WEGOVY
NOVO NORDISK
Trat. de la obesidad.
Composición.
Lapicera de 0,25 mg/dosis: Una lapicera prellenada contiene 1 mg de semaglutida* en 1,5 ml de solución. Un ml de solución contiene 0,68 mg de semaglutida*. Una lapicera prellenada contiene 4 dosis de 0,25 mg. Lapicera de 0,5 mg/dosis: Una lapicera prellenada contiene 2 mg de semaglutida* en 1,5 ml de solución. Un ml de solución contiene 1,34 mg de semaglutida*. Una lapicera prellenada contiene 4 dosis de 0,5 mg. Lapicera de 1 mg/dosis: Una lapicera prellenada contiene 4 mg de semaglutida* en 3 ml de solución. Un ml de solución contiene 1,34 mg de semaglutida*. Una lapicera prellenada contiene 4 dosis de 1 mg. Lapicera de 1,7 mg/dosis: Una lapicera prellenada contiene 6,8 mg de semaglutida* en 3 ml de solución. Un ml de solución contiene 2,27 mg de semaglutida*. Una lapicera prellenada contiene 4 dosis de 1,7 mg. Lapicera de 2,4 mg/dosis: Una lapicera prellenada contiene 9,6 mg de semaglutida* en 3 ml de solución. Un ml de solución contiene 3,2 mg de semaglutida*. Una lapicera prellenada contiene 4 dosis de 2,4 mg. *Análogo del péptido similar al glucagón tipo 1 (GLP-1) humano producido por tecnología de ADN recombinante en células de Saccharomyces cerevisiae. Excipientes: fosfato disódico dihidratado, propilenglicol, fenol, ácido clorhídrico/hidróxido de sodio (para ajuste del pH), agua para inyectables.
Indicaciones.
Adultos: Wegovy® está indicado en combinación con una dieta reducida en calorías y un aumento de la actividad física para el control del peso, incluyendo pérdida de peso y mantenimiento del peso, en adultos con un índice de masa corporal (IMC) inicial de ≥30 kg/m2 (obesidad), o ≥27 kg/m2 a < 30 kg/m2 (sobrepeso) en presencia de al menos una comorbilidad relacionada con el peso, por ejemplo, alteraciones de la glucemia (prediabetes o diabetes mellitus tipo 2), hipertensión, dislipidemia, apnea obstructiva del sueño o enfermedad cardiovascular. Para resultados de estudios relacionados con la reducción del riesgo cardiovascular, insuficiencia cardíaca relacionada con obesidad y las poblaciones estudiadas. Adolescentes (≥12 años): Wegovy® está indicado en combinación con una dieta reducida en calorías y un aumento de la actividad física para el control del peso en adolescentes de 12 años de edad en adelante con obesidad* y peso corporal superior a 60 kg. Se debe discontinuar y reevaluar el tratamiento con Wegovy® si los pacientes adolescentes no alcanzaron una reducción de al menos un 5% de su IMC tras 12 semanas en tratamiento con la dosis de 2,4 mg o la máxima dosis tolerada. *Obesidad (IMC ≥ percentil 95) según se define en las tablas de crecimiento del IMC específico por sexo y edad (CDC.gov) (ver Tabla 1).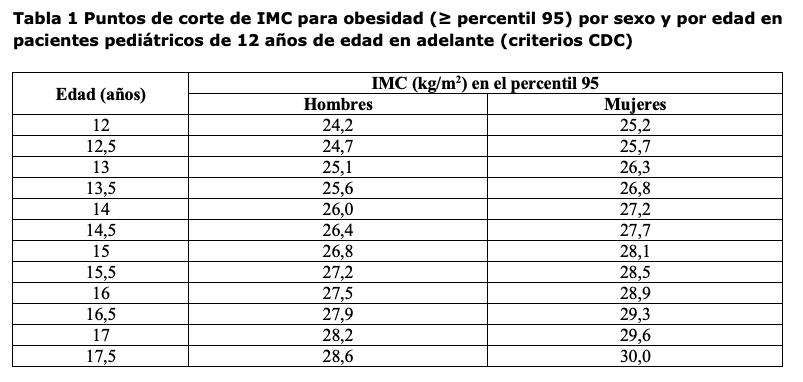
Dosificación.
Posología: Adultos: La dosis de mantenimiento de 2,4 mg de semaglutida una vez a la semana se alcanza comenzando con una dosis de 0,25 mg. Para reducir la probabilidad de síntomas gastrointestinales, la dosis debe aumentarse gradualmente durante un período de 16 semanas hasta alcanzar una dosis de mantenimiento de 2,4 mg una vez a la semana (ver Tabla 9). En caso de síntomas gastrointestinales significativos, considerar demorar el aumento gradual de la dosis o disminuir la dosis a la dosis previa hasta que los síntomas hayan mejorado. No se recomiendan dosis semanales mayores a 2,4 mg.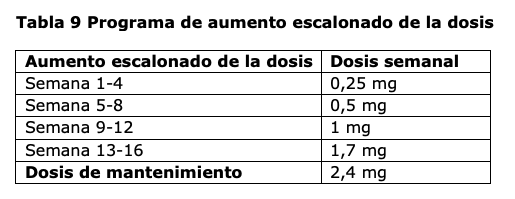
Adolescentes: Para los adolescentes de 12 años de edad en adelante, se debe aplicar el mismo esquema de aumento escalonado de la dosis de los adultos (ver Tabla 9). La dosis se debe incrementar hasta 2,4 mg (dosis de mantenimiento) o hasta que se alcance la máxima dosis tolerada. No se recomiendan dosis semanales mayores a 2,4 mg. Pacientes con diabetes tipo 2: Cuando se inicie el tratamiento con Wegovy® en pacientes con diabetes mellitus tipo 2, se debe considerar reducir la dosis de insulina o de secretagogos de insulina (como sulfonilureas) administrados en forma concomitante, para reducir el riesgo de hipoglucemia; ver sección Advertencias. Dosis omitida: Si se omite una dosis, esta se debe administrar lo antes posible dentro de los 5 días posteriores. Si han pasado más de 5 días, debe saltearse la dosis omitida, y la siguiente dosis debe administrarse el día programado habitualmente. En cada caso, los pacientes pueden luego reanudar su esquema de administración habitual de una vez a la semana. Si se omiten más dosis, se debe considerar reducir la dosis inicial para el reinicio. Poblaciones especiales: Pacientes de edad avanzada (≥65 años): No es necesario un ajuste de dosis en función de la edad. La experiencia terapéutica en pacientes de ≥85 años es limitada. Pacientes con insuficiencia renal: No es necesario un ajuste de la dosis en los pacientes con insuficiencia renal leve o moderada. La experiencia con el uso de semaglutida en pacientes con insuficiencia renal grave es limitada. No se recomienda el uso de semaglutida en pacientes con insuficiencia renal grave (TFGe < 30 ml/min/1,73m2), incluyendo pacientes con enfermedad renal terminal (ver Advertencias y Reacciones adversas). Pacientes con insuficiencia hepática: No es necesario un ajuste de dosis en pacientes con insuficiencia hepática leve o moderada. La experiencia con el uso de semaglutida en pacientes con insuficiencia hepática grave es limitada. No se recomienda el uso de semaglutida en pacientes con insuficiencia hepática grave y se debe usar con precaución en pacientes con insuficiencia hepática leve o moderada (ver Advertencias). Población pediátrica: No se requiere ajuste de dosis en adolescentes de 12 años de edad en adelante. No se ha estudiado la seguridad y la eficacia de semaglutida en niños menores de 12 años de edad. Modo de administración: Uso subcutáneo. Wegovy® se administra una vez a la semana, en cualquier momento del día, con o sin comidas. Wegovy® se debe inyectar por vía subcutánea en el abdomen, el muslo o la parte superior del brazo. Se puede cambiar el sitio de inyección. Wegovy® no debe administrarse por vía intravenosa o intramuscular. El día de la administración semanal puede cambiarse si es necesario, siempre que el tiempo entre dos dosis sea de al menos 3 días ( > 72 horas). Después de seleccionar un nuevo día de administración, se debe continuar con la administración una vez a la semana. Se debe indicar a los pacientes que lean atentamente las instrucciones de uso incluidas en el prospecto antes de administrarse el medicamento. Para más información antes de la administración, ver Precauciones especiales de descarte y manipulación.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes enumerados en la Composición.
Reacciones adversas.
Resumen del perfil de seguridad: En cuatro estudios de fase 3a, 2650 pacientes adultos fueron expuestos a Wegovy®. La duración de los estudios fue de 68 semanas. Las reacciones adversas notificadas con más frecuencia fueron trastornos gastrointestinales, incluyendo náuseas, diarrea, estreñimiento y vómitos. Tabla de reacciones adversas La Tabla 10 enumera las reacciones adversas identificadas en los estudios clínicos en adultos y en reportes post-comercialización. Las frecuencias se basan en un conjunto de estudios de fase 3a. Las reacciones adversas asociadas a Wegovy® se listan según la clasificación por órganos y sistemas y la frecuencia. Las categorías de frecuencia se definen como: muy frecuentes (≥1/10); frecuentes (≥1/100 a < 1/10); poco frecuentes (≥1/1.000 a < 1/100); raras (≥1/10.000 a < 1/1.000); muy raras ( < 1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).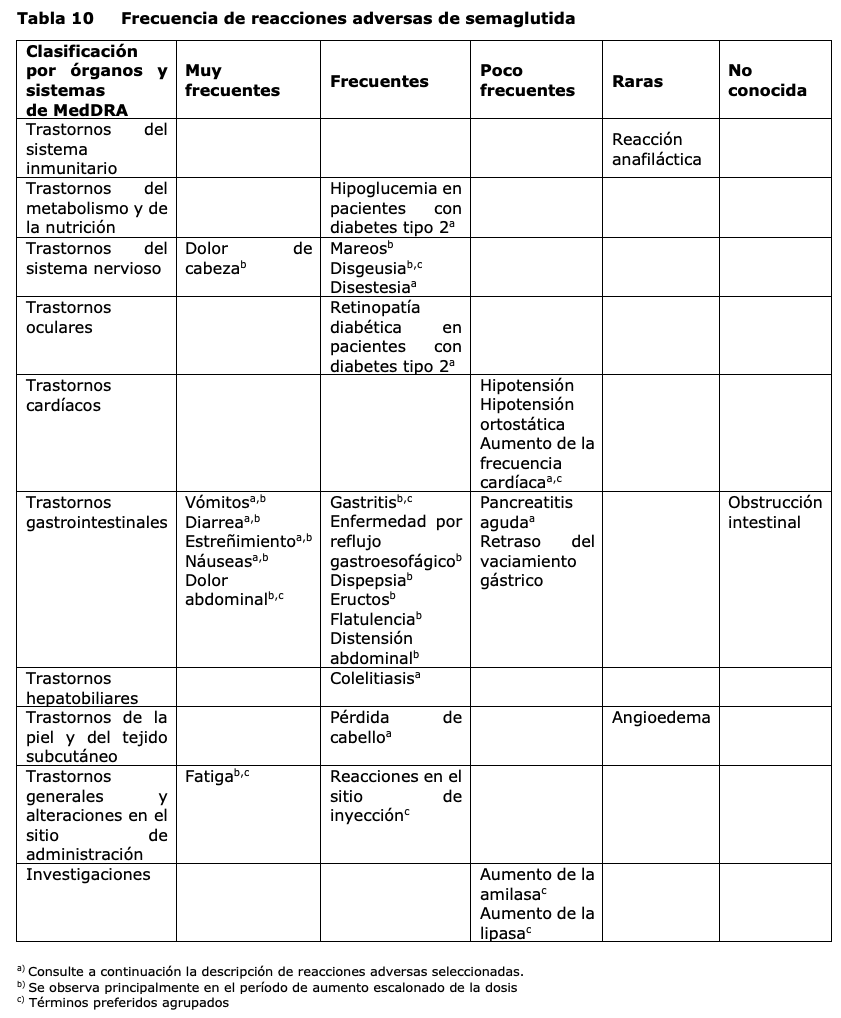
Descripción de reacciones adversas seleccionadas: La información relativa a reacciones adversas específicas, a menos que se especifique lo contrario, se refiere a los estudios de fase 3a. Reacciones adversas gastrointestinales: Durante el período de estudio de 68 semanas, se produjeron náuseas en un 43,9% de los pacientes cuando recibieron tratamiento con semaglutida (16,1% para el placebo), diarrea en un 29,7% (15,9% para el placebo) y vómitos en un 24,5% (6,3% para el placebo). La mayoría de los eventos fueron de intensidad leve a moderada y de corta duración. Se produjo estreñimiento en el 24,2% de los pacientes tratados con semaglutida (11,1% para el placebo) y fue de intensidad leve a moderada y de mayor duración. En pacientes tratados con semaglutida, la mediana de la duración de las náuseas fue de 8 días, de los vómitos 2 días, de la diarrea 3 días y del estreñimiento 47 días. Los pacientes con insuficiencia renal moderada (TFGe ≥30 a < 60 ml/min/1,73 m2) pueden experimentar más efectos gastrointestinales cuando son tratados con semaglutida. Los eventos gastrointestinales condujeron a la discontinuación permanente del tratamiento en el 4,3% de los pacientes. Pancreatitis aguda: La frecuencia de pancreatitis aguda confirmada por adjudicación informada en estudios clínicos de fase 3a fue del 0,2% para semaglutida y < 0,1% para el placebo, respectivamente. En SELECT, el estudio de resultados cardiovasculares, la frecuencia de pancreatitis aguda confirmada por adjudicación fue 0,2% para semaglutida y 0,3% para el placebo. Enfermedad aguda de cálculos biliares/colelitiasis: Se notificó colelitiasis en el 1,6% y condujo a colecistitis en el 0,6% de los pacientes tratados con semaglutida. Se notificaron colelitiasis y colecistitis en el 1,1% y el 0,3%, respectivamente, de los pacientes tratados con placebo. Pérdida de cabello: Se notificó pérdida de cabello en el 2,5% de los pacientes tratados con semaglutida y en el 1,0% de los pacientes tratados con placebo. Los eventos fueron principalmente de intensidad leve, y la mayoría de los pacientes se recuperaron mientras seguían con el tratamiento. Se notificó pérdida de cabello con más frecuencia en pacientes con una mayor pérdida de peso (≥20%). Aumento de la frecuencia cardíaca: En los estudios de fase 3a, se observó un aumento medio de 3 latidos por minuto (lpm) desde una media inicial de 72 lpm en pacientes tratados con semaglutida. Las proporciones de sujetos con un aumento del pulso desde el valor inicial ≥10 lpm en cualquier punto durante el período de tratamiento fueron del 67,0% en el grupo de semaglutida frente al 50,1% en el grupo de placebo. Inmunogenicidad: De manera consistente con las propiedades potencialmente inmunogénicas de los medicamentos que contienen proteínas o péptidos, los pacientes pueden desarrollar anticuerpos luego del tratamiento con semaglutida. La proporción de pacientes con un resultado positivo en el análisis de anticuerpos antisemaglutida en cualquier punto temporal posterior al inicio del estudio fue baja (2,9%) y al final del estudio ningún paciente presentó anticuerpos neutralizantes antisemaglutida o anticuerpos antisemaglutida con efecto neutralizante del GLP-1 endógeno. Durante el tratamiento, las altas concentraciones de semaglutida podrían haber disminuido la sensibilidad de los ensayos, por lo que no puede excluirse el riesgo de resultados falsos negativos. Sin embargo, en los sujetos que dieron positivo en el análisis de anticuerpos durante y después del tratamiento, la presencia de anticuerpos fue transitoria y sin impacto aparente en la eficacia y la seguridad. Hipoglucemia en pacientes con diabetes tipo 2: En el estudio STEP 2, se observó hipoglucemia clínicamente significativa en el 6,2% (0,1 eventos/paciente-año) de los sujetos tratados con semaglutida en comparación con el 2,5% (0,03 eventos/paciente-año) de los sujetos tratados con placebo. Se observó hipoglucemia con semaglutida tanto con como sin uso concomitante de sulfonilurea. Un episodio (0,2% de los sujetos, 0,002 eventos/paciente-año) se notificó como grave en un sujeto no tratado concomitantemente con una sulfonilurea. El riesgo de hipoglucemia fue mayor cuando semaglutida se utilizó con una sulfonilurea. En STEP-HFpEF-DM, se observó hipoglucemia clínicamente significativa en 4,2% de los sujetos, tanto en el grupo de semaglutida como en el de placebo, cuando se utilizó en combinación con sulfonilurea y/o insulina (0,065 eventos/paciente-año con semaglutida y 0,098 eventos/paciente-año con placebo). Retinopatía diabética en pacientes con diabetes tipo 2: En un estudio clínico de 2 años, se investigó semaglutida 0,5 mg y 1 mg frente a placebo en 3.297 pacientes con diabetes tipo 2, con alto riesgo cardiovascular, diabetes de larga duración y mal control de la glucemia. En este estudio, ocurrieron eventos adjudicados de complicaciones de la retinopatía diabética en más pacientes tratados con semaglutida (3,0 %) en comparación con placebo (1,8%). Esto se observó en pacientes con retinopatía diabética conocida tratados con insulina. La diferencia de tratamiento apareció tempranamente y persistió a lo largo del estudio. En el estudio STEP 2, se notificaron trastornos en la retina en el 6,9% de los pacientes tratados con Wegovy®, el 6,2% de los pacientes tratados con semaglutida 1 mg y el 4,2% de los pacientes tratados con placebo. La mayoría de los eventos se notificaron como retinopatía diabética (4,0%, 2,7% y 2,7%, respectivamente) y retinopatía no proliferativa (0,7%, 0% y 0%, respectivamente). Disestesia: Se notificaron eventos relacionados con un cuadro clínico de sensación cutánea alterada como parestesia, dolor en la piel, piel sensible, disestesia y sensación de ardor en la piel en 2,1% de los pacientes tratados con semaglutida 2,4 mg y en 1,2% de los pacientes tratados con placebo. Los eventos fueron leves a moderados en gravedad y la mayoría de los pacientes se recuperaron mientras continuaban con el tratamiento. Población pediátrica: En un estudio clínico, 133 pacientes adolescentes de 12 años hasta menos de 18 años con obesidad o con sobrepeso con al menos una comorbilidad relacionada con el peso fueron expuestos a Wegovy®. La duración del estudio fue de 68 semanas. En general, la frecuencia, el tipo y la gravedad de las reacciones adversas en adolescentes fueron comparables a las observadas en la población adulta. Se notificó colelitiasis en un 3,8% de los pacientes tratados con Wegovy® y en un 0% de los pacientes tratados con placebo. No se han encontrado efectos en el crecimiento o en el desarrollo puberal tras 68 semanas de tratamiento. Otras poblaciones especiales: En los estudios SELECT y SUSTAIN 6, el perfil de reacciones adversas en adultos con enfermedad cardiovascular establecida fue similar al observado en los estudios de control del peso de fase 3a. En los estudios HFpEF, en adultos con insuficiencia cardíaca relacionada con obesidad con fracción de eyección preservada (HFpEF), el perfil de reacciones adversas fue similar al observado en los estudios de control del peso de fase 3a.
Advertencias.
Aspiración en asociación con anestesia general o sedación profunda: Se han notificado casos de aspiración pulmonar en pacientes que recibieron agonistas del receptor de GLP-1 sometidos a anestesia general o sedación profunda. Por consiguiente, se debe considerar el aumento del riesgo de contenido gástrico residual debido al retraso del vaciamiento gástrico (ver Reacciones adversas) antes de realizar procedimientos con anestesia general o sedación profunda. Deshidratación: El uso de agonistas del receptor de GLP-1 puede asociarse a reacciones adversas gastrointestinales que pueden causar deshidratación, lo que en casos raros puede provocar un deterioro de la función renal. Se debe advertir a los pacientes sobre el posible riesgo de deshidratación en relación con los efectos secundarios gastrointestinales y aconsejar que tomen precauciones para evitar la pérdida de líquidos. Pancreatitis aguda: Se ha observado pancreatitis aguda con el uso de agonistas del receptor de GLP-1 (ver Reacciones adversas). Se debe informar a los pacientes los síntomas característicos de la pancreatitis aguda. Si se sospecha una pancreatitis, se debe interrumpir el tratamiento con semaglutida; si se confirma, no se debe reiniciar la administración de semaglutida. Se debe tener precaución en pacientes con antecedentes de pancreatitis. En ausencia de otros signos y síntomas de pancreatitis aguda, las elevaciones en las enzimas pancreáticas por sí solas no son predictivas de pancreatitis aguda. Pacientes con diabetes tipo 2: Semaglutida no debe utilizarse como sustituto de la insulina en pacientes con diabetes tipo 2. Semaglutida no debe utilizarse en combinación con otros agonistas del receptor de GLP-1, ya que no se ha evaluado y se considera probable un aumento en el riesgo de reacciones adversas relacionadas con la sobredosificación. Hipoglucemia en pacientes con diabetes tipo 2: Se sabe que la insulina y las sulfonilureas causan hipoglucemia. Es posible que los pacientes tratados con semaglutida en combinación con una sulfonilurea o insulina tengan un mayor riesgo de hipoglucemia. El riesgo de hipoglucemia puede reducirse disminuyendo la dosis de sulfonilurea o insulina al iniciar el tratamiento con un agonista del receptor de GLP-1. Retinopatía diabética en pacientes con diabetes tipo 2: Se ha observado un mayor riesgo de desarrollar complicaciones de la retinopatía diabética en pacientes con retinopatía diabética tratados con semaglutida (ver Reacciones adversas). La mejoría rápida del control de la glucemia se ha asociado con un empeoramiento temporal de la retinopatía diabética, pero no pueden excluirse otros mecanismos. Se debe controlar a los pacientes con retinopatía diabética que utilizan semaglutida y se los debe tratar de acuerdo con las guías clínicas. No hay experiencia con Wegovy® en pacientes con diabetes tipo 2 con retinopatía diabética no controlada o potencialmente inestable. En estos pacientes, no se recomienda el tratamiento con Wegovy®. Poblaciones no estudiadas La seguridad y la eficacia de Wegovy® no se ha investigado en pacientes tratados con otros productos para el control del peso; con diabetes tipo 1; con insuficiencia renal grave (ver Posología y modo de administración); con insuficiencia hepática grave (ver Posología y modo de administración); con insuficiencia cardíaca congestiva de clase IV según la Asociación Cardiológica de Nueva York (NYHA). No se recomienda el uso en estos pacientes. La experiencia con Wegovy® es limitada en pacientes: de 85 años o más (ver Posología y modo de administración); con insuficiencia hepática leve o moderada (ver Posología y modo de administración); con enfermedad inflamatoria intestinal; con gastroparesia diabética. Se debe utilizar con precaución en estos pacientes. Contenido de sodio: Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis; es decir, es esencialmente "libre de sodio". Interacción con otros medicamentos y otras formas de interacción: Semaglutida retrasa el vaciamiento gástrico y potencialmente podría influir en la absorción de medicamentos orales administrados de forma concomitante. No se observó ningún efecto clínicamente relevante en la tasa de vaciamiento gástrico con semaglutida 2,4 mg, probablemente debido a un efecto de tolerancia. Semaglutida debe utilizarse con precaución en pacientes que reciben medicamentos orales que requieren una rápida absorción gastrointestinal. Paracetamol: Semaglutida retrasa la velocidad de vaciamiento gástrico según lo determinado por la farmacocinética de paracetamol durante una prueba de comida estandarizada. El AUC0-60 min y la Cmáx de paracetamol se redujeron en un 27% y un 23%, respectivamente, tras el uso concomitante de semaglutida 1 mg. La exposición total a paracetamol (AUC0-5 h) no se vio afectada. No se observó ningún efecto clínicamente relevante sobre el paracetamol con semaglutida. No es necesario un ajuste de dosis de paracetamol cuando se administra con semaglutida. Anticonceptivos orales: No se espera que semaglutida disminuya la efectividad de los anticonceptivos orales, ya que semaglutida no modificó la exposición total al etinilestradiol y al levonorgestrel en un grado clínicamente relevante cuando un medicamento anticonceptivo combinado (0,03 mg de etinilestradiol/0,15 mg de levonorgestrel) se administró de forma concomitante con semaglutida. No se vio afectada la exposición al etinilestradiol; se observó un aumento del 20% en la exposición al levonorgestrel en estado estacionario. La Cmáx no se vio afectada para ninguno de los compuestos. Atorvastatina: Semaglutida no modificó la exposición total a la atorvastatina tras la administración de una dosis única de atorvastatina (40 mg). La Cmáx de atorvastatina disminuyó en un 38%. Esto no se consideró clínicamente relevante. Digoxina: Semaglutida no modificó la exposición total ni la Cmáx de la digoxina tras la administración de una dosis única de digoxina (0,5 mg). Metformina: Semaglutida no modificó la exposición total ni la Cmáx de la metformina tras la administración de dosis de 500 mg dos veces al día durante 3,5 días. Warfarina y otros derivados de la cumarina: Semaglutida no modificó la exposición total ni la Cmáx de la R- y S-warfarina después de una dosis única de warfarina (25 mg), y los efectos farmacodinámicos de la warfarina, según lo medido por la razón internacional normalizada (RIN), no se vieron afectados de manera clínicamente relevante. Sin embargo, se han notificado casos de RIN disminuido durante el uso concomitante de acenocumarol y semaglutida. Se recomienda el control frecuente del RIN al iniciar el tratamiento con semaglutida en pacientes tratados con warfarina u otros derivados de la cumarina. Población pediátrica: Solo se han realizado estudios de interacción en adultos. Embarazo, lactancia y fertilidad: Mujeres en edad fértil: Se recomienda que las mujeres en edad fértil utilicen métodos anticonceptivos durante el tratamiento con semaglutida (ver Interacción con otros medicamentos y otras formas de interacción). Embarazo: Los estudios en animales han mostrado toxicidad reproductiva (ver Datos preclínicos de seguridad). Se dispone de datos limitados sobre el uso de semaglutida en mujeres embarazadas. Por lo tanto, semaglutida no se debe usar durante el embarazo. Si una paciente desea quedar embarazada, o si ocurre un embarazo, se debe interrumpir el tratamiento con semaglutida. Semaglutida debe interrumpirse al menos 2 meses antes de un embarazo planificado, debido a su prolongada vida media. Lactancia: En ratas en período de lactancia, semaglutida se excretó en la leche. No es posible excluir un riesgo para el lactante. Semaglutida no debe utilizarse durante la lactancia. Fertilidad: Se desconoce el efecto de semaglutida sobre la fertilidad en seres humanos. Semaglutida no afectó la fertilidad de las ratas macho. En las ratas hembra, se observó un aumento en la duración del ciclo estral y una ligera reducción del número de ovulaciones a dosis asociadas con pérdida de peso corporal materno. Efectos sobre la capacidad para conducir y utilizar máquinas: Semaglutida tiene una influencia insignificante o nula sobre la capacidad para conducir vehículos o utilizar máquinas. Sin embargo, se pueden presentar mareos principalmente durante el período de aumento gradual de la dosis. Si se sufren mareos, la conducción o el uso de máquinas deben realizarse con precaución. Pacientes con diabetes tipo 2: Si se utiliza semaglutida en combinación con una sulfonilurea o insulina, se debe recomendar a los pacientes que tomen precauciones para evitar hipoglucemias mientras conducen vehículos o utilizan máquinas (ver Advertencias).
Incompatibilidades.
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
Conservación.
Antes del primer uso: conservar en heladera (entre 2°C y 8°C). Mantener alejado del elemento refrigerante. No congelar. Después del primer uso: conservar por hasta 6 semanas a una temperatura inferior a 30°C o en heladera (entre 2°C y 8°C). No congelar. Mantener el capuchón de la lapicera colocado cuando no está utilizándola para protegerla de la luz. Precauciones especiales de descarte y manipulación: Wegovy® no debe utilizarse si la solución no se presenta transparente e incolora. La lapicera no debe utilizarse si se ha congelado. Cualquier medicamento o material de desecho debe descartarse de acuerdo con los requerimientos locales. La lapicera es multidosis. Contiene 4 dosis. Se debe aconsejar al paciente que descarte la aguja después de cada inyección según los requerimientos locales y que almacene la lapicera de Wegovy® sin una aguja colocada. Esto puede prevenir el bloqueo de agujas, contaminación, infección, pérdida de solución y dosificación inexacta. La lapicera es para uso de una sola persona. Wegovy® puede administrarse con agujas descartables de calibre 30G, 31G o 32G y una longitud de hasta 8 mm.
Sobredosificación.
La sobredosis de semaglutida puede asociarse a trastornos gastrointestinales que podrían provocar deshidratación. En caso de sobredosis, se debe observar al paciente para detectar signos clínicos y se debe iniciar el tratamiento de soporte adecuado. Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con un centro de toxicología: Hospital de Pediatría Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital A. Posadas: (011) 4654-6648/4658-7777.
Presentación.
El envase primario consiste en un cartucho de vidrio (vidrio tipo I) contenido en una lapicera prellenada, multidosis, descartable. Hay cinco presentaciones de la lapicera prellenada Wegovy®: Wegovy® 0,25 mg/dosis: envase conteniendo 1 lapicera prellenada FlexTouch® x 1,5 ml y 4 agujas descartables NovoFine® Plus (PM-739-19). Wegovy® 0,5 mg/dosis: envase conteniendo 1 lapicera prellenada FlexTouch® x 1,5 ml y 4 agujas descartables NovoFine® Plus (PM-739-19). Wegovy® 1 mg/dosis: envase conteniendo 1 lapicera prellenada FlexTouch® x 3 ml y 4 agujas descartables NovoFine® Plus (PM-739-19). Wegovy® 1,7 mg/dosis: envase conteniendo 1 lapicera prellenada FlexTouch® x 3 ml y 4 agujas descartables NovoFine® Plus (PM-739-19). Wegovy® 2,4 mg/dosis: envase conteniendo 1 lapicera prellenada FlexTouch® x 3 ml y 4 agujas descartables NovoFine® Plus (PM-739-19).