XOLAIR®
BAGO
Otras drogas sistémicas para las enfermedades obstructivas del sistema respiratorio.
Composición.
Cada frasco ampolla de Xolair® 75 mg contiene: Omalizumab 75 mg. Excipientes: sacarosa 54,0 mg, L-histidina 0,7 mg, L-histidina clorhidrato monohidrato 1,0 mg, polisorbato 20: 0,2 mg. Cada ampolla de diluyente contiene: Agua para inyectables 2 mL. Cada frasco ampolla de Xolair® 150 mg contiene: Omalizumab 150 mg Excipientes: sacarosa 108,0 mg, L-histidina 1,3 mg, L-histidina clorhidrato monohidrato 2,1 mg, polisorbato 20: 0,4 mg. Cada ampolla de diluyente contiene: Agua para inyectables 2 mL.
Farmacología.
Características generales: Omalizumab es un anticuerpo monoclonal humanizado obtenido mediante la tecnología del ADN recombinante que se une selectivamente a la inmunoglobulina E humana (IgE). El anticuerpo es una IgG1 kappa que contiene, enmarcadas por las regiones humanas, regiones determinantes de complementariedad de un anticuerpo murino humanizado que se une a la IgE. Pacientes con asma alérgica: La cascada alérgica se inicia cuando el alérgeno interconecta las IgE que se fijan a los receptores FCERI (receptores con gran afinidad por la IgE) en la superficie de los mastocitos y los basófilos. Esto resulta en la desgranulación de estas células y la liberación de histamina, leucotrienos, citoquinas y otros mediadores. Estos mediadores están relacionados con la fisiopatología del asma alérgica incluyendo edema de las vías aéreas, contracción del músculo liso y alteraciones de la actividad celular asociada al proceso inflamatorio. Estos mediadores también contribuyen a los signos y síntomas de enfermedades alérgicas como la broncoconstricción, producción de moco, sibilancias, disnea, opresión torácica, congestión nasal, estornudos, picazón nasal, rinorrea, picazón y lagrimeo ocular. Xolair® se une a la IgE humana y previene la unión de la IgE a los receptores FCERI de alta afinidad reduciendo así la cantidad de IgE libre disponible para iniciar la cascada alérgica. El tratamiento con omalizumab de sujetos atópicos causó un pronunciado descenso del número de receptores FCERI. Además, la liberación in vitro de histamina de los basófilos aislados de individuos que habían recibido Xolair disminuyó cerca del 90% tras la estimulación con un alérgeno, en comparación con los valores previos al tratamiento. En los pacientes asmáticos de los estudios clínicos, las concentraciones séricas de IgE libre disminuyeron de forma dependiente de la dosis en la hora siguiente a la administración de la primera dosis y se mantuvieron constantes entre dos administraciones. La reducción media de las concentraciones séricas de IgE libre fue superior al 96% cuando se utilizaron las dosis recomendadas. Las concentraciones séricas de IgE total (es decir, unida y no unida a proteínas) aumentaron tras la primera dosis debido a la formación de complejos de omalizumab:IgE que se eliminan más lentamente que la IgE libre. Dieciséis semanas después de la primera dosis, las concentraciones séricas de IgE total eran, en promedio, unas cinco veces mayores que las cifras anteriores al tratamiento cuando se usaron ensayos convencionales. Tras interrumpir la administración de Xolair, el aumento de la concentración de IgE total y la disminución de la concentración de IgE libre inducidos por Xolair revirtieron sin que se observase ningún efecto de rebote en las concentraciones de IgE después del período de reposo farmacológico. Las concentraciones de IgE total no regresaron a sus niveles preterapéuticos sino hasta un año después de la discontinuación de Xolair. Pacientes con Urticaria Espontánea Crónica (UEC): Existen varias teorías sobre la etiología de la UEC; una de ellas apunta a un origen autoinmunitario de la enfermedad. Se han aislado anticuerpos autoinmunitarios contra la IgE y su receptor, FCERI, del suero de algunos pacientes que padecían UEC. Tales autoanticuerpos pueden activar los basófilos o mastocitos y provocar la liberación de histamina. Una de las hipótesis del mecanismo de acción de omalizumab en la UEC es que reduce las concentraciones de IgE libre en la sangre y posteriormente en la piel. Ello causa un descenso del número de receptores superficiales de la IgE y de ese modo se reduce la consiguiente transcripción de señales a través de la vía del FCERI, lo cual redunda en una inhibición de las reacciones inflamatorias y de activación celular. Como consecuencia, disminuyen la frecuencia y la intensidad de los síntomas de la UEC. Otra hipótesis es que la disminución de la cantidad circulante de IgE libre produce una desensibilización rápida e inespecífica de los mastocitos cutáneos. El descenso del número de FCERI puede contribuir a mantener la respuesta. En los estudios clínicos de pacientes con UEC, el tratamiento con omalizumab produjo una reducción, dependiente de la dosis, de la cantidad de IgE libre y un aumento de la concentración de IgE total en el suero, de forma similar a lo que se observa en los pacientes con asma alérgica. La máxima disminución de IgE libre se observó 3 días después de la primera dosis subcutánea. Tras la administración repetida del medicamento cada 4 semanas, las cifras séricas de IgE libre anteriores a cada administración permanecieron estables entre las semanas 12 y 24 de tratamiento. Las cifras séricas de IgE total aumentaron después de la primera dosis debido a la formación de complejos omalizumab:IgE de eliminación más lenta que la IgE libre. Al cabo de 12 semanas de administración repetida del medicamento en dosis de entre 75 y 300 mg cada 4 semanas, las cifras séricas medias de IgE anteriores a la administración eran dos o tres veces mayores que las cifras determinadas antes de la terapia (preterapéuticas) y permanecieron estables entre las semanas 12 y 24 de tratamiento. Tras la discontinuación de Xolair, durante un período de observación sin tratamiento de 16 semanas, las cifras de IgE libre aumentaron y las de IgE total disminuyeron hasta acercarse a los niveles preterapéuticos. Farmacocinética: Características generales: Absorción: Luego de la administración subcutánea, omalizumab se absorbe con una biodisponibilidad absoluta media del 62%. La farmacocinética de omalizumab es lineal a dosis mayores a 0,5 mg/Kg. Distribución: In vitro, omalizumab forma complejos de tamaño limitado con la IgE. Tanto in vitro como in vivo no se han observado complejos que precipitaran ni complejos con pesos moleculares superiores a 1x106 daltons. Los estudios de distribución tisular en monos no evidenciaron una captación específica de I125- omalizumab por parte de ningún órgano o tejido. Eliminación: La depuración de omalizumab comprende procesos de depuración de IgG y de depuración a través de uniones específicas y de formación de complejos con su ligando específico, la IgE. La eliminación hepática de IgG incluye una degradación en las células endoteliales y reticuloendoteliales del hígado. La IgG inalterada se elimina asimismo en la bilis. En los estudios con ratones y monos, los complejos de omalizumab:IgE se eliminaron a través de interacciones con los receptores Fcc en el sistema reticuloendotelial del hígado a una velocidad generalmente mayor que la depuración de las IgG. Pacientes con asma alérgica: Absorción: Después de administrar una dosis subcutánea única a pacientes adultos o adolescentes con asma, omalizumab se absorbe con lentitud y alcanza su mayor concentración sérica al cabo de 7 u 8 días, en promedio. Tras la administración de dosis repetidas de omalizumab, el área bajo la curva de concentración sérica y tiempo desde el día 0 al 14 en el estado estacionario llegó a ser hasta seis veces mayor que la obtenida después de administrar la primera dosis. Distribución: Luego de la administración subcutánea, el volumen aparente de distribución de omalizumab en los pacientes asmáticos es de 78 ± 32 ml/Kg. Eliminación: En los pacientes asmáticos, la semivida de eliminación sérica de omalizumab promedia los 26 días y su depuración aparente es de 2,4 ± 1,1 ml/Kg/día, en promedio. La duplicación del peso corporal hace que la depuración aparente sea aproximadamente el doble. Edad, raza o grupo étnico, género e índice de masa corporal: Se analizó la farmacocinética poblacional de omalizumab para determinar la influencia de las características demográficas. Los análisis de estos datos indican que no es necesario ajustar la dosis en los pacientes asmáticos en función de la edad (6-76 años), la raza, el grupo étnico, el género o el índice de masa corporal. Pacientes con urticaria espontánea crónica (UEC): Absorción: Tras la administración de una dosis subcutánea única a pacientes adultos o adolescentes con UEC, el omalizumab se absorbió con lentitud y alcanzó su mayor concentración sérica al cabo de 6 u 8 días, en promedio. En pacientes con UEC, el omalizumab presenta una farmacocinética lineal en la gama de dosis de 75 a 600 mg administradas una sola vez por vía subcutánea. Luego de la administración de dosis de 75, 150 o 300 mg cada 4 semanas, la concentración sérica mínima de omalizumab aumenta de forma proporcional a la dosis. Distribución: Según un estudio de farmacocinética poblacional, la distribución de omalizumab en los pacientes con UEC es similar a la de los pacientes con asma alérgica. Eliminación: Las simulaciones farmacocinéticas poblacionales indicaron que, en los pacientes con UEC, la semivida de eliminación sérica del omalizumab promedia los 24 días en el estado estacionario y la depuración aparente en el estado estacionario, los 240 ml/día (que corresponde a 3,0 ml/kg/día en un paciente de 80 kg). Edad, raza o grupo étnico, género e índice de masa corporal, IgE basal, autoanticuerpos anti- FCERI, medicación concomitante: Los efectos de las covariables demográficas y de otros factores sobre la exposición a omalizumab se evaluaron mediante análisis de farmacocinética poblacional. También se evaluaron los efectos de covariables analizando la relación que existía entre las distintas concentraciones de omalizumab y las respuestas clínicas. Tales análisis han revelado que no hace falta ajustar la dosis en los pacientes con UEC en función de la edad (12 a 75 años), raza o grupo étnico, género, peso corporal, índice de masa corporal, niveles de IgE basal, autoanticuerpos anti-FCERI o el uso simultáneo de antihistamínicos H2 o de antagonistas de los receptores de leucotrienos. Alteración hepática o renal: No se dispone de datos farmacocinéticos ni farmacodinámicos en pacientes con disfunción renal o hepática que padezcan asma alérgica o urticaria espontánea crónica (Ver Precauciones). Estudios Clínicos: Asma alérgica: Adultos y adolescentes > 12 años de edad: Se evaluó la seguridad y la eficacia de Xolair® en cinco ensayos multicéntricos, controlados con placebo, con diseño de doble ciego y distribución aleatoria. En los dos estudios idénticos de 16 semanas de duración (estudios 1 y 2), quedaron demostradas la seguridad y la eficacia de omalizumab como tratamiento complementario en 1071 pacientes asmáticos alérgicos, que eran sintomáticos pese al tratamiento con corticoesteroides inhalados (dipropionato de beclometasona de 500 mg a 1200 mg/día). En ambos ensayos omalizumab fue superior a placebo con respecto al criterio principal de agudización asmática (agravamiento del asma con necesidad de corticoesteroides sistémicos o una duplicación de la dosis de beclometasona inicial del paciente). El número de agudizaciones del asma fue significativamente inferior en el grupo de omalizumab (p=0,006 y p < 0,001 en los estudios 1 y 2, respectivamente). Menos pacientes tratados con omalizumab experimentaron agudizaciones del asma (14,6% frente al 23,3%, p=0,009 en el estudio 1 y 12,8% frente al 30,5%, p < 0,001 en el estudio 2). En las fases de extensión doble ciego de ambos estudios de hasta un año de duración, se siguió registrando una menor frecuencia de agudizaciones asmáticas en los pacientes tratados con omalizumab con respecto a placebo. En los estudios 1 y 2, se pudo demostrar una mejoría clínicamente importante de la calidad de vida del paciente asmático -valorada por medio del «Cuestionario de calidad de vida en pacientes con asma» de Juniper- al final del ensayo principal de 28 semanas de duración en el grupo de Xolair, en comparación con placebo (diferencia con respecto a placebo p ≤0,001 en los estudios 1 y 2). En el estudio 3, se demostró la seguridad y el efecto de «evitación de corticoesteroides» de omalizumab en 246 pacientes que padecían de asma alérgica aguda que necesitaban tratamiento diario con corticoesteroides inhalados en dosis altas (fluticasona ≥1000 mg/día) y en quienes se permitieron agonistas b2 de acción prolongada. El estudio incluyó una fase estable de 16 semanas con corticoesteroides a los que se añadió la medicación de estudio, seguida por una fase de reducción de corticoesteroides de 16 semanas. La reducción porcentual de la dosis de corticoesteroides inhalados al final de la fase de tratamiento fue notoriamente mayor en los pacientes tratados con omalizumab que en los que recibieron placebo (mediana del 60% frente al 50%, p=0,003). La proporción de pacientes tratados con omalizumab que pudieron disminuir su dosis de fluticasona a ≤500 mg/día fue del 60,3%, frente al 45,8% en el grupo de placebo (p > 0,05). En el estudio 4, se pudo demostrar la seguridad y la eficacia de omalizumab en 405 pacientes que padecían rinitis alérgica perenne y asma alérgica concurrente. Los pacientes del estudio padecían tanto de asma alérgica sintomática como de rinitis alérgica perenne. Dichos pacientes recibieron omalizumab o placebo durante 28 semanas como tratamiento complementario de ≥400 mg de budesonida administrada con el dispositivo Turbohaler. Se permitió el uso de agonistas b2 inhalados de acción prolongada (39%) y corticoesteroides nasales (17%). En el estudio 4, otros criterios igualmente importantes de valoración fueron la incidencia de agudizaciones del asma (agravamiento del asma que necesitaba corticoesteroides sistémicos o la duplicación de la dosis de budesonida inicial del paciente) y la proporción de pacientes de cada grupo terapéutico que habían mejorado ≥1,0 desde el inicio al final de la fase de tratamiento en ambas evaluaciones de la calidad de vida específicas del asma y la rinitis (Evaluación de la calidad de vida de Juniper). Los pacientes tratados con omalizumab presentaban una incidencia significativamente menor de agudizaciones asmáticas que los pacientes del grupo de placebo (20,6% con omalizumab, frente a 30,1% con placebo, p=0,02) y hubo una proporción notoriamente mayor de pacientes tratados con omalizumab que con placebo que mejoraron ≥1,0 puntos según ambas evaluaciones de la calidad de vida específicas del asma y la rinitis (57,7% con omalizumab, frente a 40,6% con placebo, p < 0,0001). La reducción de las agudizaciones y las mejoras de la calidad de vida de los pacientes tratados con omalizumab se observaron en el contexto de mejorías estadísticamente significativas en los síntomas del asma y la rinitis y la función pulmonar, en comparación con placebo. El estudio 5, de 28 semanas de duración, demostró la eficacia y la seguridad de Xolair en 419 pacientes de 12 a 79 años de edad con asma alérgica grave que presentaban una reducción de la función pulmonar (Volumen Espiratorio Máximo en el Primer Segundo [FEV1]: 40-80% del previsto) y un control insuficiente de los síntomas asmáticos a pesar de recibir tratamiento con > 1000 mg de dipropionato de beclometasona (o su equivalente) asociado a un agonista b2 de acción prolongada. Los pacientes admitidos en el estudio habían padecido múltiples agudizaciones del asma que exigían tratamiento con corticoesteroides sistémicos o habían sido hospitalizados o atendidos en un Servicio de urgencias a causa de una agudización asmática intensa el año anterior, pese a su tratamiento continuo con corticoesteroides inhalados en dosis elevadas y agonistas b2 de acción prolongada. Se administró Xolair o placebo por vía subcutánea como tratamiento complementario de > 1000 mg (o cantidad equivalente) más un agonista b2 de acción prolongada. Se permitió la administración de un tratamiento de mantenimiento a base de corticoesteroides orales (22%), teofilina (27%) o antileucotrienos (35%). En la fase de tratamiento no se modificó la terapia antiasmática concomitante. El porcentaje de agudizaciones del asma que exigían tratamiento con tandas cortas de corticoesteroides sistémicos fue el criterio principal de valoración. Omalizumab redujo el porcentaje de agudizaciones asmáticas en un 19% (p=0,153). En otras evaluaciones que pusieron de manifiesto una significación estadística a favor de Xolair® (p < 0,05) se registraron reducciones de las exacerbaciones agudas (en las que la función pulmonar del paciente había sido inferior al 60% del óptimo individual, lo cual necesitó corticoesteroides sistémicos), así como de las consultas de urgencia relacionadas con el asma (consultas médicas no programadas, consultas al Servicio de urgencias y hospitalizaciones), y se apreciaron mejoras en la evaluación general del médico de la eficacia del tratamiento, la calidad de vida del paciente asmático, los síntomas asmáticos y la función pulmonar. Los cinco estudios mencionados anteriormente incluyeron una evaluación general del médico como medida amplia de control del asma. El médico tomó en consideración el Pico de Flujo Expiratorio (PEF), los síntomas diurnos y nocturnos, el uso de una medicación de rescate, la espirometría y las agudizaciones. En los cinco estudios sin excepción una proporción significativamente mayor de pacientes tratados con Xolair® logró alcanzar una marcada mejoría o un control completo del asma en comparación con los pacientes que utilizaron placebo. Niños de 6 años a < 12 años: La principal confirmación de la seguridad y la eficacia de Xolair® en el grupo de edad de 6 años a < 12 años se obtuvo en un ensayo multicéntrico, aleatorizado, controlado con placebo, doble ciego (estudio 6) y en otro estudio confirmatorio (estudio 7). El estudio 6 fue un estudio de 52 semanas de duración que evaluó la seguridad y la eficacia de Xolair® como tratamiento complementario en 628 pacientes con asma alérgica que no estaban bien controlados pese a seguir tratamiento regular con corticoesteroides inhalados (≥200 mg/d de fluticasona en inhalador de polvo seco o su equivalente), con o sin otros tratamientos antiasmáticos. Los pacientes seleccionados para participar en el estudio padecían asma desde hacía más de un año, tenían resultados positivos de al menos un aeroalérgeno perenne en las pruebas alérgicas cutáneas y presentaban antecedentes de manifestaciones clínicas de asma persistente moderada o grave (diurnas o nocturnas), así como antecedentes de agudizaciones en el año previo a su admisión en el estudio. Se permitió el uso de agonistas b2 de acción prolongada (67,4%), antileucotrienos (36,6%) y corticoesteroides orales (1,3%) como tratamiento de fondo. Durante las 24 primeras semanas de tratamiento se mantuvieron constantes las dosis iniciales de corticoesteroides de cada paciente y durante las 28 semanas siguientes se permitió un ajuste de los corticoesteroides inhalados. Se definió una agudización clínicamente significativa como un empeoramiento de los síntomas asmáticos, según el criterio clínico del investigador, que exigió duplicar la dosis inicial de corticoesteroides inhalados al menos durante 3 días, usar tratamiento de rescate con corticoesteroides sistémicos (orales o intravenosos) durante al menos 3 días o ambos. Las tasas de agudizaciones asmáticas durante el período de tratamiento doble ciego de 52 semanas en los pacientes tratados con Xolair® que presentaban un FEV1 > 80% al inicio del estudio mostraron reducciones relativas del 43% en comparación con placebo (p < 0,001). En los pacientes tratados con Xolair® se observó, en comparación con aquellos que recibieron placebo, una reducción estadísticamente significativa de la tasa de agudizaciones asmáticas, independientemente del uso concomitante de agonistas b2 de acción prolongada al inicio del estudio, que supuso una reducción del 45% en los pacientes que usaron agonistas b2 de acción prolongada y una disminución del 42% en quienes no los utilizaron (p < 0,001 y p=0,011, respectivamente). El estudio 7 fue un estudio de 28 semanas de duración, controlado, doble ciego, que evaluó principalmente la seguridad en 334 pacientes que estaban bien controlados con corticoesteroides inhalados. Durante las 16 primeras semanas se mantuvieron las mismas dosis de esteroides que al principio del estudio; a continuación tuvo lugar un período de reducción de la dosis de esteroides de 12 semanas. El estudio evaluó la reducción porcentual de la dosis de dipropionato de beclometasona y la proporción de pacientes en los que se disminuyó la dosis de dipropionato de beclometasona a las 28 semanas. La reducción porcentual de la dosis de dipropionato de beclometasona a las 28 semanas fue mayor en el grupo de Xolair® que en el grupo de placebo (mediana de la reducción: 100% frente al 66,7%; p=0,001), así como la proporción de pacientes con una reducción de la dosis de dipropionato de beclometasona (p=0,002). La frecuencia e incidencia de episodios de agudización asmática durante la fase de reducción de la dosis de esteroides también fue menor en el grupo de omalizumab (tasa media de 0,42 frente a 0,72, p < 0,001; porcentaje de pacientes con agudizaciones del 18% frente al 39%, p < 0,001). Durante las 16 primeras semanas del período de tratamiento de 24 semanas fue evidente la tendencia a la superioridad de omalizumab con respecto a la reducción de la frecuencia y la incidencia de agudizaciones. El 55,7% de los pacientes tratados con omalizumab tuvieron una reducción completa (100%) de la dosis de corticoesteroides al final del período de tratamiento de 28 semanas, en comparación con el 43,2% en los pacientes que recibieron placebo. Además, el número de pacientes en los que se redujo la dosis de corticoesteroides ≥50% fue mayor en el grupo de omalizumab que en el grupo de placebo (80,4% frente al 69,5%, p=0,017). Los dos estudios mencionados anteriormente (6 y 7) incluyeron una evaluación general realizada por el médico terapista como medida amplia del control del asma. El médico tuvo en cuenta el flujo espiratorio máximo, los síntomas diurnos y nocturnos, el uso de tratamientos de rescate, la espirometría y las agudizaciones. En ambos estudios, la proporción de pacientes que presentaron una mejoría importante o un control completo del asma fue significativamente mayor en los pacientes tratados con Xolair que en los que recibieron placebo. Urticaria espontánea crónica (UEC): El programa de desarrollo clínico de fase III en urticaria espontanea crónica comprendió los siguientes tres estudios comparativos con placebo, con grupos paralelos, aleatorizados, doble ciegos y multicéntricos: Q4881g, Q4882g y Q4883g. En los estudios Q4881g y Q4882g, se evaluaron la eficacia y la seguridad de la administración de 75, 150 ó 300 mg de Xolair cada 4 semanas durante 24 y 12 semanas, respectivamente, con un período de observación sin tratamiento de 16 semanas, en pacientes (12-75 años) con UEC resistente pese a su tratamiento con antihistamínicos H1. En el estudio Q4883g, se evaluaron la seguridad y la eficacia de la administración de 300 mg de Xolair cada 4 semanas durante 24 semanas, con un período de observación sin tratamiento de 16 semanas, en pacientes (12-75 años) con UEC resistente pese a su tratamiento con antihistamínicos H1 o H2 o con un antagonista del receptor de leucotrienos.
En los estudios Q4881g y Q4882g, la dosis de 75 mg no siempre permitió satisfacer el criterio principal de eficacia (variación de la PIP semanal con respecto al inicio al cabo de 12 semanas) u otros criterios secundarios. Se estimó que no era eficaz y por eso mismo no se abordará más aquí. Variación de la PIP semanal con respecto al inicio al cabo de 12 semanas: Ambas dosis de 150 y 300 mg satisficieron el criterio de eficacia principal (variación de la PIP semanal con respecto al inicio al cabo de 12 semanas) en los estudios Q4881g y Q4882g, y lo mismo sucedió con la dosis de 300 mg en el estudio Q4883g (ver Tabla 2).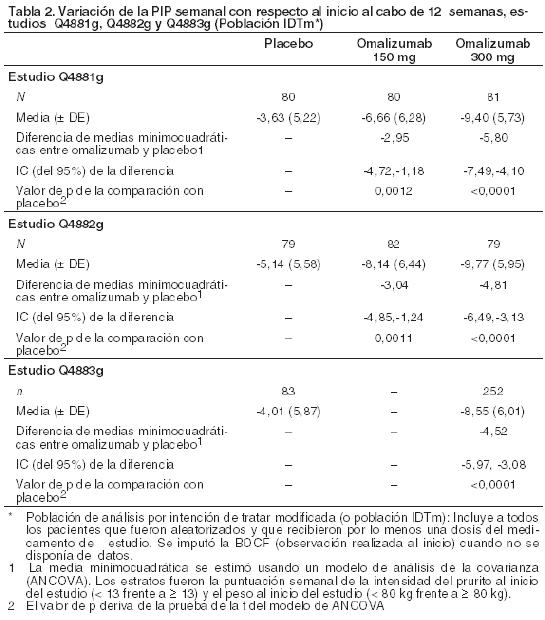
La Figura 1 presenta la puntuación semanal media de la intensidad del prurito obtenida a lo largo del estudio Q4881g. Las puntuaciones semanales medias de la intensidad del prurito disminuyeron considerablemente en ambos grupos terapéuticos; el efecto máximo se registró alrededor de la semana 12 y fue constante durante el período de tratamiento de 24 semanas. Los estudios Q4883g (administración de 300 mg durante el período de tratamiento de 24 semanas) y Q4882g (administración de 150 ó 300 mg durante el período de tratamiento de 12 semanas) arrojaron resultados similares a los del estudio Q4881g. En los tres estudios (ver la Figura 1 correspondiente al estudio Q4881g), la puntuación semanal media de la intensidad del prurito aumentó gradualmente con ambas dosis de omalizumab durante el período de observación sin tratamiento de 16 semanas, lo cual es indicativo de una recidiva de los síntomas. Los valores medios al final del período de observación fueron similares a los del grupo del placebo, pero menores que los respectivos valores medios iniciales.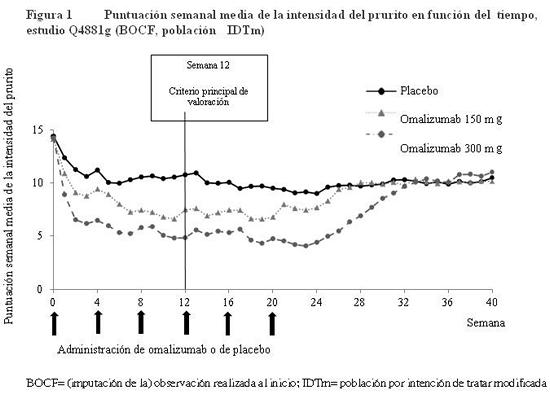
Tiempo transcurrido hasta observar una DMI en la PIP semanal durante el período de 12 semanas: En los estudios Q4881g y Q4882g, el tiempo mediano transcurrido hasta observar una DMI de 5 puntos en la puntuación semanal de la intensidad del prurito fue de 2 semanas en el grupo de 150 mg (p=0,0301 en el estudio Q4881g; p=0,0101 en el estudio Q4882g), de 1 semana (p < 0,0001) en el grupo de 300 mg y de 4 semanas con placebo. Similares resultados se obtuvieron en el estudio Q4883g, en el que la DMI se observó al cabo de una mediana de 2 semanas en el grupo de 300 mg (p < 0,0001) o de 5 semanas en el grupo del placebo. Variación de la PAU7 con respecto al inicio al cabo de 12 semanas: En los estudios de fase III, se apreció una diferencia estadísticamente significativa entre los grupos tratados con omalizumab (150 o 300 mg) y el grupo de placebo en la variación media con respecto al inicio de la PAU7 al cabo de 12 semanas (Figura 2 del estudio Q4881g). Se obtuvieron resultados estadísticamente significativos (p < 0,0001) en el grupo de 300 mg de los tres estudios y en el grupo de 15 mg de los estudios Q4881g (p=0,0008) y Q4882g (p=0,0001). La Figura 2 muestra la PAU7 media a lo largo del estudio Q4881g y revela una disminución significativa con respecto a los valores iniciales en ambos grupos terapéuticos, con un efecto máximo alrededor de la semana 12. La magnitud del efecto se mantuvo durante el período de tratamiento de 24 semanas. En los estudios Q4883g (300 mg administrados durante 12 semanas) y Q4882g (150 ó 300 mg administrados durante 24 semanas) se obtuvieron resultados similares a los del estudio Q4881g. En los tres estudios (ver la Figura 2 correspondiente al estudio Q4881g), la PAU7 aumentó gradualmente en ambos grupos del omalizumab durante el período de observación sin tratamiento de 16 semanas, lo cual era indicativo de una recidiva de los síntomas. Los valores medios al final del período de observación fueron similares a los del grupo del placebo, pero menores que los respectivos valores medios iniciales.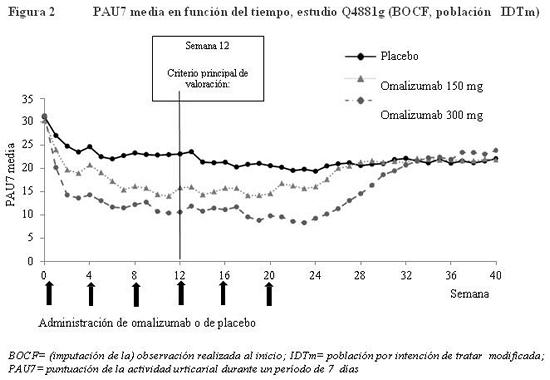
Proporción de pacientes con una PAU7 ≤ 6 al cabo de 12 semanas: Los porcentajes de respuesta PAU7 ≤6 al cabo de 12 semanas fueron todos estadísticamente significativos, de 52-66% en el grupo de 300 mg (51,9% en el estudio Q4881g, 65,8% en el estudio Q4882g y 52,4% en el estudio Q4883g; p < 0,0001) o de 40-43% en el grupo de 150 mg (40,0% en el estudio Q4881g, 42,7% en el estudio Q4882g; p < 0,001), en comparación con 11-19% del grupo de placebo (11,3% en el estudio Q4881g, 19,0% en el estudio Q4882g y 12,0% en el estudio Q4883g). Proporción de pacientes con una PAU = 0 al cabo de 12 semanas: La proporción de pacientes que habían respondido por completo al tratamiento (PAU7 = 0) fue estadísticamente significativa en el grupo de 300 mg al cabo de 12 semanas -a saber, de 34- 44% (35,8% en el estudio Q4881g, 44,3% en el estudio Q4882g y 33,7% en el estudio Q4883g, todos con una p < 0,0001)- y mayor que la observada en el grupo de 150 mg, que fue de 15,0% en el estudio Q4881g y de 22,0% en el estudio Q4882g, en comparación con 5-9% del grupo de placebo (8,8% en el estudio Q4881g, 5,1% en el estudio Q4882g y 4,8% en el estudio Q4883g). Variación de la puntuación semanal del número de ronchas con respecto al inicio al cabo de 12 semanas: En los tres estudios de fase III, la variación media de la puntuación semanal del número de ronchas con respecto al inicio al cabo de 12 semanas en los grupos de tratamiento con 300 mg fue estadísticamente significativa (p < 0,001); todos ellos presentaban un descenso de la puntuación del número de ronchas en comparación con el grupo de placebo (-11,35 en el estudio Q4881g, -11,97 en el estudio Q4882g y -10,46 en el estudio Q4883g y de -4,37, -5,22 y -4,49 en los respectivos grupos de placebo). En los grupos de tratamiento con 150 mg, la variación media fue de -7,78 (p=0,0017) en el estudio Q4881g y de -9,75 (p < 0,001) en el estudio Q4882g. Proporción de días transcurridos sin angioedema desde la semana 4 hasta la semana 12: En los tres estudios de fase III, desde la semana 4 hasta la semana 12 de tratamiento, los pacientes del grupo de 300 mg gozaron sistemáticamente de la mayor proporción media de días transcurridos sin angioedema (96,1% en el estudio Q4881g; 95,5% en el estudio Q4882g; 91% en el estudio Q4883g; todos los valores p < 0,001) en comparación con el grupo de placebo (88,2%, 89,2%, 88,1%, respectivamente). En los grupos de tratamiento con 150 mg, durante el mismo período, la proporción media de días transcurridos sin angioedema en los estudios Q4881g y Q4882g fue de 89,6% y 91,6% respectivamente, y no se apreció ninguna diferencia estadísticamente significativa con respecto a placebo. Variación del ICVD (Índice de Calidad de Vida en Dermatología) general con respecto al inicio al cabo de 12 semanas: En los tres estudios de fase III, la variación media con respecto al inicio del ICVD general en los grupos de tratamiento con 300 mg fue mayor que la observada en el grupo de placebo, con una diferencia estadísticamente significativa (p < 0,001) entre ambos grupos. En el estudio Q4881g se apreció una mejora de 10,3 puntos, en el Q4882g, de 10,2, y en el Q4883g, de 9,7, mientras que la mejora en los correspondientes grupos de placebo fue de 6,1, 6,1 y 5,1, respectivamente. En los grupos de tratamiento con 150 mg, la variación media fue de 8,0 puntos (p=0,2286) en el estudio Q4881g, de 8,3 puntos (p=0,0215) en el estudio Q4882g y de 6,1 en cada uno de los respectivos grupos de placebo. Eficacia tras 24 semanas de tratamiento: La Tabla 3 recoge los resultados obtenidos al cabo de 24 semanas de tratamiento. La magnitud de la respuesta al tratamiento fue similar a la que se apreció tras 12 semanas de tratamiento.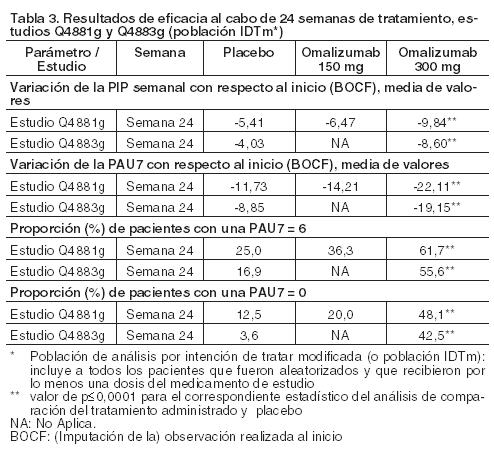
Datos de seguridad preclínicos: No se han observado signos de reacciones anafilácticas sistémicas por desgranulación de los mastocitos en monos adultos o jóvenes. En todos los estudios con primates se hallaron complejos circulantes de omalizumab:IgE, pero tras la administración de omalizumab no se encontraron signos de enfermedad mediada por inmunocomplejos en órgano alguno (incluido el riñón). Los complejos de omalizumab:IgE no fijan complemento, ni median la citotoxicidad dependiente del complemento. La administración repetida de dosis de hasta 250 mg de omalizumab/Kg (al menos 14 veces la dosis clínica máxima recomendada) fue bien tolerada en primates adultos y jóvenes, salvo la disminución dosis-dependiente del número de trombocitos observada en algunas especies con concentraciones séricas generalmente superiores a la exposición humana máxima que se utilizó en los estudios clínicos fundamentales. Los monos jóvenes fueron más sensibles que los adultos a estos efectos trombocíticos. Además, los monos mostraban signos de inflamación y hemorragia aguda en el lugar de la inyección, indicativos de una reacción inmunitaria local a la administración subcutánea repetida de una proteína heteróloga. No se han realizado estudios formales del poder cancerígeno de omalizumab. Se han detectado anticuerpos anti-omalizumab en algunos monos tras la administración subcutánea o intravenosa. Cabe esperar que ello suceda al administrar una proteína heteróloga. Algunos animales no pudieron ser evaluados debido a las concentraciones séricas elevadas de omalizumab, a las concentraciones elevadas de IgE o a ambas a la vez. No obstante, los animales mantuvieron las elevadas concentraciones séricas de omalizumab durante el período de tratamiento de los estudios, y no hubo signos evidentes de toxicidad debido a la presencia de anticuerpos contra omalizumab. Los estudios de la función reproductora, lactancia y fecundidad se describen en el apartado Embarazo y lactancia.
Indicaciones.
Asma alérgica: Xolair® está indicado para el tratamiento del asma alérgica persistente moderada o severa en pacientes adultos y niños (de 6 años en adelante) cuyos síntomas no sean adecuadamente controlados con corticoides inhalatorios. En estos pacientes Xolair® ha demostrado que disminuye la incidencia de exacerbaciones. No se ha demostrado la inocuidad o la eficacia del medicamento en otros procesos alérgicos. Urticaria Espontánea Crónica: Xolair® está indicado como terapia de combinación para el tratamiento de urticaria espontanea crónica en pacientes adultos y adolescentes (de 12 años en adelante) con respuesta inadecuada al tratamiento de antihistamínicos H1.
Dosificación.
Este producto debe administrarse únicamente por vía subcutánea. No debe administrarse por vía intravenosa ni intramuscular. Posología en asma alérgica: La dosificación y frecuencia de administración de Xolair® se determina por los niveles de Inmunoglobulina E (IgE) (UI/mL) basales medidos antes del inicio del tratamiento y el peso corporal (Kg). Antes de la dosis inicial se debe determinar el nivel de IgE sérico con cualquier método comercial para determinación de IgE libre total. Según esta determinación 75-600 mg de Xolair® en 1 a 4 inyecciones pueden ser necesarias para cada administración. Ver tabla 4 para la conversión de dosis a número de ampollas y volumen a administrar y tablas 5 y 6 para la determinación de dosis en dosis en niños (6 años a < 12 años) y en adultos y adolescentes (≥12 años). Para dosis de 225, 375 ó 525 mg Xolair® 150 mg debe utilizarse en combinación con Xolair® 75 mg. No se debe administrar Xolair® a pacientes cuyo nivel basal de IgE o cuyo peso corporal excedan los límites indicados en la tabla de administración.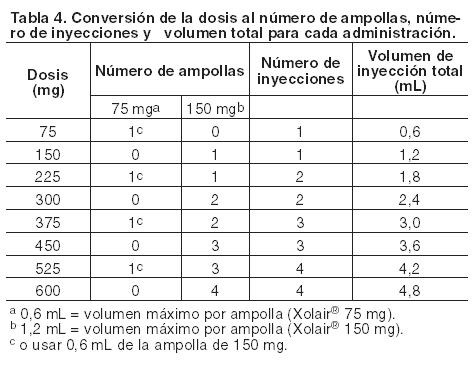
Duración del tratamiento, monitorización terapéutica y ajuste de dosis: En los estudios clínicos hubo una reducción de las exacerbaciones y utilización de medicación de rescate acompañado de una reducción de la sintomatología durante las primeras 16 semanas de tratamiento. Se requieren al menos 12 semanas para evaluar adecuadamente si un paciente responde al tratamiento con Xolair. Xolair® es un tratamiento de largo plazo. La discontinuación generalmente resulta en un retorno a niveles elevados de IgE y síntomas asociados. Los niveles de IgE total (IgE libre + complejos omalizumab:IgE) se elevan durante el tratamiento y permanecen elevados hasta 1 año después de la discontinuación del tratamiento. Por lo tanto una nueva determinación de la IgE durante el tratamiento con Xolair® no puede utilizarse como guía para establecer la dosis. La dosificación luego de una interrupción del tratamiento menor a 1 año debe estar basada en los niveles de IgE séricos obtenidos al momento de la determinación inicial de la dosis. Los niveles de IgE séricos pueden ser medidos nuevamente para establecer la dosis si el tratamiento con Xolair® ha sido interrumpido durante 1 año o más tiempo. Las variaciones significativas del peso corporal exigen una adaptación posológica (ver las tablas 5 y 6).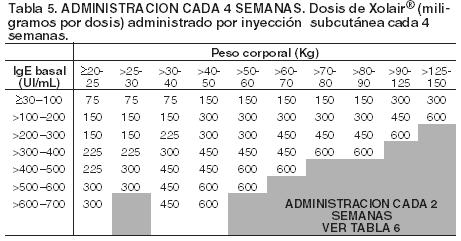
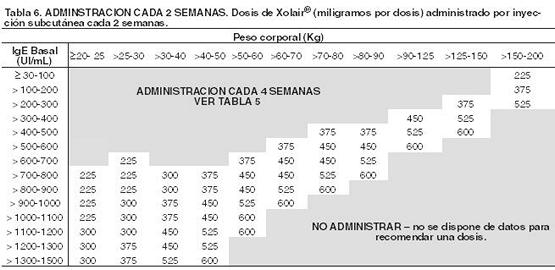
Posología en Urticaria Espontánea Crónica (UEC): La dosis recomendada es de 300 mg, que se administra cada cuatro semanas por inyección subcutánea. Algunos pacientes pueden lograr un control de sus síntomas con una dosis de 150 mg administrada cada cuatro semanas. Se recomienda a los médicos prescriptores una reevaluación periódica de la necesidad de continuar con la terapia. Poblaciones especiales: Insuficiencia renal y hepática: No se han realizado estudios sobre el efecto de la insuficiencia renal o hepática en la farmacocinética de Xolair; no obstante, es improbable que la insuficiencia renal o insuficiencia hepática afecten a la depuración de omalizumab administrado en dosis clínicas, dado que ésta se realiza fundamentalmente a través del sistema reticuloendotelial. Aunque no se recomiende hacer ningún ajuste de dosis en particular, Xolair® debe administrarse con precaución en estos pacientes (Ver Advertencias y Precauciones). Pacientes pediátricos: En el asma alérgica, No se ha determinado la seguridad ni la eficacia de Xolair® en pacientes menores de 6 años, por lo que no se recomienda utilizarlo en esta población. En la urticaria espontánea crónica, no se ha determinado la inocuidad ni la eficacia del medicamento en pacientes menores de 12 años de edad. Pacientes de edad avanzada: Aunque se dispone de escasos datos sobre el uso de Xolair® en pacientes mayores de 65 años, no existen pruebas de que los pacientes de edad avanzada requieran una dosificación diferente de la de los pacientes adultos más jóvenes. Instrucciones de uso y manipulación: Xolair® 75 mg y 150 mg polvo para solución inyectable se suministra en viales para uso único y no contienen conservantes antibacterianos. Desde el punto de vista químico y físico, el producto reconstituido es estable durante 8 horas entre 2 y 8°C y durante 4 horas a 30°C. Desde el punto de vista microbiológico, el producto debe utilizarse de inmediato después de la reconstitución. Si no se usa de inmediato, tanto el tiempo de conservación durante el uso como las condiciones de conservación antes del uso son responsabilidad del usuario; por lo general, la solución no debe conservarse más de 8 horas entre 2 y 8°C, a m