Xeloda®
BIOPAS
Capecitabina.
Citostático - antimetabolito.
Composición.
Cada comprimido recubierto de 150 mg contiene 150 mg de capecitabina, en un excipiente compuesto por lactosa anhidra 15,60 mg, croscarmelosa sódica 6,00 mg, hipromelosa 4,50 mg, celulosa microcristalina 7,20 mg, estearato de magnesio 2,70 mg y mezcla para recubrimiento Opadry® Pink 03A14309: 8,50 mg. Cada comprimido recubierto de 500 mg contiene 500 mg de capecitabina, en un excipiente compuesto por lactosa anhidra 52,00 mg, croscarmelosa sódica 20,00 mg, hipromelosa 15,00 mg, celulosa microcristalina 24,00 mg, estearato de magnesio 9,00 mg y mezcla para recubrimiento Opadry® Pink 03A14380: 18,00 mg.
Farmacología.
Código ATC: L01BC06. Grupo farmacoterapéutico: Citostático (antimetabolito). Propiedades farmacodinámicas: La capecitabina es un carbamato de fluoropirimidina no citotóxico que, administrado por vía oral, actúa como un precursor del citotóxico 5-fluorouracilo (5-FU). La capecitabina se activa a través de varios pasos enzimáticos (ver Farmacología, Propiedades farmacocinéticas). La enzima responsable de la conversión final a 5-FU, la timidina fosforilasa (ThyPasa), se encuentra tanto en tejidos tumorales como normales, aunque con niveles generalmente más bajos. En modelos de xenotrasplante de cáncer humano, capecitabina mostró un efecto sinérgico en combinación con docetaxel, lo cual puede estar relacionado con la estimulación (upregulation) de la timidina fosforilasa producida por el docetaxel. Existen pruebas de que el metabolismo de 5-FU por vía anabólica bloquea la reacción de metilación del ácido desoxiuridílico hacia el ácido timidílico, por ello interfiere con la síntesis del ácido desoxirribonucleico (ADN). La incorporación del 5-FU también conduce a la inhibición del ARN y síntesis proteica. Dado que tanto el ADN como el ARN son esenciales para la división y el crecimiento celular, el efecto de 5-FU puede crear una deficiencia de timidina que provoca un desarrollo no equilibrado y la muerte celular. Los efectos de la privación del ADN y el ARN se acentúan en las células que proliferan más rápidamente y que metabolizan 5-FU con mayor velocidad. Cáncer colorrectal y de colon: Terapia adyuvante en cáncer de colon con Xeloda en monoterapia: Se llevó a cabo un ensayo clínico (XACT; M66001) de Fase III, multicéntrico, aleatorizado, controlado, para estudiar el empleo de Xeloda en el tratamiento adyuvante de pacientes con cáncer de colon estadio III (estadio C de Dukes). En este ensayo, se aleatorizaron 1.987 pacientes para ser tratados con Xeloda (1.250 mg/m2 dos veces por día durante 2 semanas, seguidas de 1 de descanso, administrándose en ciclos de 3 semanas durante 24 semanas) o 5-FU y leucovorina (régimen de la Clínica Mayo: 20 mg/m2 de leucovorina por vía intravenosa, seguidos de 425 mg/m2 de 5-FU en bolo por vía intravenosa, los días 1 a 5, cada 28 días durante 24 semanas). Xeloda fue por lo menos equivalente a 5-FU/LV intravenoso en cuanto a la sobrevida libre de enfermedad en la población incluida en el protocolo (HR [hazard ratio] 0,92; IC 95%: 0,80-1,06). En todos los pacientes aleatorizados, las pruebas para diferenciar la sobrevida libre de enfermedad y sobrevida global de Xeloda versus 5-FU/LV dieron HR de 0,88 (IC 95%: 0,77-1,01; p = 0,068) y 0,86 (IC 95%: 0,74-1,01; p = 0,060), respectivamente. La mediana de seguimiento en el momento del análisis fue de 6,9 años. En un examen multivariante de Cox predefinido se demostró la superioridad de Xeloda frente a 5-FU/LV administrado en bolo. En el plan de análisis estadístico se preespecificaron los siguientes factores para su inclusión en el modelo: edad, tiempo desde la cirugía hasta la aleatorización, sexo, niveles basales de antígeno carcinoembriogénico (CEA), nódulos linfáticos al inicio, y país. Para toda la población aleatorizada, Xeloda demostró ser superior a 5FU/LV en sobrevida libre de progresión (HR 0,849; IC 95%: 0,739 - 0,976; p = 0,0212), así como en sobrevida global (HR 0,828; IC 95%: 0,705-0,971; p = 0,0203). Terapia adyuvante en cáncer de colon en combinación: Xeloda en combinación con oxaliplatino (XELOX) para el tratamiento adyuvante de pacientes con cáncer de colon se estudió en un ensayo clínico (NO16968) de Fase III, aleatorizado, multicéntrico y controlado, en pacientes con cáncer de colon estadio III (estadio C de Dukes). En este estudio clínico, 944 pacientes fueron aleatorizados para recibir ciclos de 3 semanas durante 24 semanas con Xeloda (1.000 mg/m2 dos veces por día durante 2 semanas, seguido de un período de descanso de 1 semana) en combinación con oxaliplatino (130 mg/m2 en infusión intravenosa durante 2 horas, administrado el día 1, cada 3 semanas); 942 pacientes fueron aleatorizados para recibir 5-FU en bolo y leucovorina. En el análisis primario de sobrevida libre de enfermedad en población por intención de tratar, XELOX mostró ser significativamente superior a 5-FU/LV (HR 0,80, IC 95%: 0,69-0,93; p = 0,0045). El valor de sobrevida libre de enfermedad a los 3 años fue 71% para XELOX frente a 67% para 5-FU/LV. El análisis del objetivo secundario de sobrevida libre de recaída avala estos resultados con un HR de 0,78 (IC 95%: 0,67-0,92; p = 0,0024) para XELOX frente a 5-FU/LV. XELOX mostró una tendencia hacia una sobrevida global superior con un HR de 0,87 (IC 95%: 0,72-1,05; p = 0,1486), que se traduce en un 13% de reducción del riesgo de muerte. El valor de sobrevida global a los 5 años fue 78% para XELOX frente a 74% para 5-FU/LV. Los datos de eficacia están basados en la mediana del tiempo de observación de 59 meses para sobrevida global y 57 meses para sobrevida libre de enfermedad. En población por intención de tratar, el porcentaje de abandonos debido a eventos adversos fue mayor en el grupo del tratamiento en combinación de XELOX (21%) que en el de 5-FU/LV (9%) en monoterapia. A los 7 años de seguimiento promedio, XELOX mantiene una diferencia estadísticamente significativa superior de la sobrevida libre de enfermedad HR de 0,80 (IC 95%: 0,69 - 0,93; p = 0,0038), y la sobrevida libre de recaída HR de 0,78 (IC 95%: 0,67 - 0,91; p = 0,0015). La tasa de sobrevida global a los 7 años fue del 73% en el grupo XELOX y del 67% en el de 5-FU/LV. Los dos años de seguimiento adicional después del análisis primario muestran un incremento en la diferencia entre las tasas de sobrevida de 3% a 6%. Monoterapia en el cáncer colorrectal metastásico con Xeloda: Se llevaron a cabo dos ensayos clínicos (SO14695; SO14796) de Fase III, controlados, idénticamente diseñados, multicéntricos y aleatorizados para estudiar el uso de Xeloda para el tratamiento en primera línea del cáncer colorrectal metastásico. En estos ensayos, se distribuyeron al azar 603 pacientes en tratamiento con Xeloda (1.250 mg/m2 dos veces por día durante 2 semanas, seguidas de 1 semana de reposo, considerándose ciclos de 3 semanas). Otros 604 pacientes se aleatorizaron en el tratamiento con 5-FU y leucovorina (régimen Mayo: 20 mg/m2 de leucovorina intravenosa seguido de un bolo intravenoso de 5-FU 425 mg/m2 los días 1 y 5, cada 28 días). Los índices de respuesta global objetiva en toda la población aleatorizada (evaluación de Investigador) fueron 25,7% (Xeloda) versus 16,7% (régimen Mayo); p < 0,0002. La mediana del tiempo hasta progresión fue de 140 días (Xeloda) versus 144 días (régimen Mayo). La mediana de sobrevida fue de 392 días (Xeloda) versus 391 días (régimen Mayo). Actualmente no se dispone de datos comparativos de Xeloda en monoterapia en cáncer colorrectal con regímenes de combinación en primera línea. Terapia de combinación en el tratamiento en primera línea del cáncer colorrectal metastásico: Los datos procedentes de un ensayo clínico Fase III (NO16966) controlado, multicéntrico y aleatorizado apoyan el uso de Xeloda en combinación con oxaliplatino o con oxaliplatino y bevacizumab para el tratamiento en primera línea del cáncer colorrectal metastásico. El ensayo tuvo dos etapas: una inicial con 2 brazos donde 634 pacientes fueron aleatorizados a dos grupos de tratamiento diferentes, que incluyeron XELOX o FOLFOX-4, y una posterior con un diseño factorial 2 x 2 en la cual 1.401 pacientes fueron distribuidos al azar a cuatro grupos de tratamiento diferentes que incluyeron XELOX + placebo, FOLFOX-4 + placebo, XELOX + bevacizumab, y FOLFOX-4 + bevacizumab. En la Tabla 1 se incluyen los distintos regímenes de tratamiento.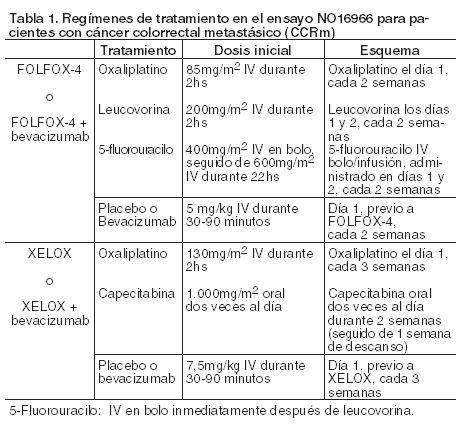
En la comparación global realizada sobre la población de pacientes aptos y sobre la población por intención de tratar se demostró la no-inferioridad en términos de sobrevida libre de progresión de los brazos que contenían XELOX frente a los brazos con FOLFOX-4 (ver Tabla 2). Los resultados indican que XELOX es equivalente a FOLFOX-4 en sobrevida global (ver Tabla 2). Se realizó un análisis exploratorio preespecificado comparando XELOX + bevacizumab frente a FOLFOX-4 + bevacizumab. En esta comparación por subgrupos de tratamiento, XELOX + bevacizumab fue similar a FOLFOX-4 + bevacizumab en sobrevida libre de progresión (HR 1,01; IC 97,5%: 0,84-1,22). El seguimiento medio en el momento de los análisis principales en la población por intención de tratar fue de 1,5 años; los datos procedentes de la investigación realizada después de un año adicional de seguimiento también se han incluido en la Tabla 2. Sin embargo, los datos de la sobrevida libre de progresión durante el tratamiento no confirmaron los resultados del análisis general de sobrevida libre de progresión y del de sobrevida global: el índice de riesgo (HR) de XELOX frente a FOLFOX-4 fue de 1,24 con un intervalo de confianza del 97,5%: de 1,07 a 1,44. Aunque los estudios de sensibilidad muestran que las diferencias en los esquemas de régimen y el momento de la evaluación del tumor influyen en la estimación de la sobrevida libre de progresión durante el tratamiento, no se ha encontrado una explicación completa de este resultado.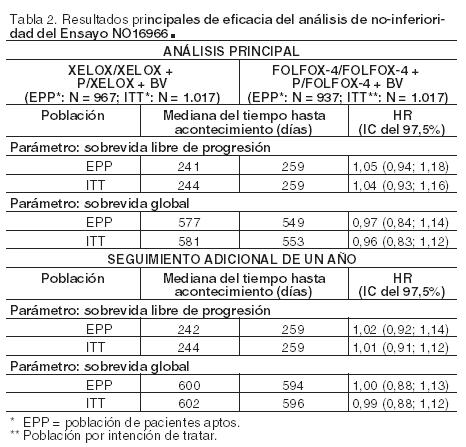
CAIRO fue un ensayo aleatorizado, controlado, de Fase III, para estudiar el uso de Xeloda a una dosis inicial de 1.000 mg/m2 durante 2 semanas en combinación con irinotecán cada 3 semanas para el tratamiento en primera línea de pacientes con cáncer colorrectal metastásico. Se aleatorizaron 820 pacientes para ser tratados en forma secuencial (n = 410) o combinada (n = 410). La primera consistía en tratamiento en primera línea con Xeloda (1.250 mg/m2 dos veces por día durante 14 días), en segunda línea con irinotecán (350 mg/m2 en el día 1), y combinada en tercera línea de capecitabina (1.000 mg/m2 dos veces por día durante 14 días) con oxaliplatino (130 mg/m2 en el día 1). El tratamiento combinado consistió en administración en primera línea de Xeloda (1.000 mg/m2 dos veces por día durante 14 días) e irinotecán (250 mg/m2 en el día 1) (XELIRI) y en segunda línea con capecitabina (1.000 mg/m2 dos veces por día durante 14 días) junto con oxaliplatino (130 mg/m2 en el día 1). Todos los ciclos de tratamiento fueron administrados a intervalos de 3 semanas. En el tratamiento en primera línea la mediana de sobrevida libre de progresión en la población por intención de tratar fue de 5,8 meses (IC 95%; 5,1-6,2 meses) con Xeloda en monoterapia y de 7,8 meses (IC 95%; 7,0-8,3 meses; p = 0,0002) con XELIRI. Sin embargo, estas cifras se asociaron con una mayor incidencia de toxicidad gastrointestinal y neutropenia durante el tratamiento en primera línea con XELIRI (26% y 11% para XELIRI y primera línea de Xeloda, respectivamente). XELIRI se ha comparado con el 5-FU + irinotecán (FOLFIRI) en tres ensayos aleatorizados en pacientes con cáncer colorrectal metastásico. Los régimenes de XELIRI, incluyen 1.000 mg/m2 de Xeloda dos veces por día durante 14 días, en un ciclo de 3 semanas en combinación con 250 mg/m2 de irinotecán en el día 1. En el ensayo mayor (BICC-C), los pacientes fueron aleatorizados para recibir en forma abierta FOLFIRI (n = 144), bolo de 5-FU (mIFL) (n = 145) o XELIRI (n = 141) y, además, fueron distribuidos al azar para ser tratados en forma doble-ciega con celecoxib o placebo. La mediana de la sobrevida libre de progresión fue 7,6 meses en FOLFIRI, 5,9 meses en mIFL (p = 0,004 en comparación con FOLFIRI) y 5,8 meses en XELIRI (p = 0,015). La mediana de la sobrevida global fue 23,1 meses en FOLFIRI, 17,6 meses en mIFL (p = 0,09) y 18,9 meses en XELIRI (p = 0,27). Los pacientes tratados con XELIRI experimentaron excesiva toxicidad gastrointestinal en comparación con FOLFIRI (diarrea 48% y 14% en XELIRI y FOLFIRI, respectivamente). En el ensayo EORTC los pacientes fueron aleatorizados para recibir en forma abierta FOLFIRI (n = 41) o XELIRI (n = 44) con una distribución al azar adicional para ser tratados en forma doble-ciega con celecoxib o placebo. La mediana de la sobrevida libre de progresión y de la sobrevida global fue menor para XELIRI que para FOLFIRI (sobrevida libre de progresión 5,9 frente a 9,6 meses y sobrevida global 14,8 comparado con 19,9 meses); además, se notificaron tasas excesivas de diarrea en los pacientes que recibían XELIRI (41% XELIRI, 5,1% FOLFIRI). En el estudio publicado por Skof y col., los pacientes fueron aleatorizados para recibir el tratamiento de FOLFIRI o de XELIRI. La tasa de respuesta global fue del 49% en el grupo de XELIRI y del 48% en el de FOLFIRI (p = 0,76). Al final del tratamiento, el 37% de los pacientes del grupo de XELIRI y el 26% de los de FOLFIRI no tenían la enfermedad (p = 0,56). La toxicidad entre los tratamientos fue similar, a excepción de la neutropenia, de la cual se informaron más casos en pacientes tratados con FOLFIRI. Monatgnani y col., utilizaron los resultados de los tres ensayos anteriores para brindar un análisis global de los estudios aleatorizados comparando las pautas de FOLFIRI y XELIRI en el tratamiento del cáncer colorrectal metastásico. Una reducción significativa en el riesgo de progresión se asoció con FOLFIRI (HR 0,76; IC 95 %: 0,62-0,95; p < 0,01), como resultado en parte debido a la escasa tolerancia a los regímenes usados de XELIRI. Los datos de un ensayo clínico aleatorizado (Souglakos y col., 2012) que comparó el tratamiento de FOLFIRI + bevacizumab con el de XELIRI + bevacizumab, no mostraron diferencias significativas en la sobrevida libre de progresión o en la sobrevida global entre ambos tratamientos. Los pacientes fueron aleatorizados para recibir FOLFIRI + bevacizumab (grupo A, n = 167) o XELIRI + bevacizumab (grupo B, n = 166). En el grupo B, el régimen de XELIRI utilizaba Xeloda 1.000 mg/m2 dos veces por día durante 14 días + irinotecán 250 mg/m2 en el día 1. La mediana de la sobrevida libre de progresión fue 10,0 y 8,9 meses, p = 0,64, la sobrevida global 25,7 y 27,5 meses, p = 0,55 y la tasa de respuesta 45,5 y el 39,8 %, p = 0,32 para FOLFIRI + bevacizumab y XELIRI + bevacizumab, respectivamente. Los pacientes tratados con XELIRI + bevacizumab registraron una incidencia significativamente más alta de diarrea, neutropenia febril y síndrome mano-pie que aquéllos tratados con FOLFIRI + bevacizumab con un incremento significativo en reducciones de dosis, retrasos e interrupciones del tratamiento. Xeloda (a la dosis inicial de 800 mg/m2 durante 2 semanas cada 3 semanas) en combinación con irinotecán y bevacizumab fue estudiado para el tratamiento en primera línea de pacientes con cáncer colorrectal metastásico en un ensayo Fase II (AIO KRK 0604) controlado, multicéntrico y aleatorizado. Se distribuyeron al azar 128 pacientes para recibir tratamiento con Xeloda asociado con irinotecán (XELIRI modificado) y bevacizumab: Xeloda (800 mg/m2 dos veces por día durante dos semanas seguido de un período de descanso de 7 días), irinotecán (200 mg/m2 en infusión durante 30 minutos, el día 1 cada 3 semanas), y bevacizumab (7,5 mg/kg en infusión durante 30 a 90 minutos el día 1 cada 3 semanas); un total de 127 pacientes fueron aleatorizados para ser tratados con Xeloda en combinación con oxaliplatino (XELOX) + bevacizumab: Xeloda (1.000 mg/m2 dos veces por día durante dos semanas seguido de un período de descanso de 7 días), oxaliplatino (130 mg/m2 en infusión durante 2 horas, el día 1 cada 3 semanas), y bevacizumab (7,5 mg/kg en infusión de 30 a 90 minutos, el día 1 cada 3 semanas). Las respuestas al tratamiento después de una duración media de seguimiento de la población en estudio de 26,6 meses se mencionan en la Tabla 3. La tasa de respuesta global (respuesta parcial más respuesta completa) fue del 56% (XELIRI más bevacizumab) en comparación con 53% (XELOX más bevacizumab).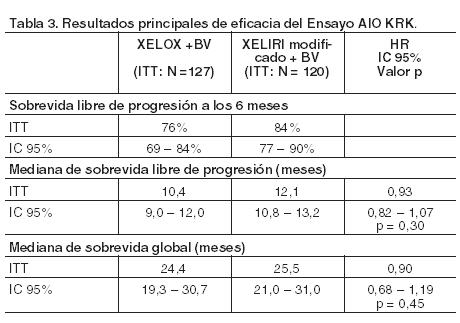
Terapia de combinación en el tratamiento en segunda línea del cáncer colorrectal metastásico: NO16967 fue un ensayo de Fase III, controlado, multicéntrico y aleatorizado que estudió el uso de Xeloda en combinación con oxaliplatino para el tratamiento en segunda línea del cáncer colorrectal metastásico. En este ensayo, 627 pacientes con carcinoma colorrectal metastásico que habían sido tratados previamente con irinotecán en combinación con un régimen de fluoropirimidina como tratamiento en primera línea, fueron aleatorizados para ser tratados con XELOX o FOLFOX-4. Para el esquema de dosificación de XELOX y FOLFOX-4 (sin la adición de placebo o bevacizumab), ver Tabla 1. En la población por protocolo y en la población por intención de tratar se demostró la no-inferioridad de XELOX frente a FOLFOX-4 en sobrevida libre de progresión (ver Tabla 4). Los resultados indicaron que XELOX es equivalente a FOLFOX-4 en sobrevida global (ver Tabla 4). La mediana de seguimiento en los análisis principales en la población por intención de tratar fue de 2,1 años; en la Tabla 4 también se incluyen datos procedentes de las investigaciones realizadas después de un período de seguimiento adicional de 6 meses.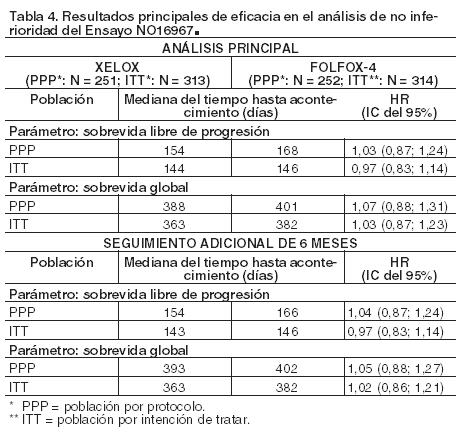
Cáncer gástrico avanzado: Un ensayo clínico (ML17032) de Fase III, multicéntrico, aleatorizado, controlado, en pacientes con cáncer gástrico avanzado o metastásico estudió el empleo de Xeloda para el tratamiento en primera línea de pacientes con cáncer gástrico avanzado o metastásico. En este ensayo, se aleatorizaron 160 pacientes para recibir tratamiento con Xeloda (1.000 mg/m2 dos veces por día durante dos semanas, seguido por un período de descanso de 7 días) y cisplatino (80 mg/m2 durante una infusión de 2 horas cada 3 semanas). Se distribuyeron al azar un total de 156 pacientes a un tratamiento con 5-FU (800 mg/m2 por día, en infusión continua los días 1 - 5 cada 3 semanas) y cisplatino (80 mg/m2 durante una infusión de 2 horas el día 1, cada 3 semanas). Xeloda en combinación con cisplatino no fue inferior a 5-FU asociado con cisplatino en sobrevida libre de progresión en el análisis por protocolo (HR 0,81; IC 95%: 0,63-1,04). La mediana de la sobrevida libre de progresión fue de 5,6 meses (Xeloda + cisplatino) versus 5,0 meses (5-FU + cisplatino). El HR de la duración de sobrevida (sobrevida global) fue similar al índice de la sobrevida libre de progresión (HR 0,85; IC 95%: 0,64-1,13). La mediana de la duración de sobrevida fue 10,5 meses (Xeloda más cisplatino) versus 9,3 meses (5-FU + cisplatino). Un estudio (REAL-2) de Fase III, multicéntrico, aleatorizado, donde se comparaba capecitabina con 5-FU y oxaliplatino con cisplatino en pacientes con cáncer gástrico avanzado se llevó a cabo para el tratamiento en primera línea del cáncer gástrico avanzado. En este ensayo, mediante un diseño factorial 2 x 2, se aleatorizaron 1.002 pacientes a cada uno de los siguientes cuatro brazos: ECF: epirrubicina (50 mg/m2 como bolo en el día 1, cada 3 semanas), cisplatino (60 mg/m2 como infusión de 2 horas en el día 1 cada 3 semanas) y 5-FU (200 mg/m2 por día administrado como infusión continua a través de una vía central). ECX: epirrubicina (50 mg/m2 como bolo en el día 1 cada 3 semanas), cisplatino (60 mg/m2 como infusión de 2 horas en el día 1 cada 3 semanas) y Xeloda (625 mg/m2, dos veces por día en forma continuada). EOF: epirrubicina (50 mg/m2 como bolo en el día 1 cada 3 semanas), oxaliplatino (130 mg/m2 administrado como una infusión de 2 horas el día 1, cada 3 semanas) y 5-FU (200 mg/m2 por día por infusión continua a través de una vía central). EOX: epirrubicina (50 mg/m2 como bolo en el día 1 cada 3 semanas), oxaliplatino (130 mg/m2 administrado como una infusión de 2 horas en el día 1, cada 3 semanas) y Xeloda (625 mg/m2 dos veces por día en forma continuada). Los análisis principales de eficacia en la población por protocolo demostraron la no inferioridad de la capecitabina frente a los regímenes basados en 5-FU (HR 0,86; IC 95%: 0,8 - 0,99) y del oxaliplatino versus esquemas basados en cisplatino (HR 0,92; IC 95%: 0,80 - 1,1). La mediana de la sobrevida global fue 10,9 meses en los regímenes basados en capecitabina y de 9,6 meses en aquéllos basados en 5-FU. La mediana de la sobrevida global fue de 10,0 meses en los regímenes basados en cisplatino y de 10,4 meses con aquéllos basados en oxaliplatino. Xeloda fue también utilizado en combinación con oxaliplatino para el tratamiento del cáncer gástrico avanzado. Los estudios con Xeloda en monoterapia indicaron que ejerce actividad sobre el cáncer gástrico avanzado. Cáncer de colon, colorrectal y gástrico avanzado: metanálisis: Un metanálisis de seis ensayos clínicos (SO14695, SO14796, M66001, NO16966, NO16967, ML17032) investigó si Xeloda puede reemplazar a 5-FU en monoterapia y en el tratamiento de combinación en cáncer gastrointestinal. El análisis conjunto incluyó 3.097 pacientes tratados con regímenes que contienen Xeloda y 3.074 tratados con esquemas con 5-FU. La mediana del tiempo de sobrevida global fue de 703 días (IC 95%; 671-745) en los pacientes tratados con regímenes con Xeloda y de 683 días (IC 95%; 646-715) en aquéllos que recibieron esquemas con 5-FU. El HR para la sobrevida global fue de 0,94 (IC 95%: 0,89-1,00; p = 0,0489) con regímenes con Xeloda indicando que éstos son no inferiores a los que contienen 5-FU. Cáncer de mama: Terapia combinada con Xeloda y docetaxel en el cáncer de mama localmente avanzado o metastásico: Xeloda en combinación con docetaxel para el tratamiento de pacientes con cáncer de mama localmente avanzado o metastásico después del fracaso de la terapia citotóxica que incluya una antraciclina fue estudiado en un ensayo clínico (SO14999) de Fase III, multicéntrico, aleatorizado y controlado. En ese ensayo, se aleatorizaron 255 pacientes en tratamiento con Xeloda (1.250 mg/m2 dos veces por día durante 2 semanas seguidas de 1 semana de descanso y docetaxel en dosis de 75 mg/m2 en infusión intravenosa durante 1 hora cada 3 semanas). Otros 256 fueron distribuidos al azar para ser tratados con docetaxel solo (100 mg/m2 en infusión intravenosa durante 1 hora cada 3 semanas). La sobrevida resultó mayor en la rama de tratamiento combinado con Xeloda + docetaxel (p = 0,0126). La mediana de sobrevida fue de 442 días (Xeloda + docetaxel) versus 352 días (docetaxel solo). Los índices de respuesta objetiva globales en toda la población aleatorizada (evaluación del Investigador) fueron del 41,6% (Xeloda + docetaxel) versus 29,7% (docetaxel solo); p = 0,0058. El tiempo hasta la progresión de la enfermedad fue superior en el brazo tratado con la combinación Xeloda + docetaxel (p < 0,0001). La mediana de tiempo hasta la progresión fue de 186 días (Xeloda + docetaxel) versus 128 días (docetaxel solo). Monoterapia con Xeloda después del fracaso con taxanos, quimioterapia que contenga antraciclinas o para aquellos pacientes en los que la terapia con antraciclinas no esté indicada: Dos ensayos clínicos Fase II, multicéntricos, se llevaron a cabo para determinar el empleo de Xeloda en monoterapia para el tratamiento de pacientes después del fracaso con taxanos y con un régimen de quimioterapia con antraciclinas o para aquellos en los que la terapia con antraciclinas no estaba indicada. En estos ensayos, fueron tratados un total de 236 pacientes con Xeloda (1.250 mg/m2 dos veces por día durante 2 semanas seguido de 1 semana de descanso). Los índices de respuesta objetiva globales (evaluación del Investigador) fueron del 20% (primer ensayo) y 25% (segundo ensayo). La mediana del tiempo hasta progresión fue de 93 y 98 días. La mediana de sobrevida fue de 384 y 373 días. Generales: El examen de los datos de seguridad realizado en pacientes con insuficiencia renal basal tratados con Xeloda en monoterapia (cáncer colorrectal) mostró un incremento en la incidencia de las reacciones adversas de Grados 3 y 4 relacionadas con el tratamiento si se compara con los pacientes con función renal normal (36% en aquéllos sin insuficiencia renal n = 268, versus 41% en leves n = 257 y 54% en moderados n = 59, respectivamente) (ver Farmacología, Propiedades farmacocinéticas). En los pacientes con función renal moderadamente alterada se registró un aumento en la reducción de dosis (44%) versus 33% y 32% en aquéllos sin insuficiencia renal o insuficiencia leve, así como un incremento en los abandonos prematuros del tratamiento (21% durante los primeros dos ciclos) versus el 5% y 8% de los pacientes sin insuficiencia renal o insuficiencia leve. El examen de los datos de seguridad entre los pacientes de ≥ 60 años tratados con Xeloda en monoterapia y un análisis de los tratados con la combinación Xeloda más docetaxel, indicó una mayor incidencia de reacciones adversas de Grados 3 y 4 y de reacciones adversas graves ambas vinculadas con el tratamiento, si se compara con pacientes menores de 60 años. Los mayores de 60 años tratados con Xeloda más docetaxel tuvieron asimismo que abandonar el tratamiento en forma prematura debido a reacciones adversas, si se compara con pacientes menores de esa edad. Todas las indicaciones: En un metanálisis de 14 ensayos clínicos con los datos de más de 4.700 pacientes tratados con Xeloda en monoterapia o Xeloda en combinación con diferentes regímenes de quimioterapia en múltiples indicaciones (cáncer de colon, colorrectal, gástrico y mama) se demostró que los pacientes tratados con Xeloda que desarrollaron el síndrome mano-pie tuvieron una mayor sobrevida global comparada con aquéllos que no lo desarrollaron: sobrevida media global de 1.100 días (IC 95%; 1.007-1.200) frente a 691 días (IC 95%; 638-754) con un índice de riesgo del 0,61 (IC 95%; 0,56-0,66). Población pediátrica: La Agencia Europea de Medicamentos ha eximido al titular de la obligación de llevar a cabo estudios con Xeloda en todos los subgrupos de la población pediátrica en el adenocarcinoma de colon y recto, adenocarcinoma gástrico y carcinoma de mama (ver Dosificación para el uso en pediatría). Propiedades farmacocinéticas: La farmacocinética de capecitabina se ha evaluado en el intervalo posológico de 502 - 3.514 mg/m2/día. Los parámetros de capecitabina, 5'-deoxi-5-fluorocitidina (5'-DFCR) y 5'-deoxi-5-fluorouridina (5'-DFUR) medidos el día 1 y 14 fueron similares. El ABC de 5-FU aumentó un 30-35% el día 14. La reducción de dosis de capecitabina disminuye la exposición sistémica a 5-FU en forma mayor que la proporción de dosis, debido a una farmacocinética no lineal del metabolito activo. Absorción: Después de la administración oral, la capecitabina atraviesa la mucosa intestinal en forma de molécula intacta y se absorbe de modo rápido y extenso, transformándose posteriormente en forma amplia en los metabolitos 5'-DFCR y 5'-DFUR. La administración con los alimentos reduce la velocidad de absorción de la capecitabina, pero sólo modifica mínimamente el valor del ABC de 5'-DFUR y del ABC del metabolito subsiguiente, 5-FU. A la dosis de 1.250 mg/m2 en el día 14 administrada después de tomar alimentos, las concentraciones plasmáticas máximas (Cmáx en mg/ml) para capecitabina, 5'-DFCR, 5'-DFUR, 5-FU y FBAL fueron 4,67, 3,05, 12,1, 0,95 y 5,46, respectivamente. El tiempo para las concentraciones plasmáticas máximas (Tmáx en horas) fue 1,50, 2,00, 2,00, 2,00 y 3,34. Los valores del ABC0 - a en mg•h/ml fueron 7,75, 7,24, 24,6, 2,03 y 36,3. Distribución: Los estudios in vitro con plasma humano han revelado que la capecitabina, el 5'-DFCR, el 5'-DFUR y el 5-FU se unen a las proteínas, sobre todo a la albúmina, en un 54%, 10%, 62% y 10%, respectivamente. Biotransformación: En primer lugar, la capecitabina es metabolizada por la carboxilesterasa hepática en 5'-DFCR, que se transforma después en 5'-DFUR por la citidina deaminasa, localizada fundamentalmente en el hígado y en los tejidos tumorales. Después, la activación catalítica de 5'-DFUR tiene lugar mediante la timidina fosforilasa (ThyPase). Las enzimas que intervienen en la activación catalítica se localizan tanto en los tejidos tumorales como en los sanos, normalmente en niveles más bajos. La biotransformación enzimática secuencial de capecitabina a 5-FU conduce a concentraciones más altas dentro de los tejidos tumorales. En el caso de tumores colorrectales, la generación de 5-FU está localizada en mayor medida en las células del estroma tumoral. Después de la administración oral de capecitabina a pacientes con cáncer colorrectal, la relación entre concentración de 5-FU en los tumores colorrectales y los tejidos adyacentes fue 3,2 (osciló de 0,9 a 8,0). La relación de concentración de 5-FU en tumor frente a plasma fue de 21,4 (osciló de 3,9 a 59,9, n = 8), mientras que la relación entre los tejidos sanos y plasma fue de 8,9 (osciló de 3,0 a 25,8, n = 8). La actividad de la timidina fosforilasa fue medida y se encontró que era 4 veces más alta en el tumor colorrectal primario que en el tejido normal adyacente. De acuerdo con los estudios inmunohistoquímicos, la timidina fosforilasa está localizada en mayor medida en las células del estroma tumoral. Posteriormente, el 5-FU se cataboliza por la enzima dihidropirimidina dehidrogenasa (DPD) a dihidro-5-fluorouracilo (FUH2), el cual es mucho menos tóxico. La dihidropirimidinasa rompe el anillo de pirimidina y produce el ácido 5-fluoro-ureidopropiónico (FUPA). Finalmente, la b-ureido-propionasa transforma el FUPA a a-fluoro-b-alanina (FBAL), la cual es eliminada por la orina. La actividad dihidropirimidina dehidrogenasa (DPD) es el paso limitante. La deficiencia en DPD puede conducir a un aumento de la toxicidad de capecitabina (ver Contraindicaciones; y Precauciones y advertencias). Eliminación: La vida media de eliminación (t1/2 en horas) de capecitabina, 5'-DFCR, 5'-DFUR, 5-FU y FBAL fue 0,85, 1,11, 0,66, 0,76 y 3,23, respectivamente. Los metabolitos de la capecitabina se eliminan fundamentalmente por excreción urinaria. El 95,5% de la dosis administrada de capecitabina se recoge en orina. La excreción fecal es mínima (2,6%). El principal metabolito excretado en la orina es FBAL, que representa un 57% de la dosis administrada. Alrededor del 3% de la dosis administrada se excreta inalterada por la orina. Terapia combinada: Los ensayos de Fase I para evaluar el efecto de Xeloda sobre la farmacocinética de docetaxel o paclitaxel y viceversa mostraron que Xeloda no afecta la farmacocinética de estos fármacos (Cmáx y ABC), ni que éstos alteran la farmacocinética del 5'-DFUR. Farmacocinética en poblaciones especiales: Se ha realizado un análisis de farmacocinética en la población después del tratamiento con Xeloda de 505 pacientes con cáncer colorrectal a dosis de 1.250 mg/m2 dos veces por día. El sexo, presencia o ausencia de metástasis hepáticas basales, el índice de Karnofsky, la bilirrubina total, la albúmina sérica, los niveles de ASAT y ALAT no tuvieron un efecto estadísticamente significativo sobre la farmacocinética del 5'-DFUR, 5-FU y FBAL. Pacientes con insuficiencia hepática debida a metástasis hepáticas: Según un estudio farmacocinético realizado en pacientes con cáncer con insuficiencia hepática leve a moderada causada por metástasis hepáticas, la biodisponibilidad de capecitabina y la exposición a 5-FU puede aumentarse si se compara con pacientes sin insuficiencia hepática. No se disponen de datos farmacocinéticos en aquéllos con insuficiencia hepática grave. Pacientes con insuficiencia renal: Sobre la base de un estudio farmacocinético en pacientes con cáncer con insuficiencia renal de leve a grave, no hay evidencia de que exista un efecto del clearance de creatinina sobre la farmacocinética del medicamento intacto y el 5-FU. Se observó que el clearance de creatinina influye sobre la exposición sistémica a 5'-DFUR (35% de incremento en el ABC cuando el clearance de creatinina disminuye el 50%) y a FBAL (114% de aumento en el ABC cuando el clearance de creatinina se reduce el 50%). FBAL es un metabolito sin actividad antiproliferativa. Pacientes de edad avanzada: Teniendo en cuenta los análisis farmacocinéticos en la población, realizados en un amplio rango de edades (27 a 86 años) que incluyeron 234 pacientes (46%) con edades iguales o superiores a los 65 años, la edad no influyó sobre la farmacocinética del 5'-DFUR ni del 5-FU. El ABC del FBAL se acrecentó con la edad (20% de incremento en la edad supone un 15% de aumento en el ABC del FBAL). Esta situación se debe probablemente a un cambio en la función renal. Factores étnicos: Después de la administración oral de 825 mg/m2 de capecitabina dos veces por día durante 14 días, los pacientes japoneses (n = 18) tuvieron una Cmáx inferior, alrededor de un 36%, y un ABC un 24% menor de capecitabina que los pacientes caucásicos (n = 22). Los japoneses tuvieron también una Cmáx un 25% inferior y un ABC un 34% menor de FBAL que los caucásicos. Se desconoce la relevancia clínica de estas diferencias. No se observaron variaciones significativas en la exposición a otros metabolitos (5'-DFCR, 5'-DFUR y 5-FU). Datos preclínicos sobre seguridad: En los estudios de toxicidad realizados con dosis múltiples, la administración oral diaria de capecitabina a macacos de Java y a ratones se asoció con efectos tóxicos sobre los sistemas gastrointestinal, linfoide y hematopoyético, característicos de las fluoropirimidinas, que fueron reversibles. Se ha observado con capecitabina toxicidad cutánea, caracterizada por cambios degenerativos/regresivos. La capecitabina no causó toxicidad hepática sobre el sistema nervioso central. Se ha detectado toxicidad cardiovascular (por ejemplo, prolongación de los intervalos PR y QT) en macacos de Java después de la aplicación intravenosa (100 mg/kg), pero no así después de la administración oral repetida (1.379 mg/m2/día). Un estudio de carcinogenicidad de dos años realizado en ratones no evidenció carcinogenicidad con capecitabina. Durante los estudios de fertilidad estándares, se registró una alteración de la fertilidad en ratones hembras tratadas con capecitabina; sin embargo, este efecto revertió después de un descanso terapéutico. Además, durante un estudio de 13 semanas, aparecieron cambios degenerativos y atróficos en los órganos reproductores de los ratones macho; no obstante, estos efectos fueron reversibles después de un descanso terapéutico (ver Precauciones). En los estudios sobre embriotoxicidad y teratogenia efectuados en ratones se observó un incremento en las reabsorciones fetales y en la teratogenia que guardaba relación con la dosis. Con altas dosis se informaron abortos y muertes embrionarias en los monos, pero ningún signo de teratogenia. La capecitabina no fue mutagénica in vitro para bacterias (test de Ames) o células de mamífero (ensayo de mutación génica V79/HPRT de hámster chino). No obstante, como ocurre con otros análogos de los nucleósidos (por ejemplo: 5-FU), la capecitabina mostró efecto clastogénico sobre los linfocitos humanos (in vitro) y una tendencia positiva en los tests de micronúcleo de médula ósea murina (in vivo).
Indicaciones.
Xeloda está indicado para el tratamiento adyuvante después de la cirugía en pacientes con cáncer de colon estadio III (estadio C de Dukes) (ver Farmacología, Propiedades farmacodinámicas). Xeloda está indicado para el tratamiento del cáncer colorrectal metastásico (ver Farmacología, Propiedades farmacodinámicas). Xeloda está indicado en el tratamiento de primera línea del cáncer gástrico avanzado en combinación con un esquema basado en platino (ver Farmacología, Propiedades farmacodinámicas). Xeloda en combinación con docetaxel (ver Farmacología, Propiedades farmacodinámicas) está indicado para el tratamiento de pacientes con cáncer de mama localmente avanzado o metastásico después del fracaso de la quimioterapia citotóxica. La terapia previa debe haber incluido una antraciclina. Xeloda está también indicado en monoterapia para el tratamiento de pacientes con cáncer de mama localmente avanzado o metastásico después del fracaso de la terapia con taxanos y con un régimen quimioterápico que incluya una antraciclina o bien para aquellos pacientes en los que no esté indicada una terapia posterior con antraciclinas.
Dosificación.
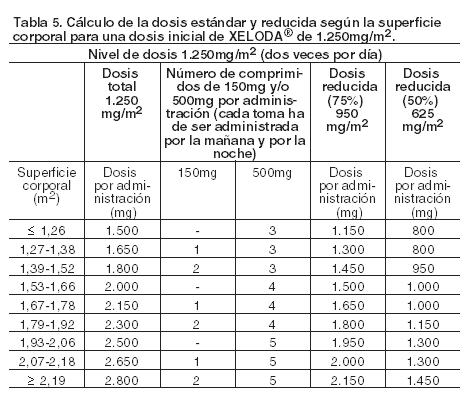
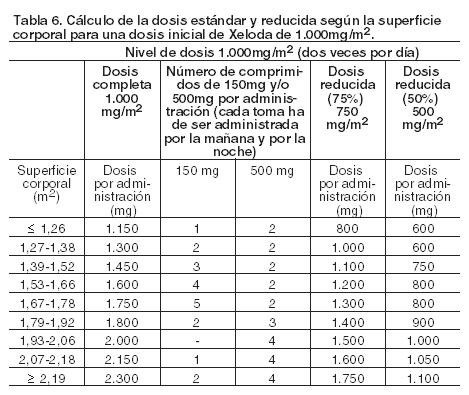
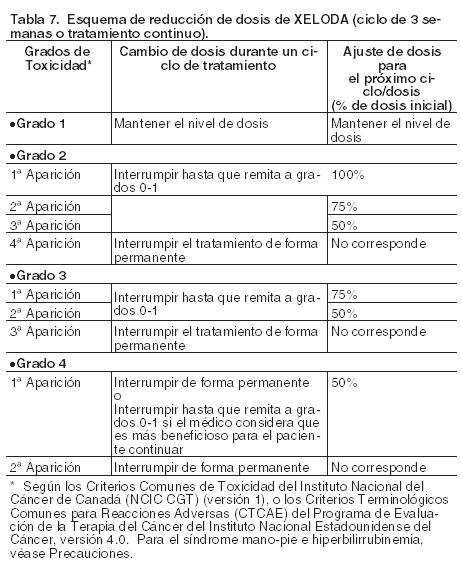
Xeloda solamente debe ser prescripto por un clínico con experiencia en el empleo de agentes antineoplásicos. Se recomienda una monitorización minuciosa para todos los pacientes durante el primer ciclo de tratamiento. El tratamiento se interrumpirá si se observa enfermedad progresiva o toxicidad intolerable. El cálculo de la dosis estándar y reducida según la superficie corporal para dosis iniciales de Xeloda de 1.250 mg/m2 y 1.000 mg/m2 se analiza en Tablas 5 y 6, respectivamente. Posología recomendada (ver Farmacología, Propiedades farmacodinámicas). Monoterapia: Cáncer de colon, colorrectal y de mama: La dosis inicial recomendada de Xeloda cuando se administra en monoterapia en el tratamiento adyuvante de cáncer de colon, en el tratamiento del cáncer colorrectal metastásico o del cáncer de mama localmente avanzado o metastásico es de 1.250 mg/m2 administrados dos veces por día (por la mañana y por la noche; equivalente a una dosis diaria total de 2.500 mg/m2) durante 14 días, seguido de un período de descanso de siete días. La duración recomendada del tratamiento adyuvante en pacientes con cáncer de colon estadio III es de 6 meses. Tratamiento en combinación: Cáncer de colon, colorrectal y gástrico: En el tratamiento en combinación (excepto con irinotecán), la dosis inicial recomendada de Xeloda es 800 - 1.000 mg/m2 administrados dos veces por día durante 14 días, seguido de un período de descanso de 7 días, o 625 mg/m2 dos veces por día cuando se administra en forma continuada (ver Farmacología, Propiedades farmacodinámicas). En la asociación con irinotecán (XELIRI), la dosis inicial recomendada de Xeloda es 800 mg/m2 administrados dos veces por día durante 14 días, seguido de un período de descanso de 7 días y 200 mg/m2 de irinotecán en el día 1 de cada una de las tres semana