113,2 ± 60,5 mcg x h/ml. No se ha establecido la biodisponibilidad absoluta de Lopinavir coformulado con Ritonavir en los seres humanos. Efectos de las comidas sobre la absorción oral: Kaletra comprimidos recubiertos: No se observaron cambios clínicamente significativos en la Cmax y AUC con la administración de Kaletra comprimidos recubiertos después de haber ingerido alimentos, comparativamente con lo comprobado cuando el fármaco fue administrado en ayunas. Con respecto a lo verificado en condiciones de ayuno, la administración de Kaletra comprimidos recubiertos, con una comida con moderado contenido graso (500-682 kcal, con un 23-25% de las calorías derivadas de las grasas) produjo un incremento en el AUC y en la Cmáx del Lopinavir, del 26,9% y el 17,6% respectivamente. Con respecto a lo verificado en condiciones de ayuno, la administración de Kaletra comprimidos recubiertos, con una comida de alto contenido graso (872 kcal, con un 56% de las calorías derivadas de las grasas) produjo un aumento aproximado del 18,9% en el AUC pero no en la Cmax. Por lo tanto, Kaletra comprimidos recubiertos puede ser tomado con las comidas o alejado de las mismas. La administración de una dosis única de la solución oral (Elixir) de KALETRA con una comida moderada en grasas (500-682 kcal, 23 a 25% de calorías provenientes de las grasas) estuvo asociada con un aumento medio del 80% y 54% en el AUC y Cmáx de Lopinavir, respectivamente, en comparación con la dosis en ayunas. En comparación con la dosis en ayunas, la administración de KALETRA con una comida rica en grasas (872 kcal-56% proveniente de grasas) aumento el AUC y la Cmáx de Lopinavir en 130% y 56%, respectivamente con la solución oral (Elixir). Para aumentar la biodisponibilidad y minimizar la variabilidad farmacocinética, KALETRA Solución Oral debe ingerirse con las comidas. Distribución: En estado de equilibrio, Lopinavir se une en un 98-99% a las proteínas plasmáticas. Si bien Lopinavir se une a la glucoproteína ácida alfa-1 (GAA) y a la albúmina, posee mayor afinidad por la GAA. En estado de equilibrio, la unión de Lopinavir a las proteínas se mantiene constante sobre el rango de concentraciones observadas después de la administración de 400/100 mg de Kaletra dos veces por día y es similar entre voluntarios sanos y pacientes HIV-seropositivos. Metabolismo: Los ensayos in vitro con microsomas hepáticos humanos indican que el metabolismo de Lopinavir es principalmente oxidativo. El Lopinavir es extensamente metabolizado por el sistema hepático del citocromo P450, casi exclusivamente por la isoenzima CYP3A. El Ritonavir es un potente inhibidor de la CYP3A que inhibe el metabolismo de Lopinavir y, en consecuencia, aumenta los niveles plasmáticos de Lopinavir. Un estudio con 14C-Lopinavir en seres humanos demostró que el 89% de la radioactividad plasmática después de una dosis única de 400/100 mg de Kaletra se debió al compuesto madre. Se han identificado por lo menos 13 metabolitos oxidativos de Lopinavir en los seres humanos. El Ritonavir ha demostrado inducir las enzimas metabólicas, provocando la inducción de su propio metabolismo. Con dosis múltiples, las concentraciones predosis de Lopinavir disminuyen con el tiempo y luego de aproximadamente 10 a 16 días se estabilizan. Eliminación: Después de una dosis de 400/100 mg de 14C-Lopinavir/ Ritonavir, aproximadamente 10,4 ± 2,3% y 82,6 ± 2,5% de la dosis administrada de 14C-Lopinavir se recupera en la orina y heces, respectivamente, después de 8 días. Alrededor de 2,2% y 19,8% de la dosis administrada de Lopinavir se elimina sin cambios por vía renal y fecal, respectivamente. Después de dosis múltiples, menos del 3% de la dosis de Lopinavir se excreta en forma inalterada en la orina. El clearence oral aparente (CL/F), de Lopinavir, es 5,98 ± 5,75 L/h (media ± DS, N=19). Dosificación de Una Toma Diaria: La farmacocinética de Kaletra de una toma diaria se ha evaluado en pacientes infectados con HIV y vírgenes de tratamiento antirretroviral. Kaletra 800/200 mg fue administrado en combinación con Emtricitabina 200 mg y Tenofovir DF 300 mg como parte de un régimen de una toma diaria. Dosis múltiples de Kaletra 800/200 mg una vez al día durante 2 semanas independientemente de las comidas (N=16) produjeron Cmáx medias de 14,8 ± 3,5 mcg/ml, que se presentaron aproximadamente 6 horas después de la administración. La concentración valle media en estado de equilibrio de Lopinavir antes de la dosis matinal fue de 5,5 ± 5,4 mcg/ml y la concentración mínima entre dosis fue de 3,2 ± 3,4 mcg/ml. El AUC de Lopinavir a lo largo del intervalo de 24 horas entre dosis promedió 206,5 ± 89,7 mcg•h/ml. Efectos sobre el electrocardiograma: El intervalo QTcF fue evaluado en un estudio randomizado, cruzado y controlado contra placebo y contra droga activa (Moxiflacina 400 mg una vez al día), en 39 adultos sanos, con 10 registros ECG a lo largo de 12 horas en el día 3 del estudio. Las diferencias máximas medias (IC 95%) del QTcF respecto al placebo fueron 3.6 (6.3) mseg y 13.1 (5.8) mseg para la dosis de 400/100 mg diarios y para la dosis supraterapéutica de 800/200 mg diarios de Lopinavir/Ritonavir, respectivamente. Los dos regímenes resultaron en exposiciones mayores a 1.5 y 3 veces que aquellas observadas con la dosis recomendada, ya sea una o dos veces diaria de Lopinavir/Ritonavir en estado de equilibrio en el día 3. Ningún sujeto experimentó un aumento del QTcF ≥ 60 mseg en relación al basal o un intervalo QTcF que excediera el umbral potencialmente relevante desde el punto de vista clínico de 500 mseg. También se observó una prolongación modesta del intervalo PR en los sujetos que recibían Lopinavir/Ritonavir en el mismo estudio en el día 3. El intervalo PR máximo fue de 286 mseg y no se observó ningún bloqueo de segundo o de tercer grado (Ver Precauciones). Poblaciones especiales: Sexo, raza y edad: No se ha evaluado la farmacocinética de Lopinavir en pacientes geriátricos. En pacientes adultos no se observaron diferencias farmacocinéticas entre hombres y mujeres. No se identificaron diferencias farmacocinéticas debidas a la raza clínicamente significativas. Pacientes pediátricos: Se evaluó la farmacocinética de Kaletra en dosis de 300/75 mg/m2 y 230/57,5 mg/m2 administradas dos veces por día a un total de 53 pacientes pediátricos con edades comprendidas entre 6 meses y 12 años. Las concentraciones plasmáticas de Lopinavir con el régimen posológico de 230/57,5 mg/m2 dos veces por día sin Nevirapina y con el régimen de 300/75 mg/m2 dos veces por día con Nevirapina fueron similares a las obtenidas en pacientes adultos con dosis de 400/100 mg dos veces por día sin Nevirapina. Kaletra una toma diaria no se ha evaluado en pacientes pediátricos. El AUC, la Cmáx y Cmín de Lopinavir en estado de equilibrio fue de 72.6 ± 31.1 mcg x h/ml, 8.2 ± 2.9 y 3.4 ± 2.1 mcg/ml, respectivamente después de la administración de 230/57,5 mg/m2 de Kaletra dos veces por día sin Nevirapina (N=12) y de 85.8 ± 36.9 mcg x h/ml, 10 ± 3.3 y 3.6 ± 3.5 mcg/ml, respectivamente luego de 300/75 mg/m2 dos veces por día con Nevirapina (N=12). La Nevirapina se administró en dosis de 7 mg/kg dos veces por día en niños de 6 meses a 8 años o en dosis de 4 mg/kg dos veces por día en niños > 8 años. Insuficiencia renal: No se ha evaluado la farmacocinética de Lopinavir en pacientes con insuficiencia renal; sin embargo, debido a que el clearance renal de Lopinavir es insignificante, no es probable que el clearance corporal total disminuya en pacientes con insuficiencia renal. Insuficiencia hepática: Lopinavir es principalmente metabolizado y eliminado por el hígado. Múltiples dosis de Lopinavir/Ritonavir 400/100 mg dos veces al día administradas a pacientes coinfectados con HIV y HCV, con compromiso hepático leve a moderado, provocaron un aumento del 30% en el AUC de Lopinavir y un 20% de aumento en la Cmax comparado con pacientes infectados con HIV con una función hepática normal (N=12). Adicionalmente, la unión a las proteínas plasmáticas de Lopinavir fue menor tanto en el compromiso hepático leve como moderado, comparado con controles (99,09 vs 99,31% respectivamente). Se debe proceder con cautela al administrar Kaletra a sujetos con insuficiencia hepática. Kaletra no ha sido estudiado en pacientes con un compromiso hepático severo (ver Precauciones). Interacciones droga-droga: Ver también Contraindicaciones e Interacciones medicamentosas en Precauciones. Kaletra es un inhibidor in vitro de la isoforma CYP3A del citocromo P450. La coadministración de Kaletra con otros agentes principalmente metabolizados por la CYP3A puede provocar una elevación de las concentraciones plasmáticas de esos agentes que podrían aumentar o prolongar sus efectos terapéuticos y adversos (ver Contraindicaciones). Kaletra no inhibe a las isoformas CYP2D6, CYP2C9, CYP2C19, CYP2E1, CYP2B6 o CYP1A2 en concentraciones clínicamente significativas. Kaletra ha demostrado inducir in vivo su propio metabolismo y elevar la biotransformación de algunas drogas metabolizadas por las enzimas del citocromo P450 y por glucuronización. Kaletra es metabolizado por la CYP3A. Es de esperar que los agentes inductores de la actividad de la CYP3A aumenten el clearance de Lopinavir y por lo tanto reduzcan su concentración plasmática. La administración concomitante de Kaletra y otros agentes inhibidores de la CYP3A puede aumentar las concentraciones plasmáticas de Lopinavir, si bien este efecto no se ha observado con la coadministración de Ketoconazol. Se llevaron a cabo estudios de interacciones farmacológicas entre Kaletra y otros agentes probablemente a ser coadministrados y algunos agentes comúnmente empleados como controles para estudiar las interacciones farmacocinéticas. La Tabla 3 resume los efectos de la coadministración de otros agentes sobre el AUC, Cmáx y Cmín de Lopinavir coformulado en Kaletra y la Tabla 4 los efectos de Kaletra sobre otros agentes. Los efectos de otros agentes sobre Ritonavir no están ilustrados, salvo por las notas al pie de la tabla, ya que generalmente se correlacionan con los observados con Lopinavir (si las concentraciones de Lopinavir disminuyen, también disminuyen las concentraciones de Ritonavir). Para información sobre recomendaciones clínicas, ver Precauciones.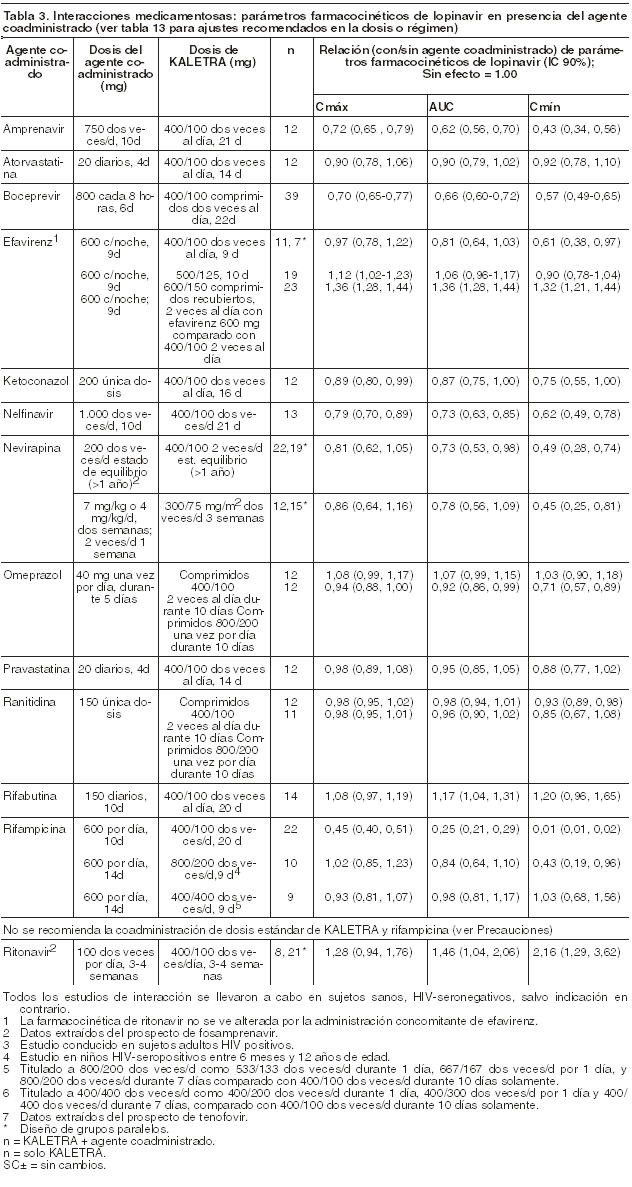
Indicaciones.
Kaletra está indicado en combinación con otros agentes antirretrovirales para el tratamiento de la infección por HIV. Esta indicación se basa en los análisis de los niveles plasmáticos de ARN HIV y recuentos de células CD4 de un estudio controlado de 48 semanas con Kaletra y de estudios de menor tamaño no controlados de rangos posológicos con Kaletra de 144-360 semanas de duración. Aún no se han publicado los resultados de los estudios controlados que evalúan el efecto de Kaletra sobre el avance clínico del HIV.
Dosificación.
Kaletra comprimidos recubiertos puede ser tomado con las comidas o alejado de las mismas. Kaletra comprimidos recubiertos deben ser tragados enteros y no deben ser masticados, partidos ni triturados. Kaletra Solución Oral debe ser ingerido con las comidas. La dosis oral recomendada de Kaletra es la siguiente: Adultos: Kaletra comprimidos recubiertos 400/100 mg (2 comprimidos recubiertos de 200/50 mg) 2 veces por día, con las comidas o alejados de las mismas. Kaletra 400/100 mg (5 ml) 2 veces por día tomados con las comidas. Kaletra comprimidos recubiertos 800/200 mg (4 comprimidos recubiertos de 200/50 mg) 1 vez por día, con las comidas o alejados de las mismas en pacientes con menos de tres mutaciones asociadas con Lopinavir. No hay suficientes datos para avalar el empleo de la administración una vez al día de Kaletra en pacientes adultos con tres o más mutaciones asociadas con Lopinavir. Kaletra 800/200 mg (10 ml) 1 vez por día tomados con las comidas en pacientes con menos de tres mutaciones asociadas con Lopinavir. No hay suficientes datos para avalar el empleo de la administración una vez al día de Kaletra en pacientes adultos con tres o más mutaciones asociadas con Lopinavir. Kaletra no debe administrarse una vez al día en combinación con Carbamazepina, Fenobarbital o Fenitoína (ver Interacciones Medicamentosas). Tratamiento concomitante: Omeprazol y Ranitidina: Kaletra Solución Oral y Comprimidos Recubiertos pueden utilizarse en combinación con agentes reductores de la acidez gástrica (Omeprazol y Ranitidina) sin ajuste de la dosis. Efavirenz, Nevirapina, Amprenavir o Nelfinavir: Comprimidos: Deberá considerarse un aumento de la dosis de Kaletra a 500/125 mg dos veces al día (por ejemplo dos comprimidos de 200/50 mg y un comprimido de 100/25) cuando se lo utilice en combinación con Efavirenz, Nevirapina, Amprenavir o Nelfinavir en el tratamiento de pacientes con experiencia previa en quienes se sospeche clínicamente una susceptibilidad reducida al Lopinavir (ya sea por la historia de tratamiento o por evidencia de laboratorio) (Ver Interacciones Medicamentosas). Solución oral: Se recomienda aumentar la dosis de Kaletra a 533/133 mg (6,5 ml) dos veces por día administrada con las comidas cuando se coadministre con Efavirenz, Nevirapina, Amprenavir o Nelfinavir en pacientes habitualmente tratados en los que clínicamente se sospeche susceptibilidad reducida al Lopinavir (por antecedentes terapéuticos o evidencia de laboratorio) (ver Interacciones Medicamentosas). Kaletra no deberá administrarse como régimen de una vez al día en combinación con Efavirenz, Nevirapina, Amprenavir o Nelfinavir. Administración durante el embarazo y en el período postparto: No se requiere el ajuste de dosis con los comprimidos de Lopinavir/ritonavir durante el embarazo y el postparto. La administración en una única toma diaria de Lopinavir/ritonavir no está recomendada para las mujeres embarazadas. Pacientes Pediátricos (Comprimidos): Los comprimidos y la solución oral de Kaletra no han sido evaluados en pacientes pediátricos, administrado una vez al día. La dosis de Kaletra comprimidos para adultos (400/100 mg dos veces al día) sin Efavirenz, Nevirapina, Nelfinavir o Amprenavir, puede ser utilizada en niños de 35 kg o más, o con una superficie corporal de 1.4 m2 o mayor. Para niños con un peso menor de 35 Kg o con una superficie corporal entre 0.6 a 1.4 m2 y que sean capaces de tragar los comprimidos, referirse a las tablas siguientes. La solución de Kaletra está disponible para niños con una superficie corporal menor de 0.6 m2 o para quienes no puedan tragar los comprimidos.
La siguiente tabla contiene las guías de dosificación para los comprimidos de Kaletra 100/25 mg basadas en el peso corporal: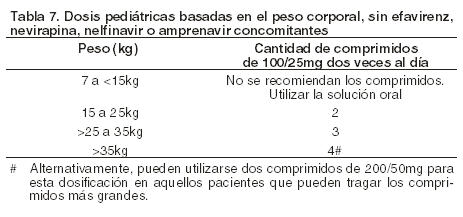
La siguiente tabla contiene las guías de dosificación para Kaletra comprimidos de 100/25 mg basadas en el peso corporal cuando se utiliza en combinación con Efavirenz, Nevirapina, Nelfinavir o Amprenavir en niños.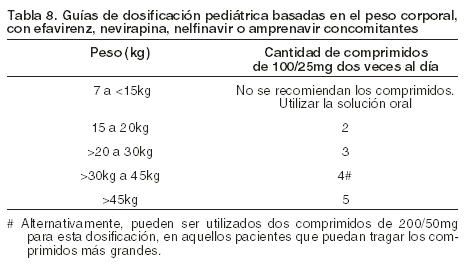
Pacientes pediátricos - Solución oral: Uso pediátrico (seis meses de edad o mayores): la dosis recomendada de Kaletra solución oral es de 12/3 mg/Kg para aquellos con un peso de 7 a menos de 15 Kg y de 10/2.5 mg/Kg para aquellos que pesen entre 15 y 40 Kg (aproximadamente equivalente a 230/57.5 mg/m2 dos veces al día tomados con las comidas, hasta una dosis máxima de 400/100 mg en niños de más de 40 Kg (5 mL) dos veces al día. Kaletra una vez al día no ha sido evaluado en pacientes pediátricos. Es preferible que el médico prescriptor calcule la dosis aproximada en mg para cada niño de 12 años o menos de edad, y determinar el volumen correspondiente de la solución o el número de cápsulas blandas. Sin embargo, como una alternativa, la siguiente tabla contiene las guías de dosificación para Kaletra solución oral, basadas en el peso corporal. Los valores totales de alcohol y propilenglicol de todos los medicamentos, incluyendo solución oral de Lopinavir/Ritonavir deben ser tenidos en cuenta para evitar toxicidad por estos excipientes al ser administrados a lactantes (ver secciones Descripción, Precauciones y Advertencias y Sobredosis).
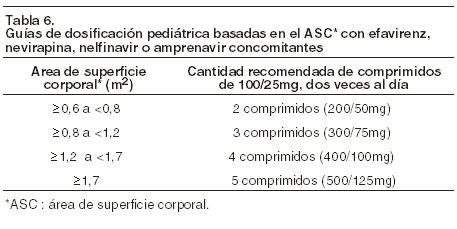
Terapia concomitante con Efavirenz, Nevirapina, Amprenavir, o Nelfinavir: Se deberá considerar un incremento de Kaletra solución oral a 13/3.25 mg/Kg para aquellos pacientes de 7 a menos de 15 Kg y 11/2.75 mg/Kg para aquellos de entre 15 a 45 Kg (aproximadamente equivalente a 300/75 mg m2) dos veces al día junto con las comidas, hasta una dosis máxima de 533/133 mg en niños de más de 45 Kg de peso dos veces al día, cuando se lo utilice en combinación con Efavirenz, Nevirapina, Amprenavir, o Nelfinavir en niños con experiencia previa de entre seis meses a 12 años de edad en los cuales se sospeche clínicamente una susceptibilidad disminuida al Lopinavir (por la historia de tratamiento o evidencia de laboratorio). La siguiente tabla contiene las guías de dosificación para Kaletra solución oral basadas en el peso corporal, cuando se lo utilice en combinación con Efavirenz, Nevirapina, Amprenavir, o Nelfinavir en niños (ver Farmacología: Interacciones medicamentosas).
Guías de dosificación basadas en el área de superficie corporal (ASC) (m2): La dosis recomendada de Kaletra es de 230/57.5 mg/m2 dos veces al día junto con las comidas, hasta una dosis máxima de 400/100 mg dos veces al día. La dosificación de 230/57.5 mg/m2 puede ser insuficiente en algunos niños cuando se coadministra con Nevirapina, Nelfinavir, Amprenavir o Efavirenz. Deberá considerarse un aumento de la dosis de Kaletra a 300/75 mg/m2 en estos pacientes. La dosis deberá ser administrada utilizando una jeringa calibrada para dosificación oral.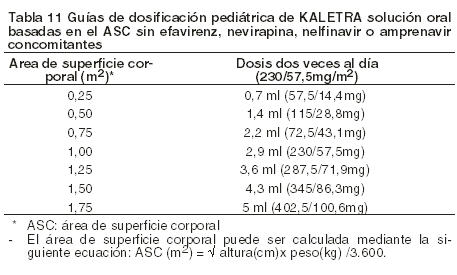
Contraindicaciones.
Kaletra está contraindicado en pacientes con hipersensibilidad conocida a Lopinavir, Ritonavir o a alguno de sus excipientes. La coadministración de Kaletra está contraindicada con agentes cuyo clearance depende fundamentalmente de la CYP3A y cuyas concentraciones plasmáticas elevadas se encuentran asociadas con episodios serios y/o potencialmente mortales. La coadministración de Kaletra está contraindicada con los inductores potentes de la CYP3A, donde las concentraciones significativamente disminuidas de Lopinavir pueden estar asociadas con la pérdida potencial de la respuesta virológica y con una posible resistencia así como también una resistencia cruzada. Estas drogas se detallan en la siguiente tabla: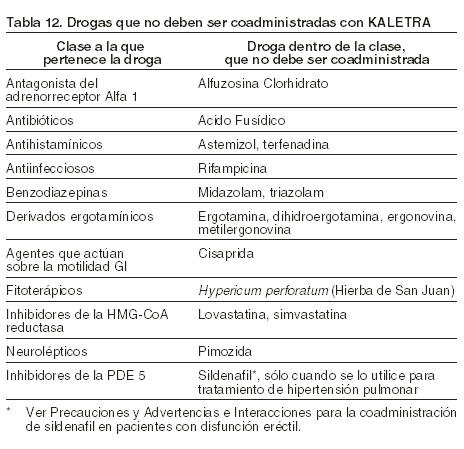
Reacciones adversas.
Adultos: Reacciones adversas asociadas con el Tratamiento: La seguridad de lopinavir/ritonavir ha sido estudiada en más de 2600 pacientes en Estudios Clínicos Fases II a IV, en los cuales más de 700 pacientes han recibido una dosis de 800/200 mg (4 comprimidos) una vez al día. En algunos estudios junto a Inhibidores nucleósidos de la transcriptasa reversa, lopinavir/ritonavir fue administrado en combinación con efavirenz o nevirapina. Las reacciones adversas a Lopinavir/Ritonavir comúnmente reportadas durante estudios clínicos incluyen diarrea, náuseas, vómitos, hipertrigliceridemia e hipercolesterolemia. Diarrea, náuseas y vómitos pueden ocurrir al comenzar el tratamiento mientras que la hipertrigliceridemia e hipercolesterolemia suelen ocurrir luego. Las siguientes han sido identificadas como reacciones adversas de intensidad moderada a severa: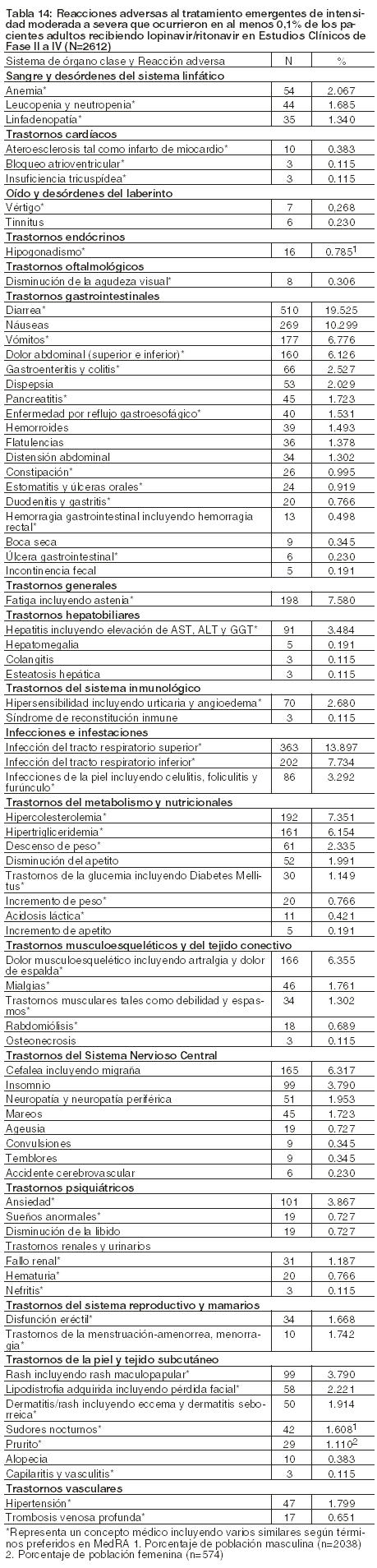
Anormalidades de Laboratorio:
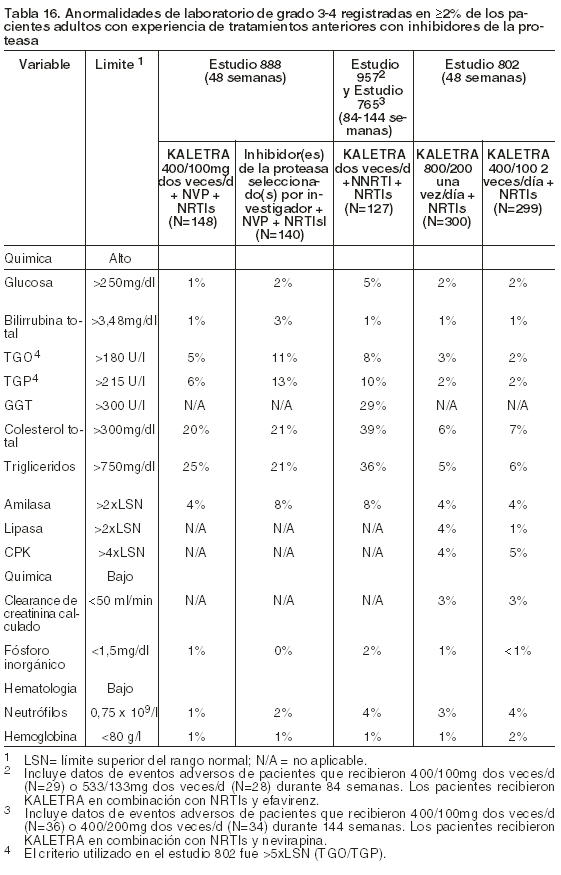
Las Tablas 15 y 16 presentan el porcentaje de pacientes adultos tratados con la terapéutica de combinación que incluyó a Kaletra, y que registraron anormalidades de laboratorio de Grado 3-4.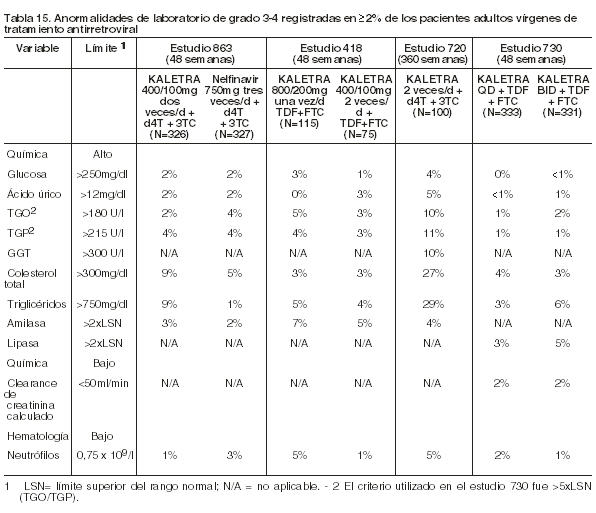
Pacientes Pediátricos: Episodios adversos emergentes durante el tratamiento: Kaletra ha sido estudiado en 100 pacientes pediátricos de 6 meses a 12 años de edad. El perfil de episodios adversos observado durante un estudio clínico fue similar al de los pacientes adultos. Durante el estudio clínico 940 pacientes pediátricos tratados con un régimen combinado que incluyó Kaletra durante un período de hasta 48 semanas, los episodios adversos más comúnmente informados relacionados con la droga, de distintos grados de intensidad, fueron disgeusia, vómitos y diarrea. Un total de 8 niños presentó episodios adversos de intensidad moderada o severa, al menos posiblemente relacionados con Kaletra. El único episodio adverso clínico relacionado con la droga de intensidad moderada a severa observado en ≥2% de los niños incluidos en el estudio fue erupción (informada en un 3%). La Tabla 17 presenta el porcentaje de pacientes pediátricos tratados con la terapéutica combinada con Kaletra que registraron anormalidades de laboratorio de Grado 3-4.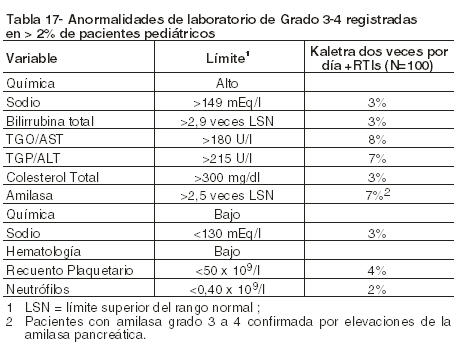
Experiencia post marketing: Se ha reportado hepatitis, Síndrome de Stevens-Johnson, necrólisis epidérmica tóxica, eritema multiforme y bradiarritmia en pacientes bajo tratamiento con Kaletra.
Precauciones.
Disfunción hepática: Debido a que Kaletra se metaboliza principalmente en el hígado, deberán extremarse las precauciones cuando se administre este agente a pacientes con disfunción hepática, porque las concentraciones de Kaletra pueden elevarse. Kaletra no ha sido estudiado en pacientes con compromiso hepático severo. Los datos farmacocinéticos sugieren aumento en las concentraciones plasmáticas de Lopinavir de aproximadamente 30% así como disminuciones en la unión a las proteínas plasmáticas en pacientes coinfectados con HIV y HCV con compromiso hepático leve a moderado (ver Farmacología: Farmacocinética). Los pacientes con hepatitis B o C subyacente o con elevaciones marcadas de las transaminasas antes del tratamiento, pueden tener el riesgo de desarrollar posteriores elevaciones de las transaminasas o descompensación hepática. Hubo informes post-marketing de disfunción hepática, incluyendo algunos casos fatales. Estos casos ocurrieron generalmente en pacientes con enfermedad avanzada por HIV que recibían varias medicaciones concomitantes en presencia de hepatitis crónica subyacente o cirrosis. No se ha establecido una relación causal con el tratamiento con Kaletra. Se deberá controlar las elevaciones de la TGO/TGP en estos pacientes, especialmente durante los primeros meses de tratamiento con Kaletra. Han sido reportadas elevaciones de transaminasas con o sin hiperbilirrubinemia en pacientes infectados con HIV-1 únicamente y pacientes no infectados tan pronto como 7 días luego de comenzar tratamiento con Lopinavir/ritonavir en conjunto con otros agentes antirretrovirales. En algunos casos, la disfunción hepática fue seria, sin embargo, una relación causal definitiva con Lopinavir/ritonavir no pudo ser establecida. Resistencia/resistencia cruzada: Se han observado distintos grados de resistencia cruzada entre los inhibidores de la proteasa. Se está actualmente investigando el efecto del tratamiento con Kaletra sobre la eficacia de los inhibidores de la proteasa administrados posteriormente (ver Microbiología). Hemofilia: Se ha informado aumento de sangrado, presencia de hematomas cutáneos espontáneos y hemartrosis en pacientes con hemofilia A y B tratados con inhibidores de la proteasa. En algunos pacientes se administró factor VIII adicional. En más de la mitad de los casos informados, se continuó o reanudó el tratamiento con los inhibidores de la proteasa. No se ha establecido una relación causal entre estos episodios y el tratamiento con inhibidores de la proteasa. Prolongación del intervalo PR: En algunos pacientes, Kaletra ha provocado una prolongación modesta y asintomática del intervalo PR. Se han informado raros casos de bloqueo aurículoventricular de segundo o de tercer grado en pacientes que recibían Kaletra y que además presentaban cardiopatía subyacente y anomalías preexistentes del sistema de conducción o en pacientes quienes recibían drogas con conocida posibilidad de prolongar el intervalo PR (tales como el Verapamilo o Atazanavir). Kaletra deberá ser utilizado con precaución en tales pacientes. (Ver Farmacología). Redistribución de la grasa corporal: En pacientes tratados con antirretrovirales se observó una redistribución/acumulación de la grasa corporal que incluyó obesidad central, aumento de la grasa dorsocervical (joroba de búfalo), consunción periférica, emaciación facial, hipertrofia mamaria y "aspecto cushingoide". Se desconoce hasta el momento el mecanismo y las consecuencias a largo plazo de estos episodios. No se ha establecido una relación causa/efecto. Elevación de lípidos: El tratamiento con Kaletra aumenta considerablemente los niveles de los triglicéridos y del colesterol total (ver Reacciones Adversas, Tablas 14 y 15 e Interacciones Medicamentosas Tabla 13). Se recomienda realizar un análisis de triglicéridos y colesterol antes de iniciar el tratamiento con Kaletra y determinaciones periódicas durante el mismo. Las alteraciones en los lípidos deberán ser clínicamente tratadas según cada caso. Ver Advertencias: Inhibidores de la HMG-CoA Reductasa e Interacciones Medicamentosas para información adicional sobre interacciones potenciales entre Kaletra e inhibidores de la HMG CoA reductasa. Síndrome de Reconstitución Inmune: El Síndrome de Reconstitución inmune ha sido reportado en pacientes infectados con HIV tratados con tratamiento antirretroviral combinado, incluyendo Kaletra. Durante la fase inicial del tratamiento antirretroviral combinado y cuando el sistema inmune responde, los