XALKORI
PFIZER
Agente antineoplásico.
Composición.
Cada cápsula de XALKORI 200 mg contiene: Crizotinib 200 mg. Dióxido de silicio coloidal 2,00 mg. Celulosa microcristalina 83,00 mg. Fosfato de calcio dibásico anhidro 83,00 mg. Almidón sódico glicolato 20,00 mg. Estearato de magnesio 12,00 mg. Cada cápsula de XALKORI 250 mg contiene: Crizotinib 250 mg. Dióxido de silicio coloidal 2,50 mg. Celulosa microcristalina 103,75 mg. Fosfato de calcio dibásico anhidro 103,75 mg. Almidón sódico glicolato 25,00 mg. Estearato de magnesio 15,00 mg.
Farmacología.
Descripción: Crizotinib es un inhibidor de receptores de las tirosina quinasas. La fórmula molecular de crizotinib es C21H22Cl2FN5O. El peso molecular es de 450,34 daltons. Crizotinib se describe químicamente como (R)-3-[1- (2,6-dicloro-3-fluorofenil)etoxi]-5-[1-(piperidin-4-il)-1H-pirazol-4-il]piridina-2-amina. La estructura química de crizotinib se muestra a continuación: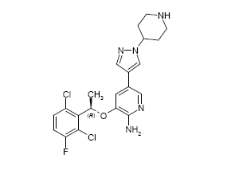
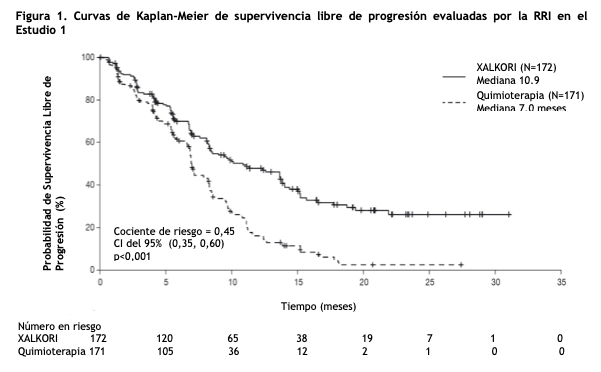
Crizotinib es un polvo blanco a amarillo claro con un pKa de 9,4 (catión piperidinio) y de 5,6 (catión piridinio). La solubilidad de crizotinib en medios acuosos se reduce en un rango de pH 1,6 a pH 8,2 de más de 10 mg/ml a menos de 0,1 mg/ml. El logaritmo del coeficiente de partición (octanol/agua) con un pH de 7,4 es de 1,65. XALKORI (crizotinib) para administración oral, se presentan como cápsulas de cubierta dura impresas que contienen 250 mg o 200 mg de crizotinib junto con dióxido de silicio coloidal, celulosa microcristalina, fosfato de calcio dibásico anhidro, glicolato sódico de almidón, estearato de magnesio y cubierta de cápsula de gelatina dura como excipientes. La cubierta opaca de color rosado de la cápsula contiene gelatina, dióxido de titanio y óxido de hierro rojo. La cubierta opaca de color blanco de la cápsula contiene gelatina y dióxido de titanio. La tinta de impresión contiene goma laca, propilenglicol, solución amoniacal potente, hidróxido de potasio y óxido de hierro negro. Propiedades farmacodinámicas: Mecanismo de acción: Crizotinib en un inhibidor de receptores de las tirosina quinasas, que incluyen ALK, el receptor del factor de crecimiento hepatocitario (HGFR, c-Met), ROS1 (c-ros) y Recepteur d'Origine Nantais (RON). Las traslocaciones pueden afectar el gen ALK con la consiguiente expresión de proteínas de fusión oncogénica. La formación de proteínas de fusión ALK tiene como consecuencia la activación y desregulación de la expresión y señalización del gen que puede contribuir a un aumento de la proliferación y supervivencia celular en tumores que expresan estas proteínas. Crizotinib demostró una inhibición dependiente de la concentración de la fosforilación de ALK, ROS1 y c-Met en ensayos basados en células en los que se utilizaron líneas celulares tumorales y demostró actividad antitumoral en ratones con xenoinjertos de tumores que expresaron proteína 4 asociada al microtúbulo de equinodermo EML4 o nucleofosmina (NPM) proteína de fusión ALK o c-Met. Farmacodinamia: Electrofisiología cardíaca: En un subestudio de ECG realizado en 52 pacientes con CPCNP ALK-positivo, la media máxima de cambio de QTcF (corregido según el método de Fridericia) desde el período basal fue de 12,3 ms (IC superior bilateral del 90%: 19,5 ms) luego de la administración de una dosis de XALKORI de 250 mg dos veces al día. Un análisis de exposición de QT indicó un aumento en el QTcF dependiente de las concentraciones plasmáticas de crizotinib (ver Advertencias). Propiedades farmacocinéticas: Luego de la administración de 250 mg de XALKORI, dos veces al día, se alcanzó un estado estacionario al cabo de 15 días, y este permaneció estable, con una razón mediana de acumulación de 4,8. El estado de equilibrio [concentración mínima observada (Cmín. y ABC)] aumentó en mayor medida que la proporcional a la dosis con un rango de dosis de 200 mg a 300 mg, dos veces al día (0,8 a 1,2 veces la dosis recomendada aprobada). Absorción: Luego de una dosis única por vía oral, crizotinib se absorbió con una mediana de tiempo hasta la concentración máxima de 4 a 6 horas y la media de la biodisponibilidad absoluta de crizotinib fue del 43% (rango: 32% a 66%). Efecto de las comidas: Una comida de alto contenido graso redujo el ABCinf y la concentración máxima observada en plasma (Cmáx) de crizotinib en aproximadamente un 14%. Distribución: La media geométrica del volumen de distribución (Vss) de crizotinib fue de 1772l luego de la administración intravenosa de una dosis. La unión de crizotinib a proteínas es de un 91% y es independiente de la concentración del medicamento in vitro. El crizotinib es un sustrato de la glicoproteína p (P-gp) in vitro. La razón de la concentración de sangre a plasma fue de aproximadamente 1. Eliminación: La vida media terminal plasmática aparente de crizotinib fue de 42 horas, luego de una dosis única de crizotinib en los pacientes. La media de la depuración aparente (CL/F) de crizotinib fue menor en el estado estacionario (60 l/h) luego de la administración de 250 mg, dos veces al día, que luego de una dosis única de 250 mg por vía oral (100 l/h). Metabolismo: Crizotinib es metabolizado predominantemente por la CYP3A4/5. Excreción: Luego de la administración en pacientes sanos de una dosis de 250 mg de crizotinib radiomarcada, el 63% (53% sin alteraciones) de la dosis administrada, se recuperó en heces y un 22% (2,3% sin alteraciones), en orina. Poblaciones especiales: No se han observado diferencias clínicas significativas en la farmacocinética del crizotinib, en base a la edad, sexo, etnia (asiáticos o no asiáticos) o peso corporal. Pacientes con deterioro de la función hepática: La media del estado estacionario de crizotinib ABC y Cmax, se redujo en un 9% en pacientes con deterioro hepático leve (AST mayor al LSN y bilirrubina total menor o igual a 1 vez el LSN o cualquier AST y bilirrubina total mayor a 1 vez el LSN, pero menor o igual a 1,5 veces el LSN), en comparación con los paciente con función hepática normal luego de una dosis de 250 mg oral de XALKORI dos veces al día. La media del estado estacionario de crizotinib ABC aumento un 14% y la Cmax aumento un 9% en pacientes con insuficiencia hepática moderada (cualquier AST y bilirrubina total mayor a 1,5 veces el LSN y menor o igual a 3 veces el LSN) luego de la administración de XALKORI 200 mg oral dos veces al día en comparación con pacientes con función hepática normal, luego de una dosis de XALKORI de 250 mg oral dos veces al día. La media del ABC se redujo en un 35% y la Cmax se redujo en un 27% en pacientes con insuficiencia hepática severa (cualquier AST y bilirrubina total mayor a 3 veces el LSN), luego de una dosis de XALKORI de 250 mg oral 1 vez al día, en comparación con pacientes con función hepática normal, luego de una dosis de XALKORI de 250 mg oral dos veces al día. (ver Posologia y metodo de administracion y Uso en poblaciones expecificas). Deterioro de la función renal: La insuficiencia renal leve o moderada (CLcr 60-89 ml/min o 30-59 ml/min, respectivamente calculado por la ecuación modificada de Cockcroft-Gault) no mostró un efecto clínicamente relevante en la exposición a crizotinib. Luego de una dosis única de 250 mg, la media ABC0-inf del crizotinib aumento un 79% y la media Cmax aumento un 34%, en pacientes con insuficiencia renal severa (CLcr < 30 ml/min) los cuales no requerían diálisis, en comparación con pacientes con función renal normal (CLcr ≥90 ml/min). Se observaron cambios similares en ABCinf y Cmáx para el metabolito activo de crizotinib (Ver Posología y forma de administración y Uso en poblaciones específicas). Estudios de Interacciones medicamentosas: Estudios clínicos: Agentes reductores de la acidez: No se han observado diferencias clínicas significativas en la farmacocinética de crizotinib, cuando se utilizó concomitantemente con esomeprazol, un inhibidor de la bomba de protones. Inhibidores potentes de la CYP3A: La administración concomitante de una dosis única de 150 mg por vía oral de crizotinib con ketoconazol, un inhibidor potente de la CYP3A, aumentó el ABCinf en un 216% y la Cmax en un 44%, en comparación con crizotinib solo. La coadministración de XALKORI 250 mg oral 1 vez al día con itraconazol, un inhibidor fuerte de la CYP3A, aumento el estado basal del ABC en un 57% y la Cmax en un 33%, en comparación con crizotinib solo (Ver Precauciones, Interacción con otros medicamentos y otras formas de interacción). Inductores potentes de la CYP3A: La administración concomitante de XALKORI 250 mg oral dos veces al día con rifampicina, un inductor potente de la CYP3A, redujo el ABC0-tau en un 84% y la Cmáx en un 79% del crizotinib, en comparación con crizotinib solo (Ver Interacción con otros medicamentos y otras formas de interacción). Sustratos de la CYP3A: La coadministración de XALKORI 250 mg oral dos veces al día durante 28 días aumentó la ABC0-inf de midazolam oral (sustrato CYP3A) 3,7 veces en comparación con midazolam solo (Ver Interacciones con otros medicamentos y otras formas de interacción). Estudios in vitro: Enzimas CYP: Crizotinib es un inhibidor del CYP2B6 in vitro. El crizotinib no inhibe a CYP1A2, CYP2C8, CYP2C9, CYP2C19 o CYP2D6. El crizotinib no induce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 o CYP3A. Glucuroniltransferasa UDP-(UGT): El crizotinib no inhibe UGT1A1, UGT1A4, UGT1A6, UGT1A9 o UGT2B7. Transportadores: Crizotinib inhibe la P-gp, al transportador de cationes orgánicos (OCT)1 y al OCT2. El crizotinib no inhibe a los aniones orgánicos que transportan poliproteínas (OATP)B1, OATP1B3, a los aniones de transporte orgánico (OAT)1, OAT3, o la bomba exportadora de sales biliares hepática (BSEP). Carcinogénesis, mutagénesis, trastornos de fertilidad: No se han realizado estudios de carcinogenicidad con crizotinib. Crizotinib fue genotóxico en un ensayo in vitro de micronúcleos en cultivos de ovarios de hámster chino, en un ensayo in vitro de aberraciones cromosómicas de linfocitos humanos, y en ensayos in vivo de micronúcleos de médula ósea en ratas. Crizotinib no fue mutagénico in vitro en el ensayo de mutación bacteriana reversa (Ames). No se han realizado estudios específicos con crizotinib en animales para evaluar su efecto sobre la fertilidad. Sin embargo, se considera que crizotinib puede potencialmente reducir la función reproductiva y la fertilidad en humanos, sobre la base de hallazgos de estudios de toxicidad con dosis repetidas en ratas. Los hallazgos observados en el tracto reproductivo de los machos incluyeron degeneración de espermatocitos paquitenos testiculares en ratas que recibieron una dosis igual o mayor de 50 mg/kg/día durante 28 días (más de 1,7 veces el ABC con la dosis humana recomendada). Los hallazgos observados en el tracto reproductivo de hembras incluyeron necrosis unicelular de folículos ováricos en una rata que recibió 500 mg/kg/día (aproximadamente 10 veces la dosis humana recomendada por mg/m2) durante 3 días. Ensayos clínicos: CPCNP metastásico ALK-positivo sin tratamiento previo - Estudio 1 (Perfil 1014; NCT01154140): Se demostró la eficacia de XALKORI para el tratamiento de pacientes con CPCNP ALK-positivo metastásico, que no hayan recibido tratamiento sistémico previo para la enfermedad avanzada, en un ensayo aleatorizado, metacéntrico, controlado con principio activo y abierto (Estudio 1). Era necesario que los pacientes tuvieran un CPCNP ALK-positivo identificado por un método aprobado por la FDA, el kit Vysis ALK Break-Apart hibridación fluorescente in situ (FISH) Probe, antes de la aleatorización. El resultado principal de eficacia fue la supervivencia libre de progresión (SLP), en base al Criterio de Evaluación de Respuesta en Tumores Sólidos (RECIST) versión 1.1, según la evaluación de la revisión radiológica independiente (RRI). Los resultados adicionales de eficacia incluyeron la tasa de respuesta objetiva (TRO) según la evaluación de la RRI, Duración de Respuesta (DDR) y la supervivencia global (SG). Los síntomas del cáncer de pulmón informados por los pacientes fueron evaluados al inicio y periódicamente durante el tratamiento. Se aleatorizaron los paciente para recibir XALKORI (n=172) o quimioterapia (n=171). La aleatorización se estratificó por el estado de desempeño según el Grupo Cooperativo Oncológico del Este (ECOG) (0-1,2), raza (asiáticos y no asiáticos) y metástasis cerebrales (presentes, ausentes). Los pacientes de la rama de XALKORI recibieron XALKORI 250 mg en forma oral dos veces al día, hasta la progresión documentada de la enfermedad, intolerancia al tratamiento o hasta que el investigador determinara que el paciente ya no experimentaba un beneficio clínico. La quimioterapia consistió en pemetrexed 500 mg/m2 combinado con cisplatino 75 mg/m2 o carboplatino a una dosis área bajo la curva (AUC) de 5 o 6 mg·min/mL por vía intravenosa cada 3 semanas hasta por 6 ciclos. No se les permitió a los pacientes en la rama de quimioterapia, recibir quimioterapia de mantenimiento. Al momento de la progresión documentada de la enfermedad y según la evaluación de la revisión radiológica independiente, a los pacientes aleatorizados a quimioterapia se les ofreció XALKORI. Las características demográficas de toda la población del estudio fueron: 62% mujeres, mediana de edad 53 años, estado de desempeño basal ECOG 0 o 1 (95%), 51% de raza blanca y 46% asiática, 4% fumadores actuales, 32% ex fumadores y 64% no fumadores. Las características de la enfermedad de la población general del estudio fueron: enfermedad metastásica en 98% de los pacientes y el 92% de los tumores de los pacientes se clasificaron como una histología de adenocarcinoma, 27 % de los pacientes tenían metástasis cerebrales y 7% de los pacientes habían recibido quimioterapia sistémica como terapia adyuvante o neo adyuvante. Al momento del análisis final de la supervivencia global, el 84% de los pacientes aleatorizados a quimioterapia, recibieron subsecuentemente XALKORI. El Estudio 1 demostró una mejora estadísticamente significativa en la SLP en pacientes tratados con XALKORI. No hubo una diferencia estadísticamente significativa en la supervivencia global entre los pacientes tratados con XALKORI y los pacientes tratados con quimioterapia. La Tabla 1 y la Figura 1 resumen los resultados de eficacia. La medición de síntomas basales y post tratamiento de disnea, tos y dolor en el pecho, reportados por los pacientes, sugirieron un retraso en el tiempo hasta la aparición o el empeoramiento de la disnea, pero no de la tos o el dolor en el pecho, en los pacientes tratados con XALKORI en comparación con quimioterapia. El retraso en el reporte del paciente en el inicio o empeoramiento de la disnea puede deberse a una sobreestimación, ya que los pacientes no estaban cegados al momento de la asignación del tratamiento.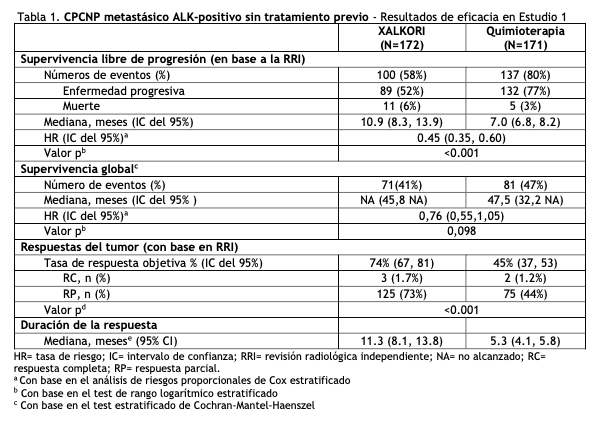
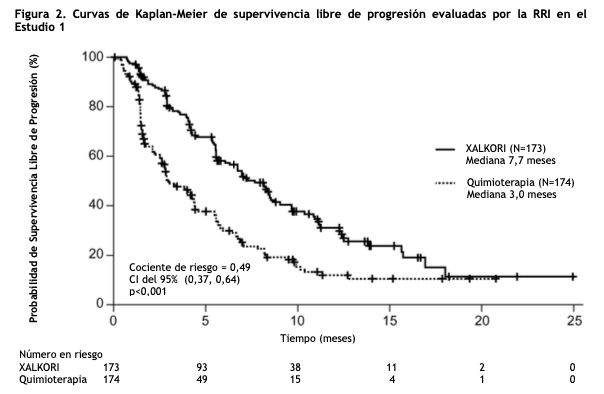
CPCNP ALK-positivo metastásico, previamente tratados - Estudio 2 (Perfil 1007, NCT00932893): Se demostró la eficacia de XALKORI como monoterapia para el tratamiento de 347 pacientes con CPCNP ALK-positivo metastásico, previamente tratados con un régimen quimioterápico basado en platino, en un estudio aleatorizado, multicéntrico, abierto, con control activo (Estudio 2). El resultado principal de eficacia fue la supervivencia libre de progresión (SLP) según RECIST versión 1.1, evaluado por la revisión radiológica independiente (RRI). Los resultados adicionales de eficacia incluyeron la tasa de respuesta objetiva (TRO) según la evaluación de la RRI, Duración de Respuesta (DDR) y supervivencia global (SG). Se aleatorizaron los pacientes para recibir XALKORI 250 mg en forma oral dos veces al día (n= 173) o quimioterapia (n= 174). La quimioterapia consistió en pemetrexed 500 mg/m2 (para pacientes sin tratamiento previo con pemetrexed, n= 99) o docetaxel 75 mg/m2 (n= 72) intravenoso (IV) cada 21 días. Los pacientes en ambas ramas de tratamiento continuaron con el tratamiento hasta la progresión documentada de la enfermedad, intolerancia al tratamiento o hasta que el investigador determinara que el paciente ya no experimentaba un beneficio clínico. La aleatorización se estratificó por el estado de desempeño ECOG (0-1, 2), metástasis cerebrales (presentes, ausentes) y tratamiento previo con un inhibidor de la tirosina quinasa EGFR (sí, no). Era necesario que los pacientes tuvieran un CPCNP ALK- positivo identificado por el ensayo aprobado de la FDA, el kit Vysis ALK Break-Apart FISH Probe, antes de la aleatorización. Las características demográficas de toda la población del estudio fueron de 56% mujeres, mediana de edad 50 años, estado de desempeño basal ECOG 0 o 1 (90%), 52% de raza blanca y 45% asiática, 4% fumadores actuales, 33% ex fumadores y 63% no fumadores. Las características de la enfermedad de la población total fue enfermedad metastásica en al menos 95% de los pacientes y al menos el 93% de los tumores de los pacientes se clasificaron como una histología de adenocarcinoma. Al momento del análisis final de la supervivencia global, 89% de los pacientes aleatorizados al grupo de quimioterapia recibió subsecuentemente XALKORI. El Estudio 2 demostró una mejora estadísticamente significativa en la SLP de los pacientes tratados con XALKORI. La Tabla 2 y la Figura 2 resumen los resultados de eficacia.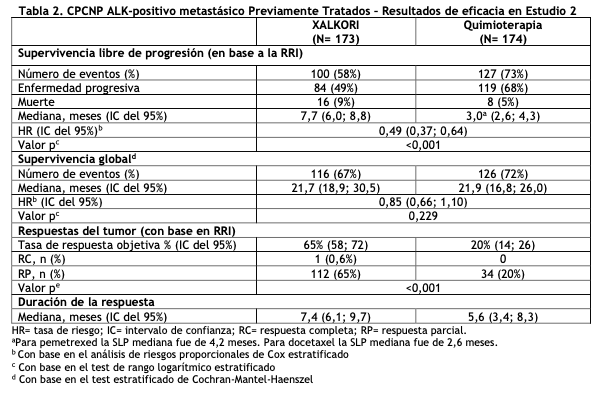
CPCNP metastásico ROS1 positivo - Estudio 3 (Perfil 1001, NCT00585195): La eficacia y seguridad de XALKORI fue investigada en un estudio multicéntrico, de una sola rama (Estudio 3), en el que pacientes con CPCNP metastásico con reordenamiento de ROS 1 recibieron XALKORI 250 mg por vía oral dos veces al día. Era necesario que los pacientes tuvieran CPCNP avanzado confirmado por histología ROS 1 positivo, 18 años o mayor, estado de desempeño basal ECOG de 0, 1 o 2, y enfermedad medible. El resultado de eficacia fue TRO y DDR de acuerdo al RECIST versión 1.0, según la evaluación de la RRI y del investigador, con imagen radiológica cada 8 semanas durante las primeras 60 semanas. Las características demográficas y de enfermedad fueron mujeres (56%), mediana de edad 53 años, valor basal del estado de desempeño (ECOG) 0 o 1 (98%), raza blanca 54% y asiática 42%, 22% ex fumadores, 78% no fumadores, 92% con enfermedad metastásica, 96% con adenocarcinoma, 14 % no recibieron quimioterapia sistémica para enfermedad metastásica y 80% recibieron quimioterapia previa basada en platino para enfermedad metastásica. El estado ROS1 fue determinado sobre las muestras de tejido de CPCNP por FISH con punto de quiebre desarrollado por un laboratorio (96%) o por RT-PCR (4%). Para la evaluación de positividad de ROS1 por FISH, se requirió ≥15% de un mínimo de 50 núcleos evaluados contenidos en un reordenamiento del gen ROS1.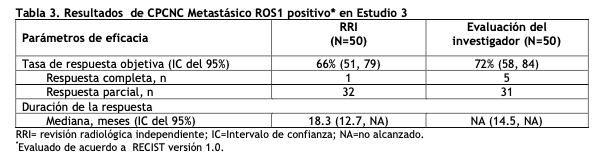
Indicaciones.
CPCNP metastásico ALK positivo: XALKORI es un inhibidor de quinasas indicado para el tratamiento de pacientes con cáncer de pulmón de células no pequeñas metastásico (CPCNP) cuyos tumores son positivos para la quinasa del linfoma anaplásico (ALK, por sus siglas en inglés), determinado mediante un test aprobado por ANMAT. CPCNP metastásico ROS1 positivo: XALKORI está indicado para el tratamiento de pacientes con CPCNP metastásico, cuyos tumores son positivos para ROS1.
Dosificación.
Posología recomendada: La dosis recomendada de XALKORI es de 250 mg por vía oral, con o sin alimentos, dos veces al día hasta la progresión de la enfermedad o hasta que el paciente no lo tolere. Las cápsulas deben ingerirse enteras. En caso de omisión de una dosis de XALKORI, compense dicha dosis, salvo que resten menos de 6 horas hasta la dosis siguiente. Si se producen vómitos luego de la ingesta de una dosis de XALKORI, tome la próxima dosis a la hora habitual. Modificación de la dosis debido a reacciones adversas: Las reducciones de dosis recomendadas se describen a continuación: Primera reducción de dosis: XALKORI 200 mg, vía oral, dos veces al día. Segunda reducción de dosis: XALKORI 250 mg, vía oral, una vez al día. Interrumpa en forma permanente si no puede tolerar XALKORI 250 mg por vía oral, una vez al día. En las tablas 4 y 5, se proporcionan las modificaciones de las dosis a causa de las reacciones adversas con XALKORI.
Se debe monitorear el hemograma completo, incluido el recuento diferencial de glóbulos blancos, cada mes y según lo indicado clínicamente, con pruebas repetidas con más frecuencia si se observan anomalías de Grado 3 o 4 o si se produce fiebre o infección.
Una vez al mes, deben realizarse hemogramas completos que incluyan un recuento diferencial de leucocitos según la indicación clínica, con un aumento de la frecuencia de repetición de análisis en caso de observarse anomalías de grado 3 ó 4, o en caso de fiebre o infección. Modificación de la dosis en insuficiencia hepática moderada y severa: La dosis recomendada de XALKORI en pacientes con deterioro hepático moderado preexistente (cualquier aspartato aminotransferasa (AST) y bilirrubina total mayor a 1,5 veces el límite superior normal (LSN) y menor o igual a 3 veces el LSN), es de 200 mg orales dos veces al día. La dosis recomendada de XALKORI en pacientes con deterioro hepático severo preexistente (cualquier AST y bilirrubina total mayor a 3 veces el LSN) es de 250 mg oral una vez al día (ver Uso en poblaciones específicas y Propiedades fármacodinámicas). Modificación de la dosis en insuficiencia renal severa: La dosis de XALKORI recomendada en pacientes con deterioro renal severo (depuración de creatinina < 30 ml/min calculada usando la ecuación modificada de Cockcroft-Gault) que no requieren diálisis es de 250 mg vía oral, una vez al día (Ver Uso en poblaciones específicas y Características farmacológicas, Propiedades farmacodinámicas). Modificación de la dosis para el uso concomitante de inhibidores potentes del CYP3A: Se debe evitar el uso concomitante de inhibidores potentes del CYP3A. Si el uso concomitante de inhibidores potentes del CYP3A es inevitable, se debe reducir la dosis de XALKORI a 250 mg por vía oral 1 vez al día (ver Interaccion con otros medicamentos y otras formas de interaccion). Luego de discontinuar el inhibidor potente del CYP3A, se debe iniciar la dosis de XALKORI, utilizada antes de la administración concomitante con el inhibidor potente del CYP3A.
Contraindicaciones.
El uso de XALKORI está contraindicado en pacientes con hipersensibilidad al crizotinib o a algún otro componente de XALKORI.
Reacciones adversas.
Las siguientes reacciones adversas clínicamente significativas se describen en otras secciones del prospecto: Hepatotoxicidad (ver Advertencias y precauciones). Enfermedad pulmonar intersticial/neumonitis (ver Advertencias y precauciones). Prolongación del intervalo QTc (ver Advertencias y precauciones). Bradicardia (ver Advertencias y precauciones). Pérdida severa de la visión (ver Advertencias y precauciones). Experiencia en ensayos clínicos: Dado que los estudios clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observados en los ensayos clínicos de un medicamento no pueden compararse directamente a las tasas en estudios clínicos de otro medicamento y es posible que no reflejen las tasas observadas en la práctica clínica. Los datos en Advertencias y precauciones reflejan la exposición a XALKORI en 1719 pacientes que recibieron XALKORI 250 mg dos veces al día incluidos en los Estudios 1 (incluyendo 109 pacientes adicionales que cruzaron desde el brazo de control), 2, 3, un ensayo de un solo brazo (n = 1063) de CPCNP ALK positivo, y un cohorte de expansión de estudio de hallazgo de dosis en pacientes con CPCNP (n = 154). La data descripta abajo se basa principalmente en 343 pacientes con CPCNP ALK-positivo metastásico que recibieron XALKORI a una dosis de 250 mg dos veces al día por vía oral, en dos ensayos abiertos aleatorizados, controlados con principio activo (Estudios 1 y 2). La seguridad de XALKORI fue también evaluada en 50 pacientes con CPCNP metastásico ROS1 positivo de un estudio de un solo brazo (Estudio 3). Las reacciones adversas más comunes (≥25%) de XALKORI son trastornos visuales, diarrea, vómitos, constipación, edema, elevación de las transaminasas, fatiga, infección respiratoria superior, disminución del apetito, mareos Y neuropatía. Estudio 1 de CPCNP Metastásico ALK-positivo Sin Tratamiento Previo (Perfil 1014): Los datos en la Tabla 6 derivan de 340 pacientes con CPCNP metastásico ALK-positivo que no hayan estado bajo tratamiento sistémico previo para la enfermedad avanzada que recibieron tratamiento en un ensayo aleatorizado, multicéntrico, controlado con principio activo y abierto (Estudio 1). Los pacientes en el grupo de XALKORI (n=171) recibieron XALKORI 250 mg oralmente dos veces por día hasta que se documentó la progresión de la enfermedad, la intolerancia al tratamiento, o que el investigador determinó que el paciente no presentaba más beneficios clínicos. Un total de 169 pacientes en el grupo de quimioterapia recibieron 500 mg/m2 de pemetrexed con cisplatino 75 mg/m2 (n=91) o carboplatino a una dosis calculada para producir un área bajo la curva de concentración-tiempo (ABC) de 5 o 6 mg·min/mL (n=78). La quimioterapia se administró mediante infusión intravenosa cada 3 semanas por hasta 6 ciclos, en ausencia de toxicidades limitantes de la dosis relacionadas con la quimioterapia. Luego de los 6 ciclos, los pacientes permanecieron en el estudio sin tratamientos anti-cáncer adicionales, y las evaluaciones de los tumores continuaron hasta que se documentó la progresión de la enfermedad. La duración mediana del tratamiento del estudio fue de 10,9 meses para los pacientes del grupo que recibió XALKORI y de 4,1 meses para los pacientes del grupo que recibió quimioterapia. La mediana de la duración del tratamiento fue de 5,2 meses para pacientes que recibieron XALKORI luego del cruce desde quimioterapia. Entre los 340 pacientes que fueron tratados en el Estudio 1, la edad mediana era de 53 años; 16% de los pacientes eran mayores de 65 años. Un total del 62% de los pacientes eran mujeres y el 46% provenían de Asia. Se informaron reacciones adversas graves en 34% de los pacientes tratados con XALKORI. Las reacciones adversas graves más frecuentes informadas en pacientes tratados con XALKORI fueron disnea (4,1%) y embolia pulmonar (2,9%). Las reacciones adversas mortales en pacientes tratados con XALKORI ocurrieron en 2,3% pacientes, que comprendían: shock séptico, insuficiencia respiratoria aguda, y cetoacidosis diabética. Se requirieron reducciones de la dosis debido a las reacciones adversas en el 6% de los pacientes tratados con XALKORI. Las reacciones adversas más frecuentes que dieron lugar a la reducción de la dosis en los pacientes tratados con XALKORI fueron náuseas (1,8%) y elevación de transaminasas (1,8%). La interrupción del tratamiento en pacientes tratados con XALKORI por reacciones adversas fue del 8%. Las reacciones adversas más frecuentes que dieron lugar a la interrupción en pacientes tratados con XALKORI fueron elevación de transaminasas (1,2%), hepatotoxicidad (1,2%) y EPI (1,2%). Las Tablas 6 y 7 resumen las Reacciones Adversas comunes y las Anormalidades del Laboratorio en pacientes tratados con XALKORI.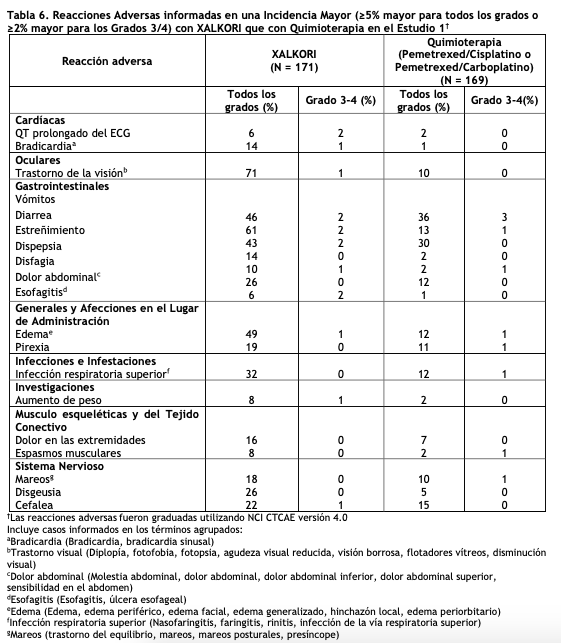
Las reacciones adversas adicionales que ocurrieron en una incidencia total entre el 1% y el 60% en pacientes tratados con XALKORI incluían náuseas (56%), disminución del apetito (30%), fatiga (29%), neuropatía (21%;trastornos en la marcha, hipoestesia, debilidad muscular, neuralgia, neuropatía periférica, parestesia, neuropatía periférica sensitiva, polineuropatía, trastornos sensoriales), erupción (11%), quiste renal (5%), EPI (1%; EPI, neumonitis), síncope (1%), y disminución de testosterona en sangre (1% hipogonadismo).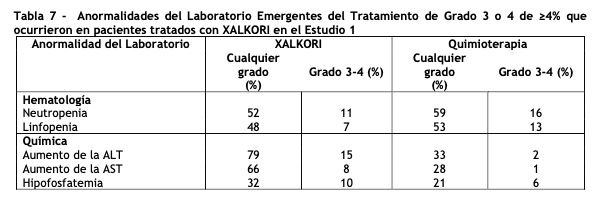
Una anormalidad adicional en pruebas de laboratorio en los pacientes tratados con XALKORI fue un aumento en la creatinina (Cualquier grado: 99%, Grado 3: 2%, Grado 4: 0%) en comparación con el grupo de quimioterapia (Cualquier grado: 92%, Grado 3: 0%, Grado 4: 1%). Estudio 2 de CPCNP ALK-positivo metastásico con tratamiento previo (Perfil 1007): Los datos en la Tabla 8 provienen de 343 pacientes con CPCNP ALK-positivo metastásico enrolados en un ensayo (Estudio 2) aleatorizado, multicéntrico, abierto y con control activo. Los pacientes en la rama de XALKORI (n= 172) recibieron XALKORI 250 mg vía oral dos veces al día hasta una progresión documentada de la enfermedad, intolerancia al tratamiento o hasta que el investigador determinó que el paciente ya no experimentaba un beneficio clínico. Un total de 171 pacientes en la rama de quimioterapia recibieron pemetrexed 500 mg/m2 (n= 99) o docetaxel 75 mg/m2 (n=72) mediante infusión intravenosa cada tres semanas hasta una progresión documentada de la enfermedad, intolerancia al tratamiento o hasta que el investigador determinó que el paciente ya no experimentaba un beneficio clínico. Los pacientes en la rama de quimioterapia recibieron pemetrexed a menos que hubieran recibido pemetrexed como parte de un tratamiento de primera línea o mantenimiento. La duración mediana del tratamiento del estudio fue de 7,1 meses para los pacientes que recibieron XALKORI y 2,8 meses para los pacientes que recibieron quimioterapia. A lo largo de los 347 pacientes que fueron aleatorizados al tratamiento del estudio (343 recibieron al menos una dosis del tratamiento del estudio), la edad mediana fue de 50 años; 14% de los pacientes fueron mayores de 65 años. Un total de 56% de los pacientes fueron mujeres y 45% de los pacientes fueron asiáticos. Se reportaron reacciones adversas serias en 37% de los pacientes tratados con XALKORI y en 23% de los pacientes en el grupo de quimioterapia. Las reacciones adversas serias más frecuentes notificadas en pacientes tratados con XALKORI fueron neumonía (4,1%), embolia pulmonar (3,5%), disnea (2,3%) y enfermedad pulmonar intersticial (EPI: 2,9%). En5% de los pacientes tratados con XALKORI en el Estudio 2, ocurrieron reacciones adversas mortales: síndrome de distrés respiratorio agudo, arritmias, disnea, neumonía, neumonitis, embolia pulmonar, EPI, insuficiencia respiratoria y sepsis. Se requirieron reducciones de dosis debidas a reacciones adversas en el 16% de los pacientes tratados con XALKORI. Las reacciones adversas más frecuentes que dieron lugar a la reducción de la dosis en los pacientes tratados con XALKORI fueron aumento (8%) de la alanina aminotransferasa (ALT) incluidos algunos pacientes con aumento concurrente de la aspartato aminotransferasa (AST), prolongación del intervalo QTc (2,9%) y neutropenia (2,3%). La discontinuación del tratamiento en pacientes tratados con XALKORI debido a reacciones adversas fue del 15,0%. Las reacciones adversas más frecuentes que condujeron a la discontinuación en pacientes tratados con XALKORI fueron EPI (1,7%), aumento de ALT y AST (1,2%), disnea (1,2%), y embolia pulmonar (1,2%). Las Tablas 8 y 9 resumen las reacciones adversas comunes y las anormalidades de laboratorio en los pacientes tratados con XALKORI.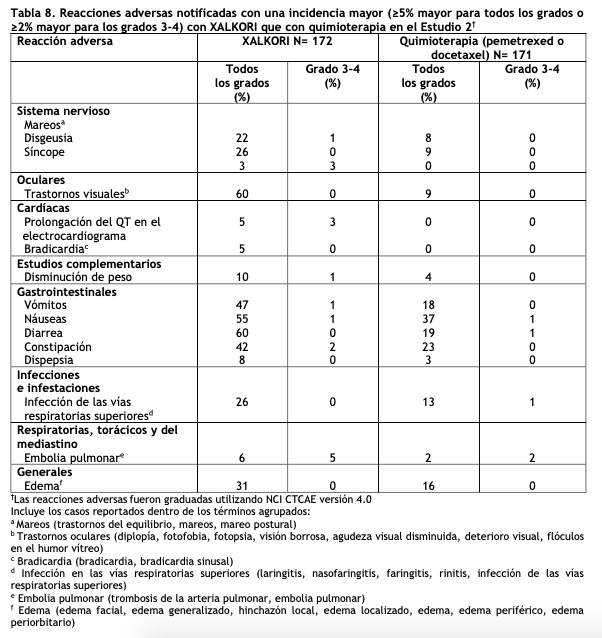
Ocurrieron otras reacciones adversas con una incidencia general de entre 1% y 30% en los pacientes tratados con XALKORI incluidos disminución de apetito (27%), fatiga (27%), neuropatía (19%; disestesia, trastornos de la marcha, hipoestesia, debilidad muscular, neuralgia, neuropatía periférica, parestesia, neuropatía sensorial periférica, polineuropatía, sensación de ardor en la piel), erupción cutánea (9%), EPI (4%; síndrome de distrés respiratorio agudo, EPI, neumonitis), quiste renal (4%), esofagitis (2%), insuficiencia hepática (1%) y disminución de testosterona en sangre (1% hipogonadismo).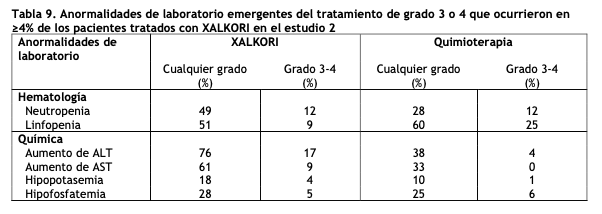
Una anormalidad adicional en pruebas de laboratorio en pacientes tratados con XALKORI fue un incremento en creatinina (Cualquier grado: 96%; Grado 3: 1%; Grado 4: 0%) en comparación con el grupo de quimioterapia (Cualquier grado: 72%; Grado 3: 0%; Grado 4: 0%). CPCNP ROS1 positivo metastásico - Estudio 3 (Perfil 1001): El perfil de seguridad de XALKORI del Estudio 3, el cual fue evaluado en 50 pacientes con CPCNP metastásico ROS1 positivo, fue consistente con el perfil de seguridad de XALKORI evaluado en pacientes con CPCNP metastásico ALK positivo (n=1669). Los trastornos visuales ocurrieron en 92% de los pacientes en el estudio 3; 90% fueron de Grado 1 y 2% Grado 2. La mediana de duración de la exposición a XALKORI fue 34,4 meses. Descripción de reacciones adversas al medicamento seleccionadas: Trastornos visuales: Los trastornos visuales más comunes, deterioros visuales, fotopsia, visión borrosa y floculos del humor vítreo ocurrieron en un63% de 1719 pacientes. La mayoría (95%) de estos pacientes tuvieron reacciones adversas visuales de grado 1. Se presentó un0,8% de pacientes con deterioro visual de Grado 3, y 0,2% de pacientes con deterioro visual de Grado 4. Basándose en el Cuestionario de Evaluación de Síntomas Visuales (VSAQ-ALK), los pacientes tratados con XALKORI en los Estudios 1 y 2 reportaron una mayor incidencia de trastornos de la visión en comparación con los pacientes tratados con quimioterapia. La aparición de trastornos de la visión generalmente ocurrió dentro de la primera semana de la administración del medicamento. La mayoría de los pacientes en la rama con XALKORI en los Estudios 1 y 2 ( > 50%) informaron disturbios visuales que ocurrieron con una frecuencia de 4-7 días cada semana, duraron hasta 1 minuto, y no tuvieron impacto o este fue leve en las actividades diarias según fue registrado en el cuestionario VSAQ-ALK para el paciente (puntajes de 0 a 3 de un puntaje máximo de 10). Neuropatía: La neuropatía, más comúnmente de naturaleza sensorial, ocurrió en un25% de 1719 pacientes. La mayoría de los eventos (95%) fue de severidad grado 1 o grado 2. Quistes renales: El 3,0% de los 1719 pacientes experimentaron quistes renales. La mayoría de los quistes renales en pacientes tratados con XALKORI eran complejos. Se produjo una invasión quística local más allá del riñón, en algunos casos con características de imagen indicati vas de la formación de abscesos. Sin embargo, en los ensayos clínicos no se confirmaron abscesos renales mediante las pruebas de microbiología. Toxicidad Renal: La tasa de filtración glomerular estimada (eGFR) disminuyó de una mediana base de 96.42 mL/min/1.73 m2 (n=1681) a una mediana de 80.23 mL/min/1.73 m2 tras 2 semanas (n=1499) en pacientes con NSCLC avanzado ALK-positivo que recibieron XALKORI en ensayos clínicos. No hubo cambios clínicamente relevantes en la mediana de eGFR desde las 12 hasta las 104 semanas de tratamiento. La mediana de eGFR aumentó ligeramente (83.02 mL/min/1.73 m2) 4 semanas después de la última dosis de XALKORI. En general, 76% de los pacientes tuvo una disminución en eGFR hasta < 90 mL/min/1.73 m2, 38% tuvo una disminución en eGFR hasta < 60 mL/min/1.73m2, y 3.6% tuvo una disminución en eGFR hasta < 30 mL/min/1.73 m2.
Advertencias.
Hepatotoxicidad: Se reportó hepatotoxicidad derivada del uso del medicamento con resultado fatal en 0,1% de los 1719 pacientes tratados con XALKORI a lo largo de estudios clínicos (ver Reacciones adversas). Se produjeron aumentos concurrentes de la ALT o AST mayor o igual a tres veces el LSN y de la bilirrubina total de más o igual a dos veces el LSN, con fosfatasa alcalina normal en < 1% tratados con XALKORI. Además, se produjeron aumentos de la ALT o de la AST de más de 5 veces el LSN en11% y 6% de los pacientes respectivamente. El 1,0 % de los pacientes requirieron discontinuar el tratamiento en forma permanente debido a elevación de las transaminasas. Las elevaciones de las transaminasas ocurrieron generalmente dentro de los primeros 2 meses de tratamiento. Realizar análisis de función hepática que incluyan ALT, AST y bilirrubina total cada 2 semanas durante los primeros 2 meses del tratamiento, luego una vez al mes y según indicación clínica, con un aumento de la frecuencia de análisis en caso de aumento de transaminasas hepáticas, fosfatasa alcalina o bilirrubina total. Se recomienda suspender temporalmente, reducir la dosis o discontinuar en forma permanente XALKORI en caso de hepatotoxicidad. (Ver Posología y forma de administración). Enfermedad pulmonar intersticial (Neumonitis): En pacientes tratados con XALKORI puede ocurrir enfermedad pulmonar intersticial (EPI)/neumonitis severa, amenazante para la vida o letal. Durante los ensayos clínicos (n= 1719), 2,9% de los pacientes tratados con XALKORI presentaron algún grado de EPI, el 1% presentó un EPI grado 3 o 4 y el 0,5% fueron casos mortales de EPI (ver Reacciones adversas). La Enfermedad Pulmonar Intersticial generalmente ocurre dentro de los tres meses luego del inicio del tratamiento con XALKORI. Debe controlarse a los pacientes para detectar síntomas pulmonares indicativos de EPI/neumonitis. Deben excluirse otras causas posibles de EPI/neumonitis y discontinuar permanentemente el uso de XALKORI en pacientes con diagnóstico de EPI/neumonitis relacionada con el medicamento. (Ver Posología y forma de administración). Prolongación del intervalo QT: Puede ocurrir una prolongación del intervalo QTc en pacientes tratados con XALKORI. Durante los ensayos clínicos, se observó en 2,1% de 1616 pacientes una prolongación del intervalo QTcF (QT corregido de acuerdo al método Fridericia) mayor o igual a 500 ms y en el 5.0% de 1582 pacientes se observó un intervalo QTcF mayor o igual a 60 ms, evaluado en una maquina automática para lectura y evaluación del ECG. Se recomienda suspender temporalmente, reducir la dosis o discontinuar en forma permanente XALKORI en caso de prolongación del intervalo QT/QTc (ver Posología y forma de administración y Características farmacológicas). Bradicardia: Puede producirse bradicardia sintomática en pacientes bajo tratamiento con XALKORI. Durante los ensayos clínicos se observó bradicardia en 13% de 1719 pacientes tratados con XALKORI. Síncope grado 3 ocurrió en 2,4% de los pacientes tratados con XALKORI y en 0,6% de los pacientes tratados con quimioterapia (ver Reacciones adversas). Evite administrar XALKORI en combinación con otros medicamentos de los cuales se sabe causan bradicardia (por ejemplo, betabloqueantes, bloqueantes cálcicos no dihidropiridínicos, clonidina y digoxina) tanto como sea posible. Monitorice la frecuencia cardíaca y la tensión arterial regularmente. En casos de bradicardia, vuelva a evaluar el uso de medicamentos de los cuales se sabe causan bradicardia. Se recomienda suspender temporalmente, reducir la dosis o discontinuar en forma permanente XALKORI en caso de bradicardia (Ver Posología y forma de administración). Pérdida severa de la visión: Durante todos los ensayos clínicos, la incidencia del defecto de campo visual de grado 4 con pérdida de la visión, fue 0,2% de 1719 pacientes (ver Reacciones adversas). La atrofia óptica y el trastorno del nervio óptico se informaron como causas potenciales de