Zelboraf®
ROCHE
Vemurafenib.
Agente antineoplásico, inhibidor de la proteína quinasa.
Composición.
Cada comprimido recubierto contiene 240 mg de vemurafenib (en forma de coprecipitado de vemurafenib y succinato acetato de hipromelosa), en un excipiente compuesto por croscarmelosa sódica 29,4 mg, sílice coloidal anhidra 10,4 mg, estearato de magnesio 5,95 mg, hidroxipropilcelulosa 4,25 mg, polivinil alcohol 8 mg, dióxido de titanio (E171) 4,982 mg, Macrogol 3350: 4,040 mg, talco 2,960 mg y óxido de hierro rojo (E172) 0,018 mg.
Farmacología.
Código ATC: L01XE15. Grupo farmacoterapéutico: Agente antineoplásico, inhibidor de la proteína quinasa. Propiedades farmacodinámicas: Mecanismo de acción y efectos farmacodinámicos: Vemurafenib es un inhibidor de la serina-treonina quinasa BRAF. Las mutaciones en el gen BRAF originan la activación constitutiva de las proteínas BRAF, que pueden causar la proliferación celular sin factores de crecimiento asociados. Resultados preclínicos generados en pruebas bioquímicas han demostrado que vemurafenib puede inhibir en forma potente las quinasas BRAF activadas por mutaciones del codón 600 (Tabla 1).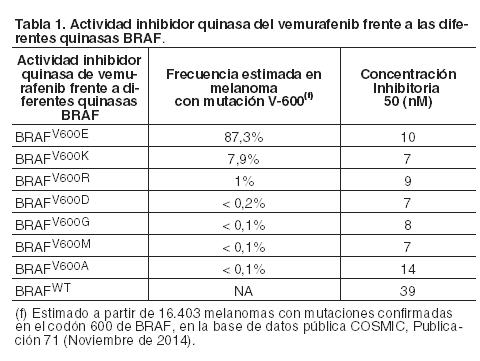
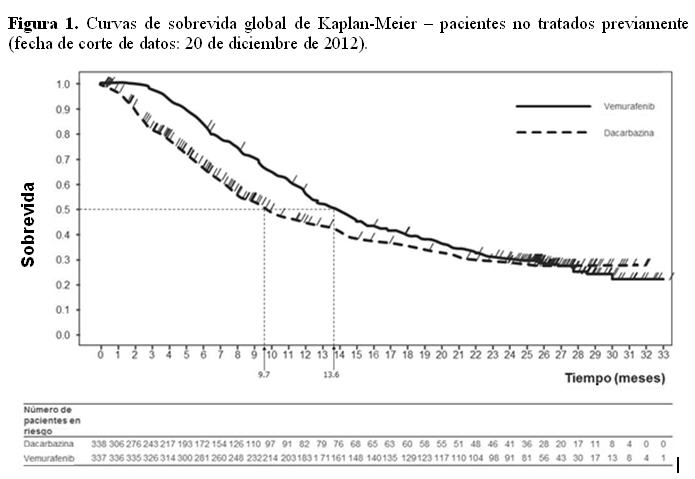
Este efecto inhibidor ha sido confirmado en las pruebas de fosforilación de ERK y de inhibición de la proliferación celular utilizando líneas celulares de melanoma que expresan mutaciones V600 del BRAF. En pruebas de inhibición de la proliferación celular, la concentración inhibitoria 50 (CI50) contra las líneas celulares V600 mutadas (líneas celulares mutadas V600E, V600R, V600D y V600K) varió desde 0,016 hasta 1,131 mM, mientras que la concentración inhibitoria 50 (CI50) frente a las líneas celulares con BRAF nativo (wild type) fueron de 12,06 y 14,32 mM, respectivamente. Determinación del estado de la mutación BRAF: Antes de comenzar el tratamiento con vemurafenib, los pacientes deben haber confirmado, por un test validado, la mutación positiva V600 de BRAF en el tumor. En los ensayos clínicos Fases II y III, los pacientes elegibles se identificaron utilizando un ensayo reacción en cadena de la polimerasa a tiempo real (Test de Cobas® 4800 para la mutación BRAF V600). Este test tiene el marcado CE y se utiliza para evaluar el status de la mutación BRAF en ADN aislado de una muestra de tejido tumoral fijada en formol e incluida en parafina (FFPE). Esta prueba se ha diseñado para detectar con alta sensibilidad la mutación predominante de BRAF V600E (hasta un 5% de secuencias de V600E en un fondo de secuencias de tipo nativo en ADN-FFPE). Estudios no clínicos y clínicos con análisis retrospectivos de secuencias han demostrado que el ensayo también reconoce las mutaciones menos comunes de BRAF V600D y V600K, con menor sensibilidad. De las muestras disponibles de los estudios clínicos y no clínicos (n = 920) que habían sido identificadas con la mutación positiva en el Test de Cobas® y que, adicionalmente fueron analizadas mediante secuenciación, no se evidenciaron muestras de tipo nativo, tanto con el método de secuenciación de Sanger como con el 454. Seguridad y eficacia clínica: La eficacia de vemurafenib ha sido evaluada en 675 pacientes de un ensayo clínico Fase III (NO25026) y 278 de dos ensayos clínicos Fase II (NP 22657 y MO25743). Se requirió que todos tuvieran melanoma avanzado con mutación BRAF V600 de acuerdo con el Test de Mutación V600 Cobas® 4800. Resultados de un estudio Fase III (NO25026) en pacientes no tratados previamente: Este ensayo abierto, aleatorizado, multicéntrico, internacional, Fase III apoya el uso de vemurafenib en pacientes con melanoma metastásico o no resecable con mutación BRAF V600E positiva no tratados previamente. Los pacientes se aleatorizaron para recibir tratamiento con vemurafenib (960 mg dos veces por día) o dacarbazina (1000 mg/m2 en el día 1 de cada 3 semanas). Se distribuyeron al azar un total de 675 pacientes para recibir vemurafenib (n=337) o dacarbazina (n=338). La asignación al azar se estratificó según el estadio de la enfermedad, LDH, estado dentro de la escala de actividad del grupo oncológico cooperativo (ECOG) y la región geográfica. Las características basales estaban bien equilibradas entre los grupos de tratamiento. La mayoría de los pacientes aleatorizados para recibir vemurafenib, fueron hombres (59%) y de raza caucásica (99%), la edad mediana fue de 56 años (un 28% fueron ≥ 65 años), todos tenían un estado ECOG de 0 ó 1, y la mayor parte (66%) tenía un estadio M1c de la enfermedad. Las variables principales finales de eficacia fueron la sobrevida global (SG) y la sobrevida libre de progresión (SLP). En un análisis interno preespecificado con fecha de corte el 30 de diciembre de 2010, se observó una mejoría significativa en las variables principales del ensayo de sobrevida global (p < 0,0001) y sobrevida libre de progresión (p < 0,0001) (test de log-rank no estratificado). El Comité de Monitorización de Datos de Seguridad (Data Safety Monitoring Board (DSMB)) recomendó que estos resultados fueran publicados en enero de 2011 y que se modificara el estudio para permitir que los pacientes con dacarbazina se cruzaran al otro grupo y pudieran recibir vemurafenib. Los análisis de sobrevida post-hoc a partir de entonces se llevaron a cabo tal y como se describe en la Tabla 2.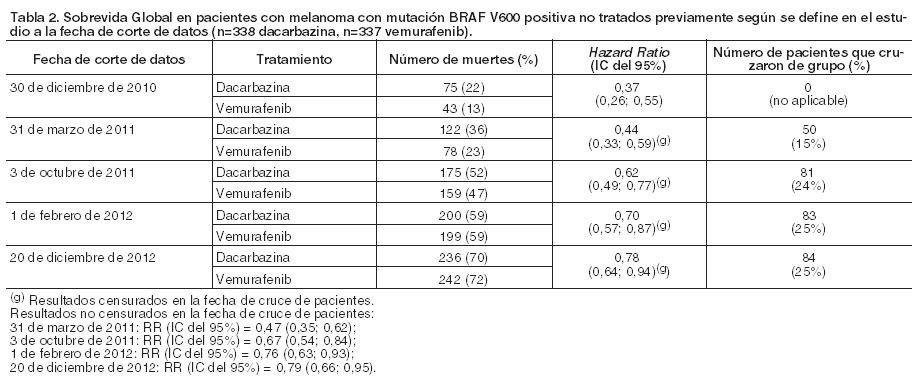
La Tabla 3 muestra el efecto del tratamiento para todas las variables de estratificación preespecificadas establecidas como factores pronóstico.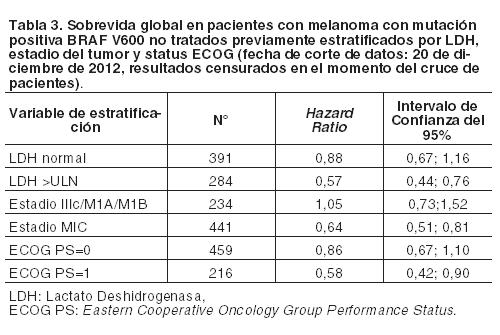
La Tabla 4 muestra la tasa de respuesta global y la sobrevida libre de progresión en pacientes con melanoma con mutación positiva BRAF V600 no tratados previamente.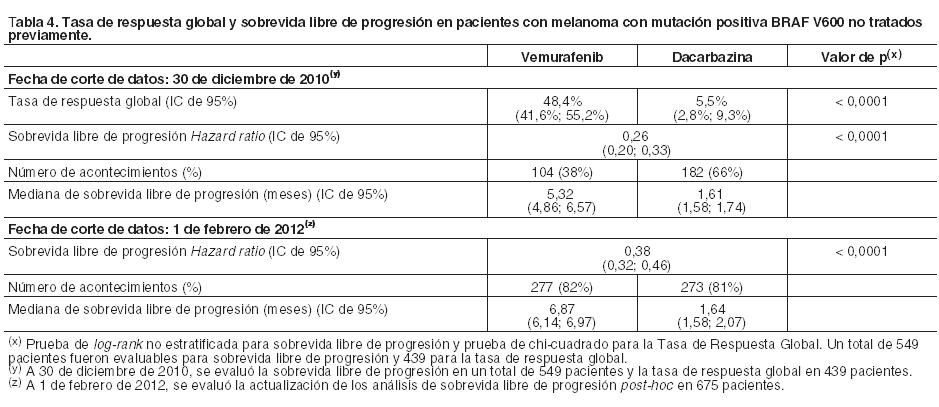
Un total de 57 pacientes de los 673 cuyos tumores fueron analizados retrospectivamente mediante secuenciación, mostraron tener melanoma con mutación positiva BRAF V600K en el ensayo NO25026. Aunque la casuística fue reducida, los análisis de eficacia de estos pacientes con tumores V600K positivo sugirieron un beneficio similar del tratamiento de vemurafenib en términos de sobrevida global, sobrevida libre de progresión y mejor respuesta global confirmada. No se dispone de datos en aquéllos con melanoma que alberguen mutaciones raras de V600 diferentes de V600E y V600K. Resultados del ensayo Fase II (NP22657) en pacientes que fracasaron al menos a un tratamiento previo: Se realizó un ensayo Fase II, de una sola rama, multicéntrico, multinacional con 132 pacientes que tenían melanoma metastásico con mutación BRAF V600E positiva de acuerdo con el Test de Cobas® 4800 para la mutación BRAF V600 y que habían recibido previamente al menos un tratamiento. La mediana de la edad fue de 52 años con un 19% de mayores de 65 años. La mayoría fue de sexo masculino (61%), de raza caucásica (99%), y con un estadio de la enfermedad M1c (61%). Un 49% había fracasado a ≥ 2 tratamientos previos. La duración media de seguimiento fue de 6,87 meses (rango, 0,6 a la 11.3). Con una mediana de seguimiento de 12,9 meses (variando desde 0,6 hasta 20,1), la variable principal de mejor tasa de respuesta global confirmada (RC+RP) tal y como fue evaluada por un Comité de Revisión Independiente (CRI) fue del 53% (IC de 95%: 44%, 62%). El tiempo medio para la respuesta fue de 1,4 meses con el 75% de las respuestas que ocurrieron a 1,6 meses de tratamiento. La duración media de la respuesta por IRC fue de 6,5 meses (IC del 95%: 5,6, no alcanzado). Se observó enfermedad estable por RECIST 1.1 en el 30% de los pacientes. La mediana de la sobrevida global fue de 15,9 meses (IC de 95%: 11,2; 19,3). La tasa de sobrevida global a los 6 meses fue del 0,77 (IC de 95%: 0,69, 0,84) y a los 12 meses del 0,58 (IC de 95%: 0,48, 0,66). La mediana de la SLP fue de 6,1 meses (IC del 95%: 5.5, 6.9), y la tasa de SLP a los 6 meses fue del 52% (IC del 95%: 43%, 61%). Nueve de los 132 pacientes incorporados en el ensayo NP22657 tuvieron tumores con mutación V600K positiva de acuerdo con el método de secuenciación de Sanger realizado en forma retrospectiva. Entre estos pacientes, 3 alcanzaron una RP, 3 tuvieron EE, 2 PE y uno no fue evaluable. Pacientes con metástasis cerebrales (MO25743): Se llevó a cabo un ensayo Fase II, de una sola rama, multicéntrico, abierto, en 146 pacientes adultos con melanoma metastásico con mutación BRAF V600 histológicamente confirmada (según el Test de Cobas® 4800 para la mutación BRAF V600) y con metástasis cerebrales. El estudio incluyó dos cohortes con reclutamiento simultáneo: Pacientes no tratados previamente (cohorte 1: N = 90): Pacientes que no habían recibido tratamiento previo para las metástasis cerebrales; estaba permitido el tratamiento sistémico previo para el melanoma metastásico, excluyendo inhibidores BRAF y MEK. Pacientes tratados previamente (cohorte 2: N = 56): Pacientes que habían sido tratados con anterioridad por sus metástasis cerebrales y que habían progresado después del tratamiento. Para pacientes tratados con radioterapia estereotáctica (SRT) o cirugía, debió desarrollarse una nueva lesión cerebral evaluable utilizando los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST) luego de esta terapia previa. Se reclutaron un total de 146 pacientes. La mediana de la edad fue de 54 años (rango 26 a 83 años) y fue similar en ambas cohortes. La mayoría de los pacientes fue de sexo masculino (61,6%) y distribuidos similarmente entre las dos cohortes. Un total de 135 (92,5%) fueron informados como de raza caucásica y no se registró la etnia de 11 pacientes (7,5%) debido a regulaciones locales. En ambas cohortes la mediana del número de lesiones cerebrales blanco al inicio fue de 2 (rango de 1 a 5). El objetivo primario del estudio fue evaluar la eficacia de vemurafenib utilizando la tasa de mejor respuesta global en el cerebro de los pacientes con melanoma metastásico con metástasis cerebrales no tratados previamente, según lo evaluado por un Comité de Revisión Independiente (CRI), utilizando los criterios RECIST versión 1.1. Los objetivos secundarios incluyeron una evaluación de la eficacia de vemurafenib usando la tasa de mejor respuesta global en el cerebro de los pacientes tratados previamente, la duración de la respuesta, la sobrevida libre de progresión y la sobrevida global en pacientes con melanoma metastásico con metástasis cerebrales (ver Tabla 5).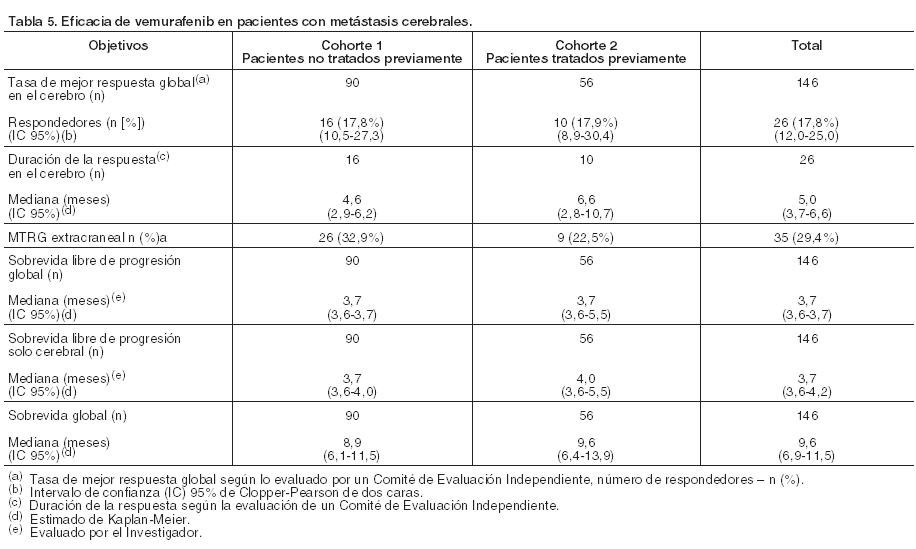
Población pediátrica: Resultados del ensayo fase I (NO25390) en población pediátrica: Se llevó a cabo un ensayo fase I con escalada de dosis que evaluó el uso de vemurafenib en seis pacientes adolescentes con melanoma en estadío IIIC o IV con mutación BRAF V600 positiva. Todos los pacientes tratados tenían al menos 15 años y pesaban 45 kg. Tres pacientes fueron tratados con 720 mg de vemurafenib dos veces al día y tres pacientes fueron tratados con 960 mg de vemurafenib dos veces al día. La dosis máxima tolerada no se pudo determinar. Aunque se observaron regresiones tumorales transitorias, la mejor tasa de respuesta global (MTRG) fue del 0% (IC 95%: 0%, 46%) basado en respuestas confirmatorias. El ensayo finalizó debido al bajo reclutamiento. Para obtener información sobre el uso pediátrico, véase Dosificación. Propiedades farmacocinéticas: Vemurafenib es un fármaco de Clase IV (baja solubilidad y permeabilidad), de acuerdo con los criterios descritos en Biopharmaceutics Classification System. Los parámetros farmacocinéticos para vemurafenib se determinaron utilizando un análisis no compartimental en un ensayo Fase I y en ensayos Fase III (20 pacientes después de 15 días de administración de dosis de 960 mg dos veces por día y 204 en el estado estacionario día 22), así como un análisis farmacocinético poblacional utilizando el conjunto de los datos de 458 pacientes. De éstos, 457 fueron de raza caucásica. Absorción: Se desconoce la biodisponibilidad absoluta de los comprimidos recubiertos de vemurafenib 240 mg. Vemurafenib se absorbe con una mediana de Tmáx de aproximadamente 4 horas después de una dosis única de 960 mg (cuatro comprimidos recubiertos de 240 mg). Vemurafenib exhibe una alta variabilidad interpaciente. En el ensayo Fase II, el ABC0-8h y la Cmáx en el día 1 fueron 22,1 ± 12,7 mg·h/ml y 4,1 ± 2,3 mg/ml. Después de la administración múltiple de dos dosis diarias de vemurafenib se produjo acumulación marcada. En el análisis no compartimental, después de dosis de 960 mg de vemurafenib dos veces por día, la relación Día 15 / Día 1 fue de 15 a 17 veces el ABC, y de 13 a 14 veces la Cmáx, obteniendo un ABC0-8h y una Cmáx de 380,2 ± 143,6 mg·h/ml y 56,7 ± 21,8 mg/ml, respectivamente, bajo condiciones de estado estacionario. La comida (con alto contenido en grasa) aumenta la biodisponibilidad relativa de una dosis única de 960 mg de vemurafenib. La relación entre las medias geométricas durante el estado de alimentación y ayuno para Cmáx y ABC fueron de 2,5 y 4,6 a 5,1 veces, respectivamente. La mediana de Tmáx incrementó de 4 a 7,5 horas cuando se administró una dosis única de vemurafenib con alimentos. Actualmente, se ignora el efecto que ejerce la alimentación en la exposición a vemurafenib en el estado estacionario. La administración constante de vemurafenib con el estómago vacío podría dar lugar a una exposición en el estado estacionario significativamente menor que cuando el fármaco se administra con o tras un corto tiempo después de la comida. Se espera que la ingesta ocasional de vemurafenib con el estómago vacío tenga una influencia limitada sobre la exposición en el estado estacionario, debido a la alta acumulación de vemurafenib en este estado. Los datos de seguridad y eficacia de los estudios pivotales se recogieron de pacientes que tomaron vemurafenib con o sin alimentos. La variabilidad en la exposición a vemurafenib puede ocurrir debido a las diferencias en el contenido del fluido gastrointestinal, volumen, pH, motilidad y tiempo de transición y composición de la bilis. En el estado estacionario alcanzado en el día 15 en el 80% de los pacientes, la exposición media de vemurafenib en plasma es estable durante el intervalo de 24 horas, según indicó el resultado de la relación promedio de 1,13 entre las concentraciones antes y 2 a 4 horas después de la dosis de la mañana. Después de la administración oral, la tasa de absorción constante para la población de pacientes con melanoma metastásico se estimó en 0,19 h-1 (con 101% de variabilidad entre pacientes). Distribución: El volumen de distribución aparente para vemurafenib en los pacientes con melanoma metastásico se estimó en 91 litros (con un 64,8% de variabilidad entre pacientes). In vitro, la unión del vemurafenib a las proteínas plasmáticas humanas es muy alta ( > 99%). Biotransformación: Las proporciones relativas de vemurafenib y de sus metabolitos se caracterizaron en un estudio de balance de masa en seres humanos con una dosis única de vemurafenib marcado con 14C administrado por vía oral. En promedio, el 95% de la dosis fue recuperada dentro de los 18 días. La mayoría (94%) en heces, con < 1% recuperado en orina. La principal enzima responsable del metabolismo de vemurafenib in vitro es CYP3A4. También se identificaron metabolitos conjugados (glucuronización y glicosilación) en seres humanos. Sin embargo, el componente predominante en plasma (95%) fue el compuesto original. Aunque el metabolismo parece que no produce una cantidad relevante de metabolitos en plasma, no se puede excluir su importancia en la excreción. La coadministración de rifampicina, un potente inductor de CYP3A4, disminuyó significativamente la exposición plasmática de vemurafenib (ABC) en aproximadamente un 40% luego de una única dosis de 960 mg de vemurafenib, sugiriendo que la vía de CYP3A4 podría ser importante en la eliminación de vemurafenib. Eliminación: El clearance aparente de vemurafenib en pacientes con melanoma metastásico se estimó en 29,3 litros/día (con un 31,9% de variabilidad entre pacientes). La vida media de eliminación poblacional estimada mediante un análisis farmacocinético poblacional para vemurafenib es de 56,9 horas (el rango del percentil 5° y 95° de la vida media estimada individual es 29,8 - 119,5 horas). En el estudio de balance de masas en seres humanos con vemurafenib administrado por vía oral, en promedio se recuperó el 95% de la dosis dentro de los 18 días. La mayoría del material relacionado con vemurafenib (94%) se recuperó en heces, y < 1% en la orina. La excreción biliar del compuesto original puede constituir una vía importante de eliminación. Sin embargo, debido a que no se conoce la biodisponibilidad absoluta, no hay certeza acerca de la importancia que puede tener la excreción renal y hepática en el clearance del compuesto original de vemurafenib. Vemurafenib es un sustrato e inhibidor in vitro de P-gp. Farmacocinética en poblaciones especiales: Pacientes pediátricos: Datos farmacocinéticos limitados de seis pacientes adolescentes de 15 a 17 años con melanoma positivo con mutación BRAF V600 en estadio IIIC o IV sugieren que las características farmacocinéticas de vemurafenib en adolescentes son similares a las de adultos. Sin embargo, no se puede hacer ninguna conclusión debido a la cantidad limitada de datos (véase Dosificación). Para obtener información sobre el uso pediátrico, véase Dosificación. Pacientes de edad avanzada: La edad no tuvo un efecto estadísticamente significativo en la farmacocinética de vemurafenib, según el análisis farmacocinético poblacional. Pacientes con insuficiencia renal: En el análisis farmacocinético poblacional utilizando los datos de los ensayos clínicos en pacientes con melanoma metastásico, la insuficiencia renal leve y moderada no influyó sobre el clearance aparente de vemurafenib (clearance de creatinina > 30 ml/min). La posible necesidad de ajustar la dosis en pacientes con insuficiencia renal grave (clearance de creatinina < 29 ml/min), no se puede determinar, ya que los datos clínicos y farmacocinéticos son insuficientes (véanse Dosificación; y Precauciones). Pacientes con insuficiencia hepática: Sobre la base de los datos preclínicos y de un estudio de balance de masa en seres humanos, la mayor parte de vemurafenib se elimina por vía hepática. En el análisis farmacocinético poblacional utilizando los datos de los ensayos clínicos en pacientes con melanoma metastásico, los aumentos en AST, ALT y bilirrubina total de hasta tres veces el límite superior del rango normal no influyeron sobre el clearance aparente de vemurafenib. La posible necesidad de ajustar la dosis en pacientes con insuficiencia hepática grave no se puede determinar, ya que los datos clínicos y farmacocinéticos son insuficientes para definir el efecto de la insuficiencia hepática a nivel metabólico o excretor sobre la farmacocinética de vemurafenib (véanse Dosificación; y Precauciones). Pacientes según su sexo: El análisis farmacocinético poblacional indicó que el clearance aparente (CL/F) era un 17% mayor y que el volumen de distribución aparente (V/F) era un 48% mayor en hombres que en mujeres. No se ha esclarecido si se trata de un efecto de sexo o de tamaño corporal. Sin embargo, las diferencias en la exposición no son lo suficientemente significativas como para justificar un ajuste de dosis en función del tamaño corporal o sexo. Datos preclínicos sobre seguridad: El perfil de seguridad preclínica de vemurafenib se evaluó en ratas, perros y conejos. Los estudios de toxicidad con dosis repetidas, identificaron el hígado y la médula ósea como órganos diana en el perro. En el ensayo llevado a cabo en perros durante 13 semanas, se observaron efectos tóxicos reversibles en el hígado (necrosis hepatocelular y degeneración hepática) con exposiciones por debajo de las previstas en la práctica clínica (en base a comparaciones de ABC). En un ensayo en perros de 39 semanas, con dosis dos veces por día, que finalizó prematuramente, se detectó necrosis de la médula ósea focal en un perro, con exposiciones similares a las previstas en la práctica clínica (en base a comparaciones de ABC). En un estudio de citotoxicidad in vitro en médula ósea, se registró una ligera citotoxicidad en algunas de las poblaciones celulares linfohematopoyéticas de ratas, perros y seres humanos, en concentraciones clínicamente relevantes. En cultivos de fibroblastos murinos después de radiación UVA, vemurafenib ha demostrado ser fototóxico in vitro, pero no en un estudio in vivo en ratas con dosis de hasta 450 mg/kg/día (con niveles inferiores a las exposiciones clínicas previstas basado en la comparación del ABC). No se han realizado investigaciones específicas con vemurafenib en animales para evaluar su efecto sobre la fertilidad. Sin embargo, en los ensayos de dosis repetidas, no se recogieron hallazgos histopatológicos en los órganos reproductores de los machos y de las hembras, en ratas y en perros en dosis de hasta 450 mg/kg/día (con niveles inferiores a las exposiciones clínicas previstas basado en la comparación de ABC). No se ha observado teratogenicidad en estudios de desarrollo embriofetal en ratas y conejos con dosis de hasta 250 mg/kg/día y 450 mg/kg/día, respectivamente, dando lugar a niveles por debajo de la exposición clínica prevista (basado en la comparación de ABC). Sin embargo, las exposiciones en los estudios de desarrollo embriofetal estuvieron por debajo de los niveles clínicos basado en la comparación de ABC, es por tanto difícil definir en qué medida estos resultados pueden ser extrapolados a seres humanos. Por consiguiente, no se puede excluir un efecto de vemurafenib en el feto. No se han realizado ensayos acerca del desarrollo pre y posnatal. No se detectaron signos de genotoxicidad en los ensayos in vitro (mutación bacteriana [test de AMES], aberraciones cromosómicas en linfocitos humanos) ni en los test in vivo en micronúcleos de médula ósea de ratas realizados con vemurafenib. No se han llevado a cabo estudios de carcinogenicidad con vemurafenib.
Indicaciones.
Vemurafenib está indicado en monoterapia para el tratamiento de pacientes adultos con melanoma no resecable o metastásico con mutación de BRAF V600 positiva (véase Farmacología; Propiedades farmacodinámicas).
Dosificación.
El tratamiento con vemurafenib debe iniciarse y ser supervisado por un médico especializado en el uso de medicamentos anticancerígenos. Antes de comenzar la administración del medicamento, los pacientes deben tener un diagnóstico de mutación BRAF V600 positiva en el tumor, confirmado por un test validado (véanse Precauciones y Farmacología, Propiedades farmacodinámicas). Posología: La dosis recomendada de vemurafenib es 960 mg (4 comprimidos de 240 mg) dos veces por día (equivalente a una dosis diaria total de 1.920 mg). La primera dosis debe tomarse por la mañana y la segunda dosis por la noche, aproximadamente 12 horas más tarde. Vemurafenib puede tomarse con o sin alimentos, pero debe evitarse el consumo constante de las dos dosis diarias con el estómago vacío (véase Farmacología; Propiedades farmacocinéticas). Duración del tratamiento: El tratamiento con vemurafenib debe continuarse hasta progresión de la enfermedad o hasta que aparezca una toxicidad no aceptable (véanse Tablas 6 y 7). Dosis olvidadas: Si se olvida tomar una dosis, puede tomarse hasta 4 horas antes de la siguiente dosis para mantener el régimen posológico de dos dosis diarias. No se deben administrar dos dosis a la vez. Vómitos: En caso de vómitos después de la administración de vemurafenib, el paciente no debe tomar una dosis adicional del medicamento, y el tratamiento se continuará como de costumbre. Ajustes de la posología: Para controlar las reacciones adversas o la prolongación del intervalo QTc puede ser necesario una reducción de la dosis, una interrupción temporal y/o la suspensión permanente del tratamiento (véanse Tablas 6 y 7). No se recomienda ajustar la posología con una dosis inferior a 480 mg dos veces por día. En caso de que el paciente desarrolle un carcinoma de células escamosas cutáneo (CCEc), se recomienda continuar el tratamiento sin modificar la dosis de vemurafenib (véanse Precauciones y Reacciones adversas).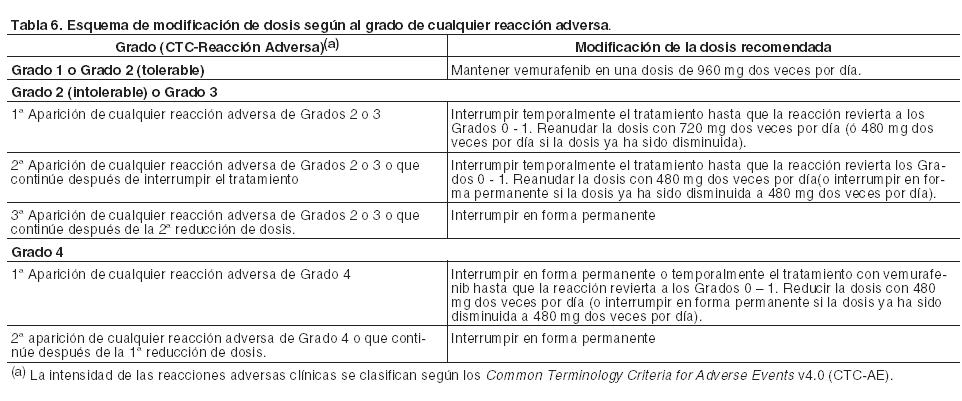
En un estudio Fase II, no controlado, abierto, en pacientes con melanoma metastásico previamente tratados, se ha observado una prolongación del intervalo QT dependiente de la exposición. El manejo de la prolongación QT puede requerir medidas específicas de monitorización (véase Precauciones).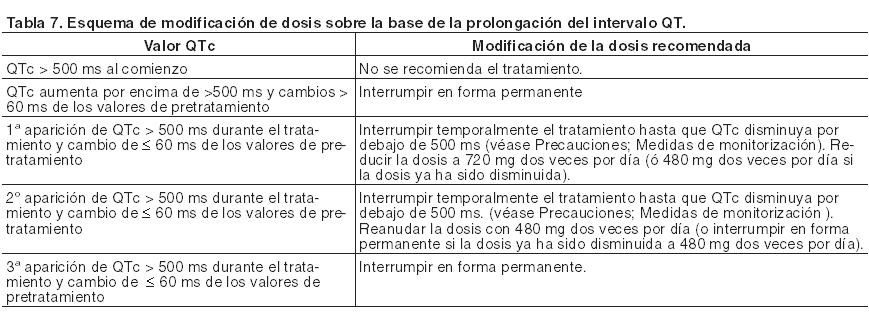
Poblaciones especiales: Pacientes pediátricos: No se ha establecido todavía la seguridad y eficacia de vemurafenib en pacientes menores de 18 años. No se dispone de datos. Vemurafenib no está aprobado para su uso en pacientes menores de 18 años (véase Farmacología; Farmacocinética en poblaciones especiales). Pacientes de edad avanzada: No se requiere un ajuste específico de la dosis en pacientes con una edad > 65 años. Pacientes con insuficiencia renal: Existe escasa información en pacientes con insuficiencia renal. No se puede descartar un aumento del riesgo de exposición en pacientes con insuficiencia renal grave, los cuales deben monitorizarse estrechamente (véanse Precauciones y Farmacología, Propiedades farmacocinéticas). Pacientes con insuficiencia hepática: Se dispone de pocos datos en pacientes con insuficiencia hepática. Los pacientes con insuficiencia hepática grave a moderada pueden tener aumentada la exposición y deben monitorizarse estrechamente, ya que el vemurafenib se elimina por el hígado (véanse Precauciones y Farmacología, Propiedades farmacocinéticas). Pacientes según su etnia: No se ha establecido todavía la seguridad y eficacia de vemurafenib en pacientes no caucásicos, dado que se carece de información. Formas de administración: Vemurafenib es para uso oral. Los comprimidos recubiertos de vemurafenib deben ingerirse enteros con agua. No deben masticarse ni partirse.
Contraindicaciones.
Zelboraf está contraindicado en pacientes con hipersensibilidad conocida a vemurafenib o a cualquiera de sus excipientes (véase Precauciones).
Reacciones adversas.
Resumen del perfil de seguridad: Las reacciones adversas más comunes ( > 30%) registradas con vemurafenib incluyen artralgia, fatiga, rash, reacciones de fotosensibilidad, náuseas, alopecia y prurito. Se notificó de manera muy frecuente carcinoma de células escamosas que en la mayoría de los casos fue tratado mediante extirpación local. Potenciación de toxicidad por radioterapia: En los reportes poscomercialización se observaron casos de hipersensibilidad y sensibilización a la radiación. Sin embargo, la frecuencia de esta reacción adversa es desconocida, dado que la información relativa a la radioterapia, incluidos los datos sobre la dosis de la radiación, no se registra en forma rutinaria en los informes espontáneos de seguridad. Las reacciones adversas que fueron informadas en los pacientes con melanoma se han enumerado bajo la clasificación de órganos del sistema MedDRA, frecuencia y grado de gravedad. La siguiente clasificación se ha utilizado para ordenarlas por frecuencia: muy frecuentes: ≥ 1/10, frecuentes: ≥ 1/100 a < 1/10, poco frecuentes: ≥ 1/1.000 a < 1/100, raras ≥ 1/10.000 a < 1/1.000 y muy raras < 1/10.000. En esta sección, las reacciones adversas a los medicamentos se basan en los resultados obtenidos en 468 pacientes adultos de un estudio abierto, aleatorizado, Fase III, con melanoma estadio IV o no resecable con mutación BRAF V600 positiva y también de un estudio Fase II, de una sola rama en pacientes con melanoma estadio IV con mutación BRAF V600 positiva, que habían fracasado previamente, por lo menos a un tratamiento sistémico anterior (véase Farmacología, Propiedades farmacodinámicas). Además, se notifican las reacciones adversas a los medicamentos procedentes de los informes de seguridad de todos los ensayos clínicos y de los reportes poscomercialización. Todos los términos incluidos están basados en los porcentajes más altos observados entre los ensayos clínicos Fases II y III. Dentro de cada categoría por órganos y sistemas, la agrupación de las reacciones adversas a los medicamentos se presentan en orden decreciente de gravedad y para la evaluación de la toxicidad se tuvo en cuenta la escala NCI-CTCAE v 4.0 (Criterios Comunes de Toxicidad).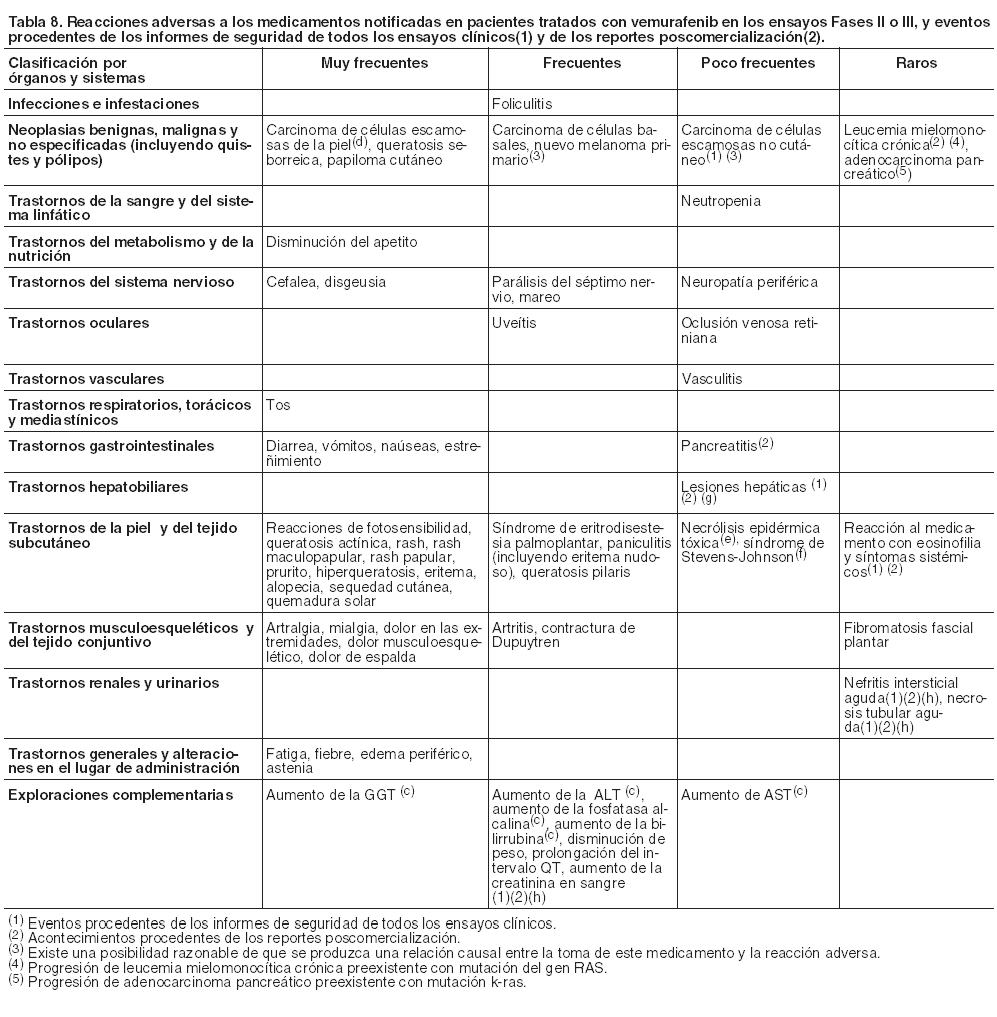
Información adicional sobre reacciones adversas seleccionadas: Aumento de las enzimas hepáticas(c): Los datos proporcionados por el ensayo clínico Fase III se expresan a continuación como la proporción de pacientes que han experimentado una modificación del valor basal al Grado 3 o 4 en las alteraciones de las enzimas hepáticas. Muy frecuentes: GGT. Frecuentes: ALT, fosfatasa alcalina, bilirrubina. Poco frecuentes: AST. La ALT, la fosfatasa alcalina o la bilirrubina, no aumentaron a Grado 4. Lesiones hepáticas(g): Sobre la base del criterio establecido por un grupo internacional de expertos clínicos y científicos, el daño hepático inducido por un fármaco quedó definido como cualquiera de las siguientes alteraciones de laboratorio: ≥ 5 x ULN ALT. ≥ 2 x ULN ALP (sin otra causa para la elevación de la ALP). ≥ 3 x ULN ALT con elevación simultánea de la concentración de bilirrubina 2 x ULN. Carcinoma cutáneo de células escamosas(d) (CCEc): Se han notificado casos de CCEc en pacientes tratados con vemurafenib. En éstos, la incidencia en todos los estudios de CCEc fue de aproximadamente el 20%. La mayoría de las lesiones extirpadas, analizadas por un laboratorio central independiente para su evaluación patológica, fueron clasificadas como un subtipo de carcinoma de células escamosas-queratoacantoma o caracterizadas como queratoacantoma mixto (52%). Muchas de las lesiones cutáneas clasificadas como "otras" (43%) fueron benignas (por ejemplo, verruga vulgar, queratosis actínica, queratosis benigna, quistes/quistes benignos). El CCEc se manifestó normalmente al comienzo del tratamiento con un tiempo mediano hasta la primera aparición de entre 7 y 8 semanas. Aproximadamente un 33% de los pacientes que sufrieron CCEc, tuvieron > 1 manifestación con un tiempo mediano entre las presentaciones de 6 semanas. Estos casos fueron normalmente tratados mediante una simple extirpación quirúrgica y los pacientes generalmente continuaron con el tratamiento sin modificar la dosis (véanse Dosificación; y Precauciones). Carcinoma de células escamosas no cutáneo (CCEnc): Se han comunicado casos de CCEnc que afectaron a pacientes tratados con vemurafenib mientras estaban participando en ensayos clínicos. La supervisión del CCEnc debe realizarse de acuerdo con lo indicado en Precauciones. Nuevo melanoma primario: En los ensayos clínicos se han informado casos de nuevos melanomas primarios, que fueron extirpados mediante cirugía, y los pacientes continuaron el tratamiento sin ajustes de dosis. La monitorización de las lesiones cutáneas debe realizarse como se indica en Precauciones. Potenciación de toxicidad por radioterapia: Los casos reportados incluyen: fenómeno evocatorio (recall), lesiones cutáneas, neumonitis, esofagitis, proctitis, hepatitis, cistitis y necrosis por radiación. Reacciones de hipersensibilidad(e): Se han notificado reacciones graves de hipersensibilidad asociadas con vemurafenib que incluyen anafilaxia. Estas manifestaciones pueden incluir Síndrome de Stevens-Johnson, rash generalizado, eritema o hipotensión. En los pacientes que experimenten reacciones graves de hipersensibilidad, se debe interrumpir permanentemente el tratamiento con vemurafenib (véase Precauciones). En un estudio clínico se registró un caso de reacción de hipersensibilidad con rash, fiebre, escalofríos e hipotensión, 8 días después del comienzo del tratamiento con 960 mg de vemurafenib dos veces por día. Síntomas similares fueron observados cuando la terapia se reinició con una dosis única de 240 mg. El paciente discontinuó permanentemente vemurafenib y se recuperó sin secuelas. Reacciones dermatológicas (f): En el ensayo clínico pivotal se han comunicado reacciones dermatológicas graves en pacientes que recibieron vemurafenib, incluyendo casos raros del Síndrome de Stevens-Johnson y de necrólisis epidérmica tóxica. En los pacientes que experimenten una reacción dermatológica grave, vemurafenib debe ser interrumpido en forma permanente. Prolongación del intervalo QT: El análisis centralizado de datos de electrocardiogramas de un subestudio QT de un estudio abierto, no controlado, Fase II con 132 pacientes con una dosis de 960 mg dos veces por día de vemurafenib (NP22657) mostró una prolongación en el intervalo QTc dependiente de la exposición. El efecto en el intervalo QTc medio permaneció estable entre 12 y 15 ms después del primer mes de tratamiento, observándose la mayor prolongación del intervalo QTc medio (15,1 ms; límite superior del IC del 95%: 17,7 ms) dentro de los 6 primeros meses (n = 90 pacientes). Dos pacientes (1,5%) desarrollaron valores absolutos de QTc > 500 ms (CTC Grado 3) desde la primera dosis del tratamiento y solamente un paciente (0,8%) experimentó un cambio en el QTc con respecto al valor basal de > 60 ms (véase Precauciones). Lesión renal aguda: Se han reportado con Zelboraf un amplio espectro de casos renales que van desde las elevaciones leves o moderadas de la creatinina a nefritis intersticial aguda y necrosis tubular aguda. Algunos de estos casos se observaron en el contexto de eventos por deshidratación, y en su mayoría, las elevaciones de creatinina fueron de leves ( > 1-1,5 x ULN) a moderadas ( > 1,5-3 x LSN) y de naturaleza reversible. En la Tabla 9 se detallan los cambios en la creatinina desde el comienzo del ensayo clínico de Fase III.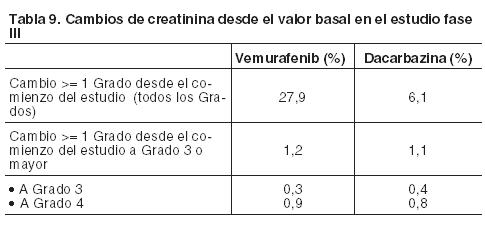
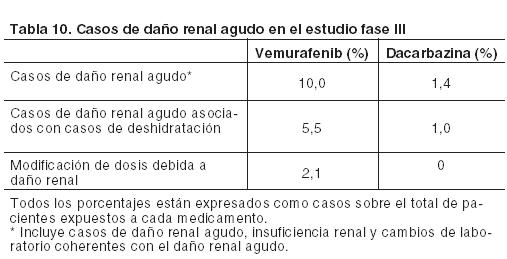
En la etapa poscomercialización se informaron alteraciones de laboratorio relacionadas con las elevaciones de la creatinina. Poblaciones especiales: Pacientes de edad avanzada: En el ensayo Fase III, 94 de los 336 pacientes (28%) con melanoma metastásico no resecable tratados con vemurafenib fueron ≥ 65 años. Los ancianos (≥ 65 años) pueden ser más susceptibles a experimentar reacciones adversas, incluyendo carcinoma cutáneo de células escamosas, disminución del apetito y alteraciones cardíacas. Pacientes según su sexo: Durante los ensayos clínicos con vemurafenib, las reacciones adversas de Grado 3 notificadas con mayor frecuencia en mujeres que en hombres, fueron rash, artralgia y fotosensibilidad. Población pediátrica: No se ha establecido la seguridad de vemurafenib en niños y adolescentes. No se observaron nuevas señales de seguridad en un estudio clínico con seis pacientes adolescentes. Comunicación de reportes de reacciones adversas: Es importante comunicar las presuntas reacciones adversas después de la autorización del medicamento. Esto permite la monitorización continua de la relación riesgo/beneficio. Se solicita a los profesionales de la salud informar sobre cualquier sospecha de eventos adversos asociados con el uso de Zelboraf al Área de Farmacovigilancia de Roche al siguiente teléfono 0800-77-ROCHE (76243). En forma alternativa, esta información puede ser reportada ante ANMAT. Ante cualquier inconveniente con el producto, el paciente puede llenar la ficha que está en la Página Web de la ANMAT: http://www.anmat.gov.ar/farmacovigilancia/Notificar.asp o llamar a ANMAT responde al 0800-333-1234.
Precauciones.
Antes de tomar vemurafenib, se debe haber confirmado por un test validado que los pacientes tienen un tumor con una mutación BRAF V600 positiva. No se ha establecido en forma convincente, la eficacia y la seguridad de vemurafenib en pacientes con tumores que expresen mutaciones BRAF V600 raras, diferentes de V600E y V600K (véase Farmacología, Propiedades farmacodinámicas). Vemurafenib no debería usarse en pacientes con melanoma maligno con BRAF de tipo nativo. Reacciones de hipersensibilidad: Se han notificado reacciones graves de hipersensibilidad asociadas con vemurafenib, incluyendo anafilaxia (véanse Contraindicaciones; y Reacciones adversas). Estas manifestaciones pueden incluir el Síndrome de Stevens-Johnson, rash generalizado, eritema o hipotensión. En los pacientes que experimenten reacciones graves de hipersensibilidad, se debe interrumpir permanentemente el tratamiento con vemurafenib. Reacciones dermatológicas: En el ensayo clínico pivotal, en pacientes que recibieron vemurafenib se han informado casos de reacciones dermatológicas graves, incluyendo casos raros del Síndrome Stevens-Johnson y necrólisis epidérmica tóxica. En la experiencia poscomercialización se comunicaron casos de erupción cutánea con eosinofilia y síntomas sistémicos (Síndrome DRESS) con el uso de Zelboraf (véase Reacciones adversas). En los pacientes que experimenten reacciones dermatológicas graves, el tratamiento con vemurafenib se debe interrumpir en forma permanente. Potenciación de la toxicidad por radioterapia: En pacientes tratados con radioterapia, ya sea antes, durante o después del tratamiento con vemurafenib, se informaron casos de hipersensibilidad y sensibilización a la radiación. En la mayoría de los casos, se observó compromiso cutáneo, pero en algunos también resultaron afectados los órganos viscerales con desenlaces mortales (véanse Interacciones y Reacciones adversas). Vemurafenib deberá usarse con precaución cuando se administre en forma simultánea o secuencial con radioterapia. Prolongación del intervalo QT: En un estudio Fase II, no controlado, abierto, en pacientes con melanoma metastásico previamente tratados, se ha observado una prolongación del intervalo QT dependiente de la exposición (véase Reacciones adversas) que puede dar lugar a un aumento del riesgo de arritmias ventriculares, incluyendo Torsade de pointes. No se recomienda el tratamiento con vemurafenib en pacientes con alteraciones no corregibles de los parámetros electrolíticos (incluyendo el magnesio), con síndrome de QT largo o que están tomando medicamentos, de los que se conoce que prolongan el intervalo QT. Antes del tratamiento con vemurafenib, después de un mes de tratamiento y después de la modificación de la dosis, se debe controlar el electrocardiograma (ECG) y los electrólitos (incluyendo el magnesio) en todos los pacientes. Se recomienda, particularmente en aquellos con insuficiencia hepática de moderada a grave, una monitorización posterior en forma mensual durante los 3 primeros meses del tratamiento, y luego con una frecuencia de 3 meses o más según se requiera clínicamente. No se aconseja iniciar el tratamiento con vemurafenib en pacientes con QTc > 500 ms. Si durante su transcurso el intervalo