XELJANZ XR
PFIZER
XELJANZ XR es la sal de citrato de tofacitinib, inhibidor de las Janus quinasas (JAK, por sus siglas en inglés).
Composición.
Cada comprimido recubierto de liberación extendida de XELJANZ XR contiene: Tofacitinib (como citrato) 11 mg. Sorbitol 152,229 mg. Hidroxietilcelulosa 16 mg. Copovidona 12 mg. Estearato de Magnesio 2 mg. Acetato de celulosa 10,440 mg. Hidroxipropilcelulosa 6,960 mg. Opadry Rosa 8 mg. Opacode Negro 0,1 mg.
Farmacología.
Descripción: XELJANZ XR es la sal de citrato de tofacitinib, un inhibidor de las JAK. El citrato de tofacitinib es un polvo blanco a blanquecino con los siguientes nombres químicos: (3R, 4R)-4-metil-3-(metil-7H-pirolo [2,3-d]pirimidina-4-ilamino)-b-oxo-1-piperidinapropanenitrilo, 2-hidroxi-1,2,3-propanetricarboxilato (1:1) o, alternativamente, 3-[(3R,4R)-4-metil-3-[metil(7H-pirolo[2,3-d] pirimidina-4-il)amino]piperidina-1-il]-3-oxopropanenitrilo, 2-hidroxi-1,2,3-propanetricarboxilato (1:1). La solubilidad del citrato de tofacitinib en agua es 2,9 mg/mL. El citrato de tofacitinib tiene un peso molecular de 504,5 Daltons (o 312,4 Daltons como la base libre de tofacitinib) y una fórmula molecular de C16H20N6O•C6H8O7. La estructura química del citrato de tofacitinib es: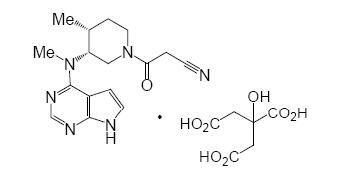
Tofacitinib es un inhibidor de las Janus quinasas (JAK). Las JAK son enzimas intracelulares que transmiten señales derivadas de las interacciones de las citocinas o del receptor del factor de crecimiento sobre la membrana celular para influir en los procesos celulares de hematopoyesis y la función celular inmune. Dentro de la vía de señalización, las JAK fosforilan y activan los Transductores de Señal y Activadores de Transcripción (STAT, por sus siglas en inglés) que modulan la actividad intracelular, incluyendo la expresión génica. Tofacitinib modula la vía de señalización en el punto de las JAK, previniendo la fosforilación y activación de los STAT. Las enzimas JAK transmiten citocinas realizando una señalización a través de emparejamiento de JAK (por ejemplo, JAK1/JAK3, JAK1/JAK2, JAK1/TyK2, JAK2/JAK2). Tofacitinib inhibió las actividades in vitro de JAK1/JAK2, JAK1/JAK3 y combinaciones de JAK2/JAK2 con IC50 de 406, 56 y 1377 nM, respectivamente. Sin embargo, se desconoce la relevancia de combinaciones específicas de JAK para la eficacia terapéutica. Farmacodinamia: El tratamiento con XELJANZ se asoció con reducciones dependientes de la dosis de linfocitos citolíticos naturales CD16/56+ circulantes, con reducciones máximas estimadas que se producen después de aproximadamente 8 a 10 semanas de comenzado el tratamiento. Estos cambios generalmente se resuelven después de 2 a 6 semanas de la suspensión del tratamiento. El tratamiento con XELJANZ se asoció con aumentos dependientes de la dosis en los recuentos de células B. Los cambios en los recuentos de linfocitos T circulantes y subconjuntos de linfocitos T (CD3+, CD4+ y CD8+) fueron pequeños e inconsistentes. Se desconoce el significado clínico de estos cambios. Los niveles totales de IgG, IgM e IgA en suero después de 6 meses de administración en pacientes con artritis reumatoidea fueron menores que el placebo; sin embargo, los cambios fueron pequeños y no dependientes de la dosis. Después del tratamiento de XELJANZ en pacientes con artritis reumatoidea se observaron disminuciones rápidas de la proteína C reactiva (CRP, por sus siglas en inglés) en suero y se mantuvieron a lo largo de toda la administración. Los cambios observados en la CRP con el tratamiento de XELJANZ no cambia completamente dentro de las 2 semanas posteriores a la suspensión, lo que indica una mayor duración de la actividad farmacodinámica en comparación con la vida media farmacocinética. Farmacocinética: Tras la administración oral de XELJANZ XR, las concentraciones plasmáticas máximas se alcanzan dentro de 4 horas, la vida media de eliminación es ~6 horas.Se obtienen concentraciones en estado estacionario en 48 horas, con una acumulación insignificante después de la administración una vez al día. XELJANZ XR 11 mg administrado una vez al dia, equivale a XELJANZ 5 mg administrado dos veces al día. Absorción: La administración concomitante de XELJANZ XR con una comida de alto contenido graso no produjo ningún cambio en el AUC mientras que la Cmáx se vio aumentada en un 27% y el Tmáx se extendió aproximadamente por 1 hora. Distribución: Después de administración intravenosa, el volumen de distribución es de 87 litros. La unión de proteínas de tofacitinib es ~40%. Tofacitinib se une principalmente a la albúmina y no parece unirse a a1-ácido glicoproteína. Tofacitinib distribuye igualmente entre los glóbulos rojos y el plasma. Metabolismo y eliminación: Los mecanismos de depuración de tofacitinib corresponden aproximadamente al 70% de metabolismo hepático y el 30% de excreción renal de droga madre. El metabolismo de tofacitinib es mediado principalmente por CYP3A4 con menor contribución de CYP2C19. En un estudio realizado en humanos con el fármaco radiomarcado, más del 65% del total de radiactividad circulante correspondió a tofacitinib sin cambios y el 35% restante se atribuyó a 8 metabolitos, representando cada uno menos del 8% de la radiactividad total. La actividad farmacológica de tofacitinib se atribuye a la molécula madre. Farmacocinética en pacientes con artritis reumatoidea: El análisis de farmacocinética poblacional realizado en pacientes con artritis reumatoidea no indicó ningún cambio clínicamente relevante en la exposición de tofacitinib, luego de tener en cuenta las diferencias en la función renal (es decir, depuración creatinina) entre pacientes, basado en la edad, peso, sexo y raza (Figura 1). Se observó una relación aproximadamente lineal entre el peso corporal y el volumen de distribución, dando como resultado un mayor pico (Cmáx) y concentraciones valle (Cmín) más bajas en los pacientes menos pesados. Sin embargo, esta diferencia no se considera clínicamente relevante. Se estima que la variabilidad entre sujetos (% de coeficiente de variación) en el AUC de tofacitinib es de aproximadamente un 27%. Poblaciones específicas: La Figura 1 indica el efecto de deterioro renal y hepático y otros factores intrínsecos en la farmacocinética de tofacitinib.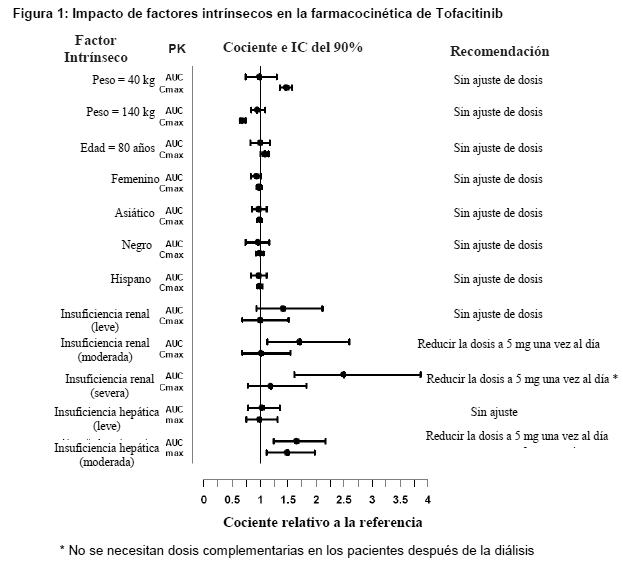
Los valores de referencia para el peso, edad, género y comparaciones de raza son 70 kg, 55 años, masculino y blanco, respectivamente;los grupos de referencia para los datos de deterioro renal y hepático son sujetos con función renal y hepática normal. Interacciones medicamentosas: Potencial de XELJANZ XR para influir sobre la farmacocinética de otros fármacos: Los estudios in vitro indican que tofacitinib no inhibe ni induce significativamente la actividad de las principales CYP metabolizadoras de fármacos en humanos (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4) en concentraciones superiores a 160 veces la Cmáx en estado estacionario de una dosis de 5 mg dos veces al día. Estos resultados in vitro fueron confirmados por un estudio de interacción medicamentosa en humanos que no demostró ningún cambio en la farmacocinética de midazolam, un sustrato de CYP3A4 altamente sensible, cuando se administró concomitantemente con XELJANZ. En pacientes con artritis reumatoide, la depuración oral de tofacitinib no varía con el tiempo, indicando que tofacitinib no normaliza la actividad de las enzimas CYP en pacientes con artritis reumatoidea. Por lo tanto, la administración concomitante con XELJANZ XR no debiera causar aumentos clínicamente relevantes en el metabolismo de los sustratos de las enzimas CYP en pacientes con artritis reumatoidea. Los datos in vitro indican que es baja la posibilidad de que tofacitinib inhiba los transportadores tales como P-glicoproteína, transportadores aniónicos o catiónicos orgánicos en concentraciones terapéuticas. La Figura 2 indica las recomendaciones posológicas para fármacos administrados concomitantemente después de la administración de Tofacitinib.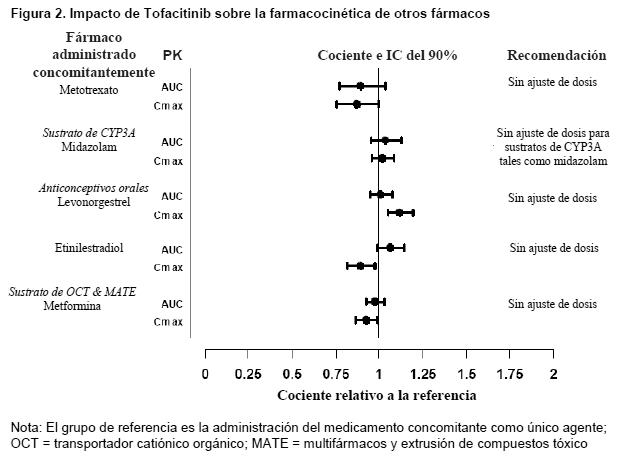
Potencial de otros fármacos para influir sobre la farmacocinética de Tofacitinib: Debido a que tofacitinib se metaboliza por CYP3A4, es probable la interacción con fármacos que inhiben o inducen CYP3A4. Es improbable que los inhibidores de CYP2C19 como único agente o P-glicoproteína alteren sustancialmente la farmacocinética de tofacitinib. La Figura 3 indica las recomendaciones posológicas para la administración de tofacitinib con inhibidores o inductores de CYP.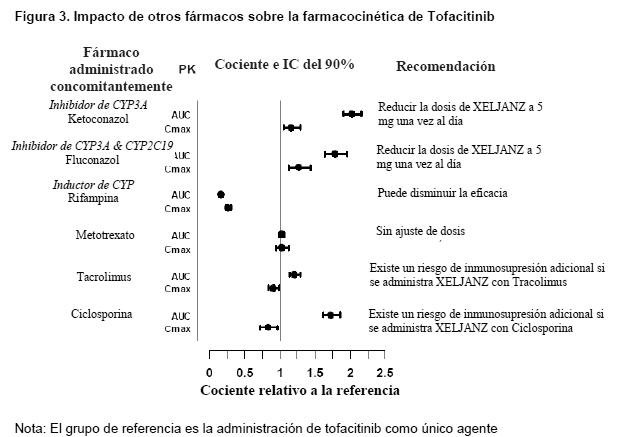
Toxicología no clínica: Carcinogénesis, mutagénesis, trastornos de fertilidad: En un estudio de toxicología de 39 semanas de duración realizado en monos, la exposición de tofacitinib produjo linfomas a niveles de aproximadamente 6 veces la MRHD (sobre una base de AUC en dosis orales de 5 mg/kg dos veces al día). No se observaron linfomas en este estudio con los niveles de exposición de 1 vez la MRHD (sobre una base de AUC en dosis orales de 1 mg/kg dos veces al día). El potencial carcinogénico de tofacitinib fue evaluado en estudios de carcinogenicidad en ratones transgénicos rasH2 de 6 meses de duración y estudios de carcinogenicidad en ratas de 2 años de duración. Tofacitinib, a niveles de exposición de aproximadamente 34 veces la MRHD (sobre una base de AUC en dosis orales de 200 mg/kg/día) no fue carcinogénico en ratones. En el estudio de carcinogenicidad oral de 24 meses realizado en ratas Sprague-Dawley, tofacitinib causó tumores benignos de células de Leydig, hibernomas (tumor del tejido adiposo pardo) y timomas benignos con dosis mayores o iguales a 30 mg/kg/día (aproximadamente 42 veces los niveles de exposición en la MRHD sobre una base de AUC). No se conoce la importancia de los tumores benignos de células de Leydig para el riesgo en humanos. Tofacitinib no fue mutagénico en el ensayo de mutación inversa de células bacterianas. Fue positivo para clastogenicidad en el ensayo de aberraciones cromosómicas in vitro con linfocitos humanos en presencia de enzimas metabólicas, pero negativo en ausencia de enzimas metabólicas. Tofacitinib fue negativo en el ensayo de micronúcleos en ratas in vivo y en el ensayo in vitro de mutación de CHO/HGPRT y el análisis in vivo de síntesis de ADN no programado en hepatocitos de rata. En ratas, tofacitinib redujo la fertilidad femenina debido a un aumento de la pérdida posterior al implante con niveles de exposición de aproximadamente 17 veces la MRHD (sobre una base de AUC en dosis orales de 10 mg/kg/día). No hubo ningún deterioro de la fertilidad de las ratas hembras con niveles de exposición de tofacitinib igual a la MRHD (sobre una base de AUC en dosis orales de 1 mg/kg/día). La exposición de tofacitinib en niveles de aproximadamente 133 veces la MRHD (sobre una base de AUC en dosis orales de 100 mg/kg/día) no tuvo efectos sobre la fertilidad de los machos, movilidad de los espermatozoides o concentración espermática. Estudios clínicos: El programa de desarrollo clínico de XELJANZ incluyó dos estudios fundamentales de determinación de la dosis entre dos y cinco ensayos confirmatorios. Aunque se estudiaron otras dosis, la dosis recomendada de XELJANZ es de 5 mg dos veces al día. Estudios de determinación de la dosis: La selección de la dosis de XELJANZ se basó en dos estudios fundamentales de determinación de la dosis. El Estudio 1 de Determinación de la Dosis fue un estudio de monoterapia de 6 meses de duración realizado en 384 pacientes con artritis reumatoidea activa que tuvieron una respuesta inadecuada a un DMARD. Se excluyó a los pacientes que previamente recibieron tratamiento con adalimumab. Los pacientes fueron aleatorizados para recibir a 1 de 7 tratamientos como monoterapia: 1, 3, 5, 10 ó 15 mg de XELJANZ dos veces al día, 40 mg de adalimumab por vía subcutánea semana por medio durante 10 semanas seguido por 5 mg de XELJANZ dos veces al día durante 3 meses, o placebo. El Estudio 2 de Determinación de la Dosis fue un estudio de 6 meses de duración en que 507 pacientes con la artritis reumatoidea activa que tuvieron una respuesta inadecuada a MTX como único agente recibieron uno de las 6 pautas posológicas de XELJANZ (20 mg una vez al día; 1, 3, 5, 10 ó 15 mg dos veces al día) o placebo agregado a MTX de base. La Figura 4 indica los resultados de los pacientes tratados con XELJANZ que alcanzaron respuestas ACR20 en los Estudios 1 y 2. Aunque en el Estudio 1 se observó una relación de respuesta a la dosis, la proporción de pacientes con una respuesta ACR20 no varió claramente entre las dosis de 10 mg y 15 mg. En el Estudio 2, una menor proporción de pacientes lograron una respuesta ACR20 en los grupos placebo y de 1 mg de XELJANZ en comparación con los pacientes tratados con las otras dosis XELJANZ. Sin embargo, no hubo diferencias en la proporción de los pacientes que respondieron al tratamiento entre los pacientes tratados con las dosis de 3, 5, 10, 15 mg de XELJANZ dos veces al día o 20 mg una vez al día.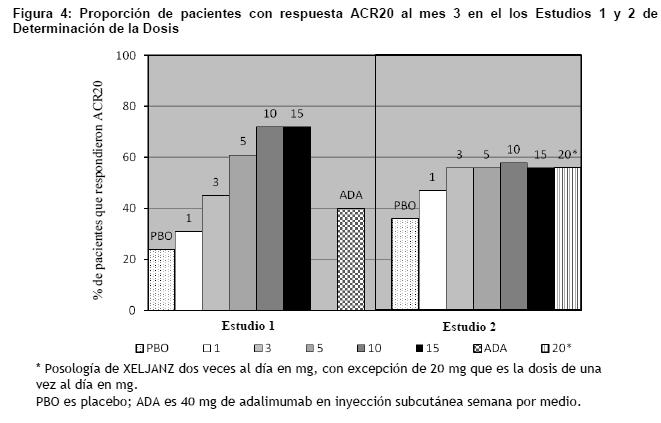
El estudio 1 fue un ensayo de rango de dosis de monoterapia, que no fue diseñado para proporcionar datos comparativos de efectividad y no se debería interpretar como evidencia de superioridad al adalimumab. Estudios de confirmación: El estudio 1 fue un estudio de monoterapia de 6 meses de duración en el que 610 pacientes con artritis reumatoidea activa de moderada a severa sin una respuesta adecuada a un DMARD (no biológico o biológico) recibieron 5 ó 10 mg de XELJANZ dos veces al día o placebo. En la visita del Mes 3, todos los pacientes aleatorizados para recibir tratamiento con placebo avanzaron de manera ciega a un segundo tratamiento predeterminado de 5 ó 10 mg de XELJANZ dos veces al día. Los criterios de valoración primaria en el Mes 3 fueron la proporción de pacientes que lograron una respuesta ACR20, cambios en el Cuestionario de Evaluación de la Salud - Índice de Discapacidad (HAQ-DI) y los índices de Puntuación de la Actividad de la Enfermedad DAS28-4(ESR) menos de 2,6. El Estudio II fue un estudio de 12 meses de duración en el que 792 pacientes con artritis reumatoide activa de moderada a severa sin una respuesta adecuada a un DMARD no biológico recibieron 5 ó 10 mg de XELJANZ dos veces al día o placebo agregado al tratamiento de base con DMARD (excluyendo potentes tratamientos inmunosupresores como azatioprina o ciclosporina). En la visita del Mes 3, los pacientes que no respondieron al tratamiento avanzaron de manera ciega a un segundo tratamiento predeterminado de 5 ó 10 mg de XELJANZ dos veces al día. Al final del Mes 6, todos los pacientes que recibieron placebo avanzaron a su segundo tratamiento predeterminado de manera ciega. Los criterios de valoración primaria fueron la proporción de pacientes que lograron una respuesta ACR20 en el Mes 6, cambios en el HAQ-DI en el Mes 3 e índices de DAS28-4(ESR) inferiores a 2,6 en el Mes 6. El Estudio III fue un estudio de 12 meses de duración realizado en 717 pacientes con artritis reumatoide activa de moderada a severa sin una respuesta adecuada a MTX. Los pacientes recibieron 5 ó 10 mg de XELJANZ dos veces al día, 40 mg de adalimumab por vía subcutánea semana por medio durante 10 semanas o placebo agregado al tratamiento de base con MTX. Los pacientes con placebo avanzaron como en el Estudio II. Los criterios de valoración primaria fueron la proporción de pacientes que lograron una respuesta ACR20 en el Mes 6, cambios en el HAQ-DI en el Mes 3 e índices de DAS28-4(ESR) inferiores a 2,6 en el Mes 6. El Estudio IV fue un estudio de 2 años de duración con un análisis planificado en 1 año en el que 797 pacientes con artritis reumatoide activa de moderada a severa sin una respuesta adecuada a MTX recibieron 5 ó 10 mg de XELJANZ dos veces al día o o placebo agregado al tratamiento de base con MTX. Los pacientes con placebo avanzaron como en el Estudio II. Los criterios de valoración primaria fueron la proporción de pacientes que lograron una respuesta ACR20 en el Mes 6, un cambio medio con respecto al inicio en la Puntuación total de Sharp modificada por van der Heijde (mTSS) en el Mes 6, HAQ-DI en el Mes 3 y DAS28-4(ESR) inferiores a 2,6 en el Mes 6. El Estudio V fue un estudio de 6 meses de duración en el que 399 pacientes con artritis reumatoidea activa de moderada a severa sin una respuesta adecuada a por lo menos un agente biológico inhibitorio de TNF aprobado recibieron 5 ó 10 mg de XELJANZ dos veces al día o placebo agregado al tratamiento de base con MTX. En la visita del Mes 3, todos los pacientes aleatorizados para recibir tratamiento con placebo avanzaron de manera ciega a un segundo tratamiento predeterminado de 5 ó 10 mg de XELJANZ dos veces al día. Los criterios de valoración primaria en el Mes 3 fueron la proporción de pacientes que lograron una respuesta ACR20, HAQ-DI y DAS28-4(ESR) inferiores a 2,6. El Estudio VI fue un estudio de monoterapia de 2 años con un análisis planificado en 1 año en el que 952 pacientes sin tratamiento con MTX con artritis reumatoide activa moderada a severa recibieron XELJANZ 5 o 10 mg dos veces al día o dosis tituladas de MTX de 20 mg semanales durante 8 semanas. Los criterios de valoración primarios fueron los cambios medios desde el valor basal en la puntuación total de Sharp (mTSS) de van der Heijde modificado en el Mes 6 y la proporción de pacientes que alcanzaron una respuesta ACR70 en el Mes 6. Respuesta clínica: La Tabla 5 indica los porcentajes de pacientes tratados con XELJANZ que lograron respuestas ACR20, ACR50 y ACR70 en los Estudios I, IV y V. Se observaron resultados similares con los Estudios II y III. En los estudios I-V, los pacientes tratados con 5 ó 10 mg de XELJANZ dos veces al día presentaron mayores tasas de respuesta ACR20, ACR50 y ACR70 en comparación con el placebo, con o sin tratamiento de base con DMARD, en el Mes 3 y Mes 6. Se observaron tasas de respuesta ACR20 dentro de 2 semanas en comparación con el placebo. En los estudios de 12 meses, las tasas de respuesta ACR de los pacientes tratados con XELJANZ fueron consistentes a los 6 y 12 meses.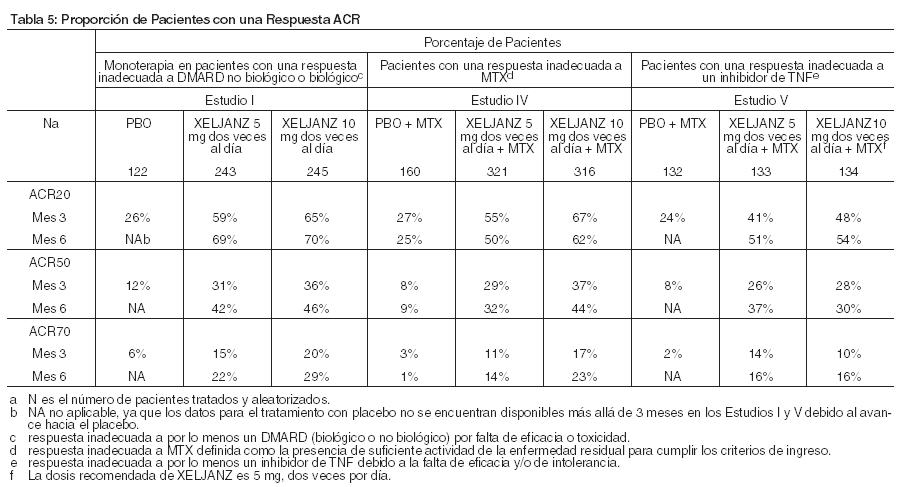
La Tabla 7 indica los resultados de los componentes de los criterios de respuesta ACR para el Estudio IV. Se observaron resultados similares para XELJANZ en los Estudios I, II, III, V y VI.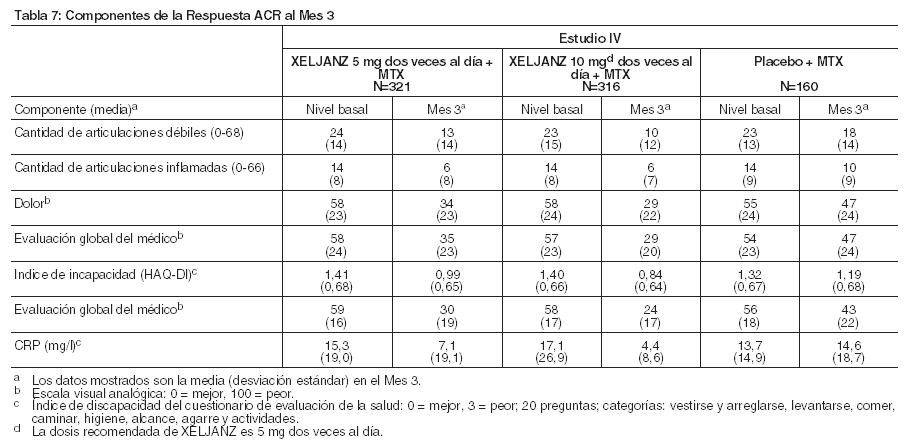
La Figura 5 indica el porcentaje de pacientes que respondieron ACR20 por visita para el Estudio IV. En los estudios I, II, III, V y VI se observaron respuestas similares para XELJANZ.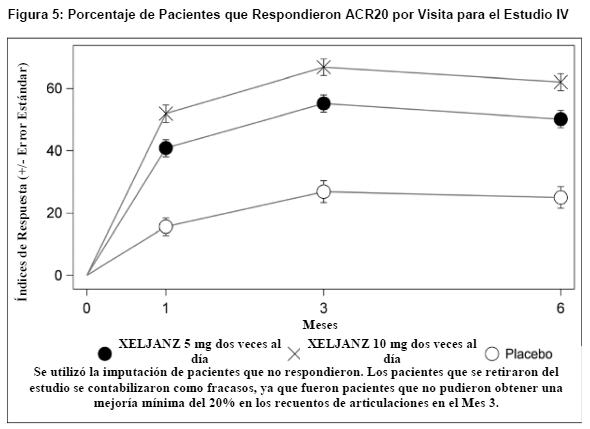
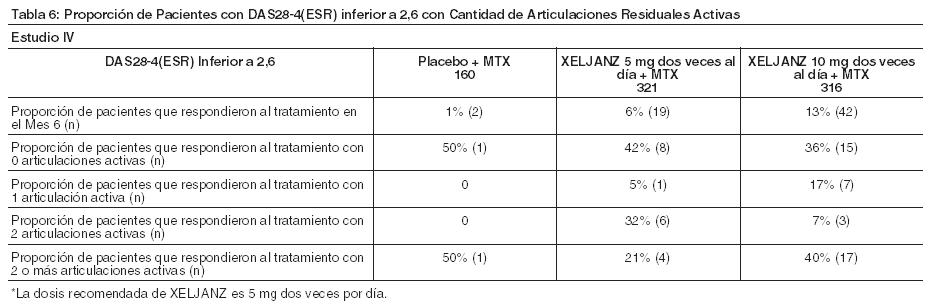
Respuesta radiográfica: Se llevaron a cabo dos estudios para evaluar el efecto de XELJANZ sobre el daño estructural de las articulaciones. En el Estudio IV y el Estudio VI, se evaluó en forma radiográfica la progresión del daño estructural de las articulaciones y se expresó como el cambio desde el valor basal en mTSS y sus componentes, la puntuación de erosión y la puntuación del estrechamiento del espacio articular, al Mes 6 y 12. También se evaluó la proporción de pacientes sin progresión radiográfica (cambio en mTSS menor o igual a 0). En el Estudio IV, XELJANZ 10 mg dos veces al día más MTX de fondo, redujeron la progresión del daño estructural en comparación con placebo más MTX al Mes 6. Cuando se administró a una dosis de 5 mg dos veces al día, XELJANZ exhibió efectos similares en la progresión media del daño estructural (sin importancia estadística). Estos resultados se muestran en la Tabla 8. Los análisis de las puntuaciones de la erosión y del estrechamiento del espacio articular fueron consistentes con los resultados generales. En el grupo de placebo más MTX, 74% de los pacientes no experimentó progresión radiográfica al Mes 6 en comparación al 84% y 79% de los pacientes tratados con XELJANZ más MTX 5 o 10 mg dos veces al día. En el Estudio VI, la monoterapia con XELJANZ inhibió la progresión del daño estructural en comparación con MTX al Mes 6 y 12 como se muestra en la Tabla 8. Los análisis de las puntuaciones de la erosión y del estrechamiento del espacio articular fueron consistentes con los resultados generales. En el grupo de MTX, el 55% de los pacientes no experimentó progresión radiográfica al Mes 6 en comparación al 73% y 77% de los pacientes tratados con XELJANZ 5 o 10 mg dos veces al día.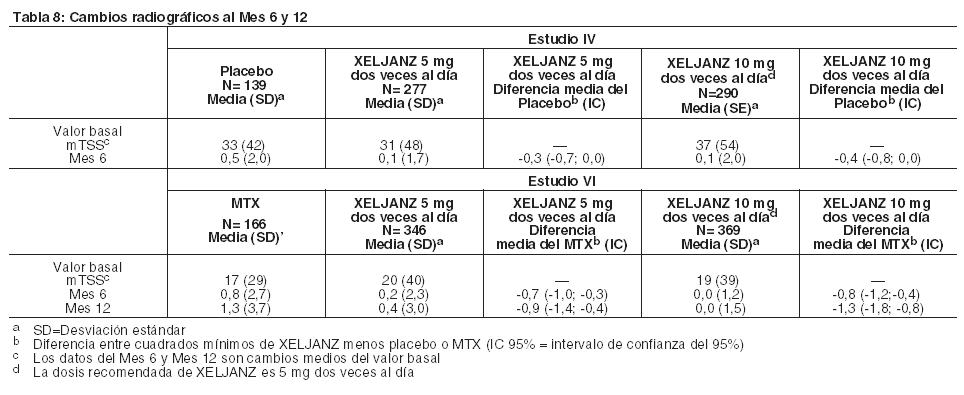
Respuesta de la función física: La mejoría en el funcionamiento físico se midió con el cuestionario HAQ-DI. Los pacientes que recibieron 5 y 10 mg de XELJANZ dos veces al día demostraron una mayor mejoría con respecto al inicio en el funcionamiento físico en comparación con placebo en el Mes 3. La diferencia de media (IC del 95%) con el placebo en la mejoría según HAQ-DI con respecto al inicio en el Mes 3 en el estudio III fue -0,22 (-0,35, -0,10) en pacientes que recibieron 5 mg de XELJANZ dos veces al día y -0.32 (-0.44, -0.19) en pacientes que recibieron 10 mg de XELJANZ dos veces al día. Se obtuvieron resultados similares en los Estudios I, II, IV y V. En los estudios de 12 meses, los resultados del HAQ-DI en pacientes tratados con XELJANZ fueron consistentes a los 6 y 12 meses. Otros resultados relacionados con la salud: El estado general de salud se evaluó mediante el cuestionario de salud Short Form (SF-36). En los Estudios I, IV, y V, los pacientes que recibieron XELJANZ 5 mg dos veces al día o XELJANZ 10 mg dos veces al día demostraron una mayor mejoría del valor basal en comparación con el placebo en la puntuación del resumen del componente físico (PCS, por sus siglas en inglés), del resumen del componente mental (MCS, por sus siglas en inglés) y en todos los 8 dominios del SF-36 al Mes 3.
Indicaciones.
XELJANZ XR está indicado para el tratamiento de pacientes adultos con artritis reumatoidea activa moderada a severa que han tenido una respuesta inadecuada o intolerancia a metotrexato. Puede utilizarse como monoterapia o en combinación con metotrexato u otros fármacos antirreumáticos no biológicos modificadores de la enfermedad (DMARD, por sus siglas en inglés). Limitaciones de uso: No se recomienda la administración de XELJANZ XR en combinación con DMARD biológicos o inmunosupresores potentes tales como azatioprina y ciclosporina.
Dosificación.
Dosis en Artritis reumatoidea: XELJANZ XR puede utilizarse como monoterapia o en combinación con metotrexato u otros fármacos antirreumáticos no biológicos modificadores de la enfermedad (DMARD, por sus siglas en inglés). La dosis recomendada de XELJANZ XR es 11 mg una vez al día. XELJANZ XR se administra en forma oral con o sin alimentos. Tomar los comprimidos recubiertos de XELJANZ XR enteras e intactas. No aplastar, escupir o masticar. Cambio de XELJANZ comprimidos recubiertos a XELJANZ XR comprimidos recubiertos: Los paciente en tratamiento con XELJANZ 5 mg dos veces al día pueden cambiar a XELJANZ XR 11 mg una vez al día, el día siguiente a la última dosis de XELJANZ 5 mg. Modificaciones de la dosis debidas a Infecciones Graves y Citopenias (ver Tablas 1, 2 y 3 a continuación): Se recomienda que XELJANZ XR no se inicie en pacientes con un recuento absoluto de linfocitos menor a 500 células/mm3, un recuento absoluto de neutrófilos (ANC, por sus siglas en inglés) menor a 1000 células/mm3 o en quienes tienen niveles de hemoglobina menores a 9 g/dL. Se recomienda una interrupción de la dosis para el abordaje de linfopenia, neutropenia y anemia (ver Advertencias y Precauciones y Reacciones Adversas). Evite administrar XELJANZ XR si un paciente desarrolla una infección grave hasta que la infección esté controlada. Modificaciones de la dosis debidas a Interacciones medicamentosas: En los pacientes que: Reciban potentes inhibidores del citocromo P450 3A4 (CYP3A4) (por ejemplo, ketoconazol). Reciban uno o más medicamentos concomitantes que producen una inhibición moderada de CYP3A4 y una inhibición potente de CYP2C19 (por ejemplo, fluconazol). La dosis recomendada es XELJANZ 5 mg una vez al día: Coadministración de inductores potentes del CYP3A4 (por ej. rifampicina) con XELJANZ XR puede resultar en una pérdida o una respuesta clínica reducida al XELJANZ XR. No se recomienda la coadministración de inductores potentes del CYP3A4 con XELJANZ XR. Modificaciones de la dosis en Pacientes con Deterioro Renal o Hepático: En pacientes: con insuficiencia renal moderada o severa, con deterioro hepático moderado. La dosis recomendada es XELJANZ 5 mg una vez al día: No se recomienda la administración de tofacitinib en pacientes con insuficiencia hepática severa.


Contraindicaciones.
El uso de XELJANZ XR está contraindicado en pacientes con hipersensibilidad al Tofacitinib o a algún otro componente de XELJANZ XR.
Reacciones adversas.
Experiencia en estudios clínicos: Como los estudios clínicos se llevan a cabo en condiciones que varían ampliamente, los índices de reacciones adversas observados en los estudios clínicos de un medicamento no pueden compararse directamente con los índices de estudios clínicos de otros medicamentos y pueden no reflejar los índices observados en la práctica clínica. Los estudios clínicos descriptos abajo fueron llevados a cabo usando XELJANZ. Aunque se estudiaron otras dosis, la dosis recomendada de XELJANZ XR es 11 mg una vez al día. Los siguientes datos incluyen dos estudios de Fase 2 y cinco estudios de Fase 3 controlados, doble ciego y multicéntricos. En estos estudios, los pacientes fueron aleatorizados para recibir dosis de 5 mg de XELJANZ dos veces al día (292 pacientes) y 10 mg dos veces al día (306 pacientes) en monoterapia, 5 mg de XELJANZ dos veces al día (1044 pacientes) y 10 mg dos veces al día (1043 pacientes) en combinación con DMARD (incluso metotrexato) y placebo (809 pacientes). Los siete protocolos incluyeron cláusulas que indican que los pacientes que reciben placebo deben recibir tratamiento con XELJANZ en el Mes 3 o Mes 6 ya sea según la respuesta del paciente (basado en la actividad de la enfermedad no controlada) o según el diseño, de manera tal que los eventos adversos no siempre pueden ser atribuidos inequívocamente a un tratamiento dado. Por consiguiente, algunos análisis que se indican a continuación incluyen pacientes que cambiaron de tratamiento por diseño o por respuesta del paciente de placebo a XELJANZ en ambos grupos de placebo y XELJANZ de un intervalo dado. Las comparaciones entre placebo y XELJANZ se basaron en los primeros 3 meses de exposición, y las comparaciones entre 5 mg de XELJANZ dos veces al día y 10 mg de XELJANZ dos veces al día se basaron en los primeros 12 meses de exposición. La población de seguridad a largo plazo incluye todos los pacientes que participaron en un estudio controlado y doble ciego (incluso los estudios anteriores de fase en desarrollo) y luego participaron en uno de dos estudios de seguridad a largo plazo. El diseño de los estudios de seguridad a largo plazo permitió la modificación de las dosis de XELJANZ según el juicio clínico. Esto limita la interpretación de los datos de seguridad a largo plazo con respecto a la dosis. Las reacciones adversas serias más frecuentes fueron infecciones serias (ver Advertencias y Precauciones). La proporción de pacientes que suspendieron el tratamiento por cualquier reacción adversa durante los meses 0 a 3 de exposición en los estudios controlados con placebo y doble ciego fue del 4% para los pacientes que recibieron XELJANZ y del 3% para los pacientes tratados con placebo. Infecciones generales: Durante los meses 0 a 3 de exposición en los siete estudios controlados, la frecuencia de infecciones fue del 20% y 22% en los grupos de 5 mg dos veces al día y 10 mg dos veces al día, respectivamente, y del 18% en el grupo placebo. Las infecciones informadas con mayor frecuencia con XELJANZ fueron infecciones de las vías respiratorias superiores, nasofaringitis, e infecciones de la vías urinarias (4%, 3% y 2% de los pacientes, respectivamente). Infecciones serias: Durante los meses 0 a 3 de exposición en los siete estudios controlados, se informaron infecciones serias en 1 paciente (0,5 eventos por 100 pacientes-año) que recibió placebo y en 11 pacientes (1,7 eventos por 100 pacientes-año) que recibieron 5 mg de XELJANZ dos veces al día o 10 mg de XELJANZ dos veces al día. La diferencia del índice entre los grupos de tratamiento (y el correspondiente intervalo de confianza del 95%) fue de 1,1 (-0,4, 2,5) eventos por 100 pacientes-año para el grupo combinado que recibió XELJANZ 5 mg dos veces al día y 10 mg dos veces al día menos placebo. Durante los meses 0 a 12 de exposición en los siete estudios controlados, se informaron infecciones serias en 34 pacientes (2,7 eventos por 100 pacientes-año) que recibieron 5 mg de XELJANZ dos veces al día y en 33 pacientes (2,7 eventos por 100 pacientes-año) que recibieron 10 mg de XELJANZ dos veces al día. La diferencia del índice entre las dosis de XELJANZ (y el correspondiente intervalo de confianza del 95%) fue de -0,1 (-1,3, 1,2) eventos por 100 pacientes-año para 10 mg de XELJANZ dos veces al día menos 5 mg de XELJANZ dos veces al día. Las infecciones serias que se informaron con mayor frecuencia con el uso de XALJANZ incluyeron neumonía, celulitis, herpes zoster e infección de las vías urinarias (ver Advertencias y Precauciones). Tuberculosis: Durante los meses 0 a 3 de exposición en los siete estudios controlados, no se informó tuberculosis en los pacientes que recibieron placebo, 5 mg de XELJANZ dos veces al día o 10 mg de XELJANZ dos veces al día. Durante los meses 0 a 12 de exposición en los siete estudios controlados, se informó tuberculosis en 0 pacientes que recibieron 5 mg de XELJANZ dos veces al día y en 6 pacientes (0,5 eventos por 100 pacientes-año) que recibieron 10 mg de XELJANZ dos veces al día. La diferencia del índice entre las dosis de XELJANZ (y el correspondiente intervalo de confianza del 95%) fue de 0,5 (0,1, 0,9) eventos por 100 pacientes-año para 10 mg de XELJANZ dos veces al día menos 5 mg de XELJANZ dos veces al día. También se informaron casos de tuberculosis diseminada. La mediana de la exposición a XELJANZ antes del diagnóstico de tuberculosis fue de 10 meses (rango de 152 a 960 días) (ver Advertencias y Precauciones). Infecciones oportunistas (excluida tuberculosis): Durante los meses 0 a 3 de exposición en los siete estudios controlados, no se informaron infecciones oportunistas en los pacientes que recibieron placebo, 5 mg de XELJANZ dos veces al día o 10 mg de XELJANZ dos veces al día. Durante los meses 0 a 12 de exposición en los siete estudios controlados, se informaron infecciones oportunistas en 4 pacientes (0,3 eventos por 100 pacientes-año) que recibieron 5 mg de XELJANZ dos veces al día y en 4 pacientes (0,3 eventos por 100 pacientes-año) que recibieron 10 mg de XELJANZ dos veces al día. La diferencia del índice entre las dosis de XELJANZ (y el correspondiente intervalo de confianza del 95%) fue de 0 (-0,5, 0,5) eventos por 100 pacientes-año para 10 mg de XELJANZ dos veces al día menos 5 mg de XELJANZ dos veces al día. La mediana de la exposición a XELJANZ antes del diagnóstico de una infección oportunista fue de 8 meses (rango de 41 a 698 días) (ver Advertencias y Precauciones.) Tumores malignos: Durante los meses 0 a 3 de exposición en los siete estudios controlados, se informaron tumores malignos excluido NMSC en 0 pacientes que recibieron placebo y en 2 pacientes (0,3 eventos por 100 pacientes-año) que recibieron 5 mg o 10 mg de XELJANZ dos veces al día. La diferencia del índice entre los grupos de tratamiento (y el correspondiente intervalo de confianza del 95%) fue de 0,3 (-0,1, 0,7) eventos por 100 pacientes-año para el grupo combinado que recibió XELJANZ 5 mg dos veces al día y 10 mg dos veces al día menos placebo. Durante los meses 0 a 12 de exposición en los siete estudios controlados, se informaron tumores malignos excluido NMSC en 5 pacientes (0,4 eventos por 100 pacientes-año) que recibieron 5 mg de XELJANZ dos veces al día y en 7 pacientes (0,6 eventos por 100 pacientes-año) que recibieron 10 mg de XELJANZ dos veces al día. La diferencia del índice entre las dosis de XELJANZ (y el correspondiente intervalo de confianza del 95%) fue de 0,2 (-0,4, 0,7) eventos por 100 pacientes-año para 10 mg de XELJANZ dos veces al día menos 5 mg de XELJANZ dos veces al día. Uno de estos tumores malignos fue un caso de linfoma que tuvo lugar durante el período de 0 a 12 meses en un paciente tratado con 10 mg de XELJANZ dos veces al día. Los tipos de tumores malignos más frecuentes, incluso los tumores malignos observados durante la extensión a largo plazo, fueron cáncer de pulmón y de mama, seguido por cáncer gástrico, colorrectal, de células renales, de próstata, linfoma, y melanoma maligno (ver Advertencias y Precauciones). Anormalidades de laboratorio: Linfopenia: En los estudios clínicos controlados, las disminuciones confirmadas en los recuentos absolutos de linfocitos por debajo de 500 células/mm3 tuvieron lugar en 0,04% de los pacientes de los grupos combinados que recibieron 5 mg de XELJANZ dos veces al día y 10 mg de XELJANZ dos veces al día durante los primeros 3 meses de exposición. Los recuentos confirmados de linfocitos inferiores a 500 células/mm3 se asociaron con un aumento en la incidencia de infecciones tratadas y serias (ver Advertencias y Precauciones). Neutropenia: En los estudios clínicos controlados, las disminuciones confirmadas en el recuento absoluto de neutrófilos por debajo de 1000 células/mm3 tuvieron lugar en 0,07% de los pacientes de los grupos combinados que recibieron 5 mg de XELJANZ dos veces al día y 10 mg de XELJANZ dos veces al día durante los primeros 3 meses de exposición. No se observaron disminuciones confirmadas en el recuento absoluto de neutrófilos por debajo de 500 células/mm3 en ningún grupo de tratamiento. No hubo una relación clara entre neutropenia y la aparición de infecciones serias. En la población de seguridad a largo plazo, el patrón y la incidencia de disminuciones confirmadas en el recuento absoluto de neutrófilos fueron congruentes con lo observado en los estudios clínicos controlados (ver Advertencias y Precauciones). Elevación de enzimas hepáticas: Se observaron aumentos confirmados en las enzimas hepáticas superiores a 3 veces el límite superior de lo normal (3x ULN, por sus siglas en inglés) en los pacientes tratados con XELJANZ. En los pacientes que tuvieron una elevación de las enzimas hepáticas, la modificación de la pauta posológica tal como la reducción de la dosis del DMARD concomitante, suspensión de XELJANZ o reducción de la dosis de XELJANZ, produjo una disminución o normalización de las enzimas hepáticas. En los estudios controlados de monoterapia (0-3 meses), no se observaron diferencias en la incidencia de elevaciones de ALT o AST entre los grupos placebo y los que recibieron 5 mg y 10 mg de XELJANZ dos veces al día. En los estudios controlados del tratamiento de base con DMARD (0-3 meses), se observaron elevaciones de ALT superiores a 3x ULN en el 1,0%, 1,3% y 1,2% de los pacientes que recibieron placebo, 5 mg y 10 mg dos veces al día, respectivamente. En estos estudios, se observaron elevaciones de ALT superiores a 3x ULN en el 0,6%, 0,5% y 0,4% de los pacientes que recibieron placebo, 5 mg y 10 mg dos veces al día, respectivamente. Se informó un caso de lesión hepática inducida por el fármaco en un paciente tratado con 10 mg de XELJANZ dos veces al día durante aproximadamente 2,5 meses. El paciente presentó elevaciones sintomáticas de AST y ALT superiores a 3x ULN y elevaciones de los niveles de bilirrubina superiores a 2x ULN que requirieron hospitalizaciones y una biopsia hepática. Elevación de los Lípidos: En los estudios clínicos controlados se observaron elevaciones relacionadas con la dosis en los parámetros lipídicos (colesterol total, colesterol LDL, colesterol HDL, triglicéridos) a un mes de exposición y permanecieron estables en adelante. A continuación se resumen los cambios en los parámetros lipídicos durante los primeros 3 meses de exposición en los estudios clínicos controlados: La media del colesterol LDL aumentó un 15% en el grupo que recibió 5 mg de XELJANZ dos veces al día y un 19% en el grupo que recibió 10 mg de XELJANZ dos veces al día. La media del colesterol HDL aumentó un 10% en el grupo que recibió 5 mg de XELJANZ dos veces al día y un 12% en el grupo que recibió 10 mg de XELJANZ dos veces al día. La media de los cocientes de LDL/HDL permanecieron esencialmente sin cambios en los pacientes tratados con XELJANZ. En un estudio clínico controlado, las elevaciones en el colesterol LDL y ApoB disminuyeron a los niveles previos al tratamiento en respuesta al tratamiento con estatina. En la población de seguridad a largo plazo, las elevaciones en los parámetros lipídicos fueron congruentes con lo observado en los estudios clínicos controlados. Elevaciones de la Creatinina sérica: En los estudios clínicos controlados se observaron elevaciones relacionadas con la dosis en la creatinina sérica con el tratamiento con XELJANZ. La media del aumento en la creatinina sérica fue < 0,1 mg/dl en el análisis de seguridad agrupado de 12 meses. Sin embargo, con el aumento en la duración de la exposición en las extensiones a largo plazo, hasta el 2% de los pacientes suspendieron el tratamiento con XELJANZ debido al criterio de suspensión especificado en el protocolo que indicaba un aumento en la creatinina de más del 50% con respecto al valor inicial. Se desconoce la significancia clínica de las elevaciones de creatinina sérica observadas. Otras reacciones adversas: En la Tabla 4 se resumen las reacciones adversas que ocurrieron en el 2% o más de los pacientes que recibieron 5 mg o 10 mg de XELJANZ dos veces al día y en al menos un 1% más que lo observado en los pacientes que recibieron placebo con o sin DMARD.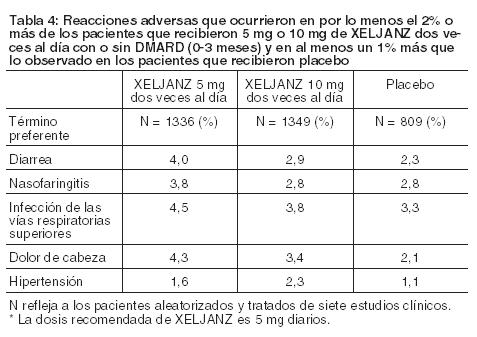
A continuación se indican otras reacciones adversas que tuvieron lugar en estudios abiertos de extensión y controlados. Trastornos de la sangre y del sistema linfático: anemia. Infecciones e infestaciones: Diverticulitis. Trastornos del metabolismo y de la nutrición: deshidratación. Trastornos psiquiátricos: insomnio. Trastornos del sistema nervioso: parestesia. Trastornos respiratorios, torácicos y mediastínicos: disnea, tos, congestión sinusal, enfermedad intersticial pulmonar (a veces fatal). Trastornos gastrointestinales: dolor abdominal, dispepsia, vómitos, gastritis, náuseas. Trastornos hepatobiliares: esteatosis hepática. Trastornos de la piel y del tejido subcutáneo: erupción cutánea, eritema, prurito. Trastornos musculoesqueléticos y del tejido conjuntivo: dolor musculoesquelético, artralgia, tendinitis, inflamación articular. Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos): cáncer de piel no melanoma. Trastornos generales y alteraciones en el lugar de administración: pirexia, fatiga, edema periférico. Experiencia Clínica en Pacientes sin Tratamiento con Metotrexato: El Estudio VI fue un e