VEKLURYTM
GADOR
Trat de enf. por Corona Virus 2019 (COVID-19).
Composición.
Cada vial contiene 100 mg de remdesivir. Después de la reconstitución, cada vial contiene 5 mg/ml de solución de remdesivir. Excipientes con efecto conocido. Cada vial contiene 3 g de éter sulfobutilico de beta ciclodextrina sódica. Polvo para concentrado para solución para perfusión (polvo para concentrado). Polvo de color entre blanco, blanquecino y amarillo.
Farmacología.
Propiedades farmacodinámicas: Grupo farmacoterapéutico: Antivirales de uso sistémico, antiviral de acción directa, otros antivirales, código ATC: J05AB. Mecanismo de acción: Remdesivir es un profármaco del nucleótido adenosina que se metaboliza en las células huésped para formar el metabolito de nucleósido trifosfato farmacológicamente activo. Remdesivir trifosfato actúa como análogo del trifosfato de adenosina (ATP) y compite con el sustrato ATP natural para la incorporación en las cadenas de ARN nacientes por la ARN polimerasa dependiente de ARN del SARS-CoV-2, lo que resulta en la terminación retardada de la cadena durante la replicación del ARN viral. Actividad antiviral: Remdesivir mostró actividad in vitro frente a un aislado clínico de SARS-CoV-2 en células epiteliales primarias de las vías respiratorias humanas a una concentración efectiva del 50 % (CE50) de 9,9 nM después de 48 horas de tratamiento. Los valores de EC50 de remdesivir frente al SARS-CoV-2 en células Vero fueron 137 nM a las 24 horas y 750 nM a las 48 horas después del tratamiento. La actividad antiviral de remdesivir fue antagonizada por el fosfato de cloroquina de forma dependiente de la dosis cuando los dos fármacos se incubaron de forma conjunta a concentraciones clínicamente relevantes en células HEp-2 infectadas con el virus sincitial respiratorio (VSR). Se observaron valores más altos de EC50 de remdesivir con concentraciones crecientes de fosfato de cloroquina. Las concentraciones crecientes de fosfato de cloroquina redujeron la formación de remdesivir trifosfato en células epiteliales bronquiales humanas normales. Resistencia: El perfil de resistencia del cultivo celular de remdesivir utilizando el virus de la hepatitis murina CoV de roedores identificó 2 sustituciones (F476L y V553L) en la ARN polimerasa dependiente de ARN viral en los residuos conservados en el CoV que confieren una susceptibilidad reducida de 5,6 veces a remdesivir. La introducción de las sustituciones correspondientes (F480L y V557L) en el SARS-CoV dio como resultado una susceptibilidad reducida 6 veces al cultivo celular de remdesivir y patogenia atenuada del SARS-CoV en un modelo de ratón. No se ha evaluado hasta la fecha el desarrollo de resistencia del SARS-CoV-2 al remdesivir en cultivos celulares. No se dispone de datos clínicos sobre el desarrollo de resistencia del SARS-CoV-2 al remdesivir. Eficacia clínica y seguridad: Ensayos clínicos en pacientes con COVID-19: Estudio ACTT-1 del NIAID (CO-US-540-5776): El ensayo clínico aleatorizado, doble ciego, controlado con placebo evaluó remdesivir 200 mg una vez al día durante 1 día seguido de remdesivir 100 mg una vez al día durante un periodo de hasta 9 días (por un total de hasta 10 días de tratamiento administrado por vía intravenosa) en pacientes adultos hospitalizados con COVID-19 con signos de afectación de las vías respiratorias bajas. En el ensayo participaron 1.063 pacientes hospitalizados: 120 pacientes (11,3 %) con enfermedad leve/moderada (definida como SpO2 > 94 % y frecuencia respiratoria < 24 respiraciones/min sin oxígeno suplementario) y 943 pacientes (88,7 %) con enfermedad grave (definida como SpO2 ≤94 % en el aire ambiente o frecuencia respiratoria ≥24 respiraciones/min y que requieren oxígeno suplementario o soporte de respiración asistida mecánica). Los pacientes fueron aleatorizados en una proporción de 1:1, estratificados en función de la gravedad de la enfermedad en el momento de la inclusión, para recibir remdesivir (n = 541) o placebo (n = 522), más el tratamiento estándar. La media de edad al inicio fue de 59 años y el 36 % de los pacientes tenían 65 años o más. El 64 % eran hombres, el 53 % eran blancos, el 21 % eran negros y el 13 % eran asiáticos. Las comorbilidades más frecuentes fueron hipertensión (49,6 %), obesidad (37,0 %), diabetes mellitus de tipo 2 (29,7 %) y coronariopatía (11,6 %). Aproximadamente el 33 % (180/541) de los pacientes recibieron un tratamiento de 10 días con remdesivir. La variable principal clínica fue el tiempo hasta la recuperación en los 28 días siguientes a la aleatorización, definido como dado de alta del hospital (con o sin limitaciones de actividad y con o sin requerimientos de oxígeno a domicilio) u hospitalizado, pero sin requerir oxígeno suplementario y que ya no requiriese asistencia médica continua. En un análisis realizado después de haber realizado el seguimiento a todos los pacientes durante 14 días, la mediana del tiempo hasta la recuperación en la población general fue de 11 días en el grupo de remdesivir en comparación con 15 días en el grupo de placebo [índice de tasa de recuperación: 1,32; (IC del 95 %: 1,12 a 1,55), p < 0,001]. El resultado difirió significativamente entre los dos estratos. En el estrato de enfermedad grave, el tiempo hasta la recuperación fue de 12 días en el grupo de remdesivir y de 18 días en el grupo de placebo [índice de tasa de recuperación: 1,37; (IC del 95 %: 1,15 a 1,63]; Tabla 1). Para el estrato de enfermedad leve/moderada, el tiempo hasta la recuperación no fue diferente entre los dos grupos (5 días para ambos, remdesivir y placebo).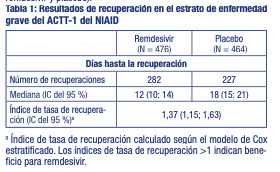
No hubo diferencias en la eficacia en pacientes aleatorizados durante los primeros 10 días después de la aparición de los síntomas en comparación con aquellos con síntomas durante más de 10 días. El beneficio clínico de remdesivir fue más evidente en pacientes que recibieron oxígeno, sin embargo, no en aquellos que recibieron ventilación, en el día 1 [índice de tasa de recuperación 1,47 (IC del 95 %: 1,17 a 1,84)]. En los pacientes que estaban recibiendo ventilación mecánica o ECMO el día 1, no se observó diferencia en la tasa de recuperación entre los grupos de tratamiento (0,95 [IC del 95 %: 0,64 a 1,42]). La mortalidad a los 29 días en la población general fue del 11,6 % en el grupo de remdesivir en comparación con el 15,4 % en el grupo de placebo (razón de riesgo (hazard ratio): 0,73; [IC del 95 %: 0,52 a 1,03]; p = 0,07). En la Tabla 2 se describe un análisis realizado a posteriori de la mortalidad a los 29 días mediante escala ordinal.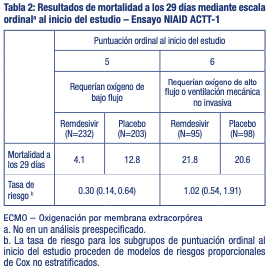
QT: Los datos clínicos y preclínicos actuales no indican un riesgo de prolongación del intervalo QT, pero la prolongación del intervalo QT no se ha evaluado completamente en los seres humanos. Propiedades farmacocinéticas Las propiedades farmacocinéticas de remdesivir se han investigado en voluntarios sanos. No se dispone de datos farmacocinéticos de pacientes con COVID-19. Absorción: Se han evaluado las propiedades farmacocinéticas de remdesivir y el metabolito circulante predominante GS-441524 en sujetos adultos sanos. Tras la administración intravenosa de la pauta posológica de remdesivir para adultos, se observó una concentración plasmática máxima observada al final de la perfusión, independientemente del nivel de dosis y disminuyó rápidamente a partir de entonces con una semivida de aproximadamente 1 hora. Se observaron concentraciones plasmáticas máximas de GS-441524 entre las 1,5 y 2,0 horas después del inicio de la perfusión de 30 minutos. Distribución: Remdesivir se une a las proteínas plasmáticas humanas en aproximadamente un 88 %. La unión a proteínas de GS-441524 fue baja (2 % de unión) en plasma humano. Después de una dosis única de 150 mg de [14C]-remdesivir en sujetos sanos, el cociente sangre/plasma de radioactividad 14C fue de aproximadamente 0,68 a los 15 minutos del inicio de la perfusión, aumentó con el tiempo alcanzando un cociente de 1,0 a las 5 horas, lo que indica una distribución diferencial de remdesivir y sus metabolitos al plasma o a los componentes celulares de la sangre. Biotransformación: Remdesivir se metaboliza ampliamente dando lugar al análogo de nucleósido trifosfato farmacológicamente activo GS-443902 (formado intracelularmente). La vía de activación metabólica implica la hidrólisis por esterasas, lo que da lugar a la formación del metabolito intermedio, GS-704277. La escisión de fosforamidato seguida de la fosforilación forma el trifosfato activo, GS-443902. La desfosforilación de todos los metabolitos fosforilados puede dar lugar a la formación del metabolito nucleósido GS-441524 que, por sí mismo, no se vuelve a fosforilar de forma eficiente. El estudio de balance de masas en seres humanos también indica la presencia de un metabolito principal (M27) no identificado actualmente en plasma. Eliminación: Después de una dosis única de 150 mg IV de [14C]-remdesivir, la media de recuperación total de la dosis fue del 92 %, que se compone de aproximadamente el 74 % y el 18 % recuperado en orina y heces, respectivamente. La mayor parte de la dosis de remdesivir recuperada en la orina fue GS-441524 (49 %), mientras que el 10 % se recuperó como remdesivir. Estos datos indican que el aclaramiento renal es la principal vía de eliminación de GS-441524. La mediana de la semivida terminal de remdesivir y GS-441524 fue de aproximadamente 1 y 27 horas, respectivamente. Otras poblaciones especiales: Sexo, raza y edad: No se han evaluado diferencias farmacocinéticas debidas al sexo, la raza o la edad. Pacientes pediátricos: No se ha evaluado la farmacocinética en pacientes pediátricos. Insuficiencia renal: No se ha evaluado la farmacocinética de remdesivir y GS-441524 en insuficiencia renal. Remdesivir no se elimina inalterado en orina de forma considerable, pero su metabolito principal GS-441524 se elimina por vía renal y las concentraciones de metabolitos en plasma pueden aumentar teóricamente en pacientes con insuficiencia renal. El excipiente éter sulfobutilico de beta ciclodextrina sódica se elimina por vía renal y se acumula en pacientes con función renal disminuida. No se debe utilizar VEKLURY® en pacientes con una TFGe < 30 ml/min. Insuficiencia hepática: No se ha evaluado la farmacocinética de remdesivir y GS-441524 en insuficiencia hepática. Se desconoce la función del hígado en el metabolismo de remdesivir. Interacciones: No se estudió el potencial de interacción de remdesivir como víctima con respecto a la inhibición de la vía hidrolítica (esterasa). Se desconoce el riesgo de interacción clínicamente relevante. Remdesivir inhibió CYP3A4 in vitro. A concentraciones fisiológicamente adecuadas (estado estacionario), remdesivir o sus metabolitos GS-441524 y GS-704277 no inhibieron CYP1A2, 2B6, 2C8, 2C9, 2C19 y 2D6 in vitro. Sin embargo, remdesivir puede inhibir transitoriamente CYP2B6, 2C8, 2C9 y 2D6 el primer día de administración. No se estudió la relevancia clínica de esta inhibición. No se estudió el potencial de inhibición dependiente del tiempo de las enzimas CYP450 por remdesivir. Remdesivir indujo CYP1A2 y potencialmente CYP3A4, pero no CYP2B6 in vitro. Los datos in vitro indican que no se produce inhibición clínicamente relevante de UGT1A1, 1A3, 1A4, 1A6, 1A9 o 2B7 por remdesivir o sus metabolitos GS-441524 y GS-704277. Remdesivir inhibió OATP1B1 y OATP1B3 in vitro. No hay datos disponibles para la inhibición de OAT1, OAT3 u OCT2 por remdesivir. A concentraciones fisiológicamente adecuadas, remdesivir y sus metabolitos no inhibieron la gp-P y el BCRP in vitro. Datos preclínicos sobre seguridad: Toxicología: Tras la administración intravenosa (bolo lento) de remdesivir a monos Rhesus y ratas, se produjo toxicidad renal grave después de tratamientos de corta duración. En monos Rhesus machos a niveles de dosis de 5, 10 y 20 mg/kg/día durante 7 días dio como resultado, en todos los niveles de dosis, un aumento en el nitrógeno ureico medio y un aumento en la creatinina media, atrofia tubular renal y basofilia y cilindros, y la muerte imprevista de un animal a un nivel de dosis de 20 mg/kg/día. En ratas, los niveles de dosis de > 3 mg/kg/día durante hasta 4 semanas dieron como resultado datos indicativos de lesión y/o disfunción renal. Las exposiciones sistémicas (AUC) del metabolito circulante predominante de remdesivir (GS-441524) fueron 0,1 veces (monos a 5 mg/kg/día) y 0,3 veces (rata a 3 mg/kg/día) la exposición en humanos a la dosis recomendada en humanos (DRH). Se demostró que el metabolito principal no identificado (M27) estaba presente en el plasma humano. Se desconoce la exposición a M27 en monos Rhesus y ratas. Por lo tanto, los estudios en animales pueden no ser informativos de los posibles riesgos asociados a este metabolito. Carcinogénesis: No se han realizado estudios a largo plazo en animales para evaluar el potencial carcinogénico de remdesivir. Mutagénesis: Remdesivir no fue genotóxico en una batería de ensayos, incluidos mutagenicidad bacteriana, aberración cromosómica utilizando linfocitos de sangre periférica humana y ensayos de micronúcleos de rata in vivo. Toxicidad para la reproducción: En ratas hembras, se observó disminución del número de cuerpos lúteos, número de lugares de implantación y embriones viables, cuando se administró remdesivir por vía intravenosa a diario a una dosis tóxica sistémica (10 mg/kg/día) 14 días antes del apareamiento y durante la fecundación; Las exposiciones del metabolito circulante predominante (GS-441524) fueron 1,3 veces la exposición en humanos a la DRH. No se produjeron efectos sobre los resultados reproductivos femeninos (apareamiento, fertilidad y fecundación) a este nivel de dosis. En ratas y conejos, remdesivir demostró no tener ningún efecto adverso sobre el desarrollo embriofetal cuando se administró a animales preñados en las exposiciones sistémicas (AUC) del metabolito circulante predominante de remdesivir (GS-441524) que fueron hasta 4 veces la exposición en humanos a la DRH. En ratas no se produjo ningún efecto adverso sobre el desarrollo pre y posnatal en las exposiciones sistémicas (AUC) del metabolito circulante predominante de remdesivir (GS-441524) que fueron similares a la exposición en humanos a la DRH. Se desconoce si el análogo de nucleósido trifosfato activo GS443902 y el metabolito humano principal no identificado M27 se forman en ratas y conejos. Por lo tanto, los estudios de toxicidad para la reproducción pueden no ser informativos de los posibles riesgos asociados a estos metabolitos.
Indicaciones.
VEKLURY® está indicado para el tratamiento de la enfermedad por coronavirus 2019 (COVID-19) en adultos y adolescentes (de 12 años de edad y mayores con un peso corporal de al menos 40 kg) con neumonía que requieren oxígeno suplementario (oxígeno de alto o bajo flujo u otra ventilación no invasiva al inicio del tratamiento).
Dosificación.
El uso de remdesivir se limita a los centros sanitarios en los que los pacientes pueden ser controlados adecuadamente Posología: La dosis recomendada de remdesivir en pacientes de 12 años de edad y mayores y que pesen al menos 40 kg es: Día 1: una dosis única de carga de remdesivir de 200 mg administrada mediante perfusión intravenosa. A partir del día 2: 100 mg administrados una vez al día mediante perfusión intravenosa. La duración total del tratamiento debe ser de al menos 5 días y no más de 10 días. Poblaciones especiales: Pacientes de edad avanzada: No se requiere un ajuste de la dosis de remdesivir en pacientes mayores de 65 años Insuficiencia renal: No se ha evaluado la farmacocinética de remdesivir en pacientes con insuficiencia renal. Los pacientes con una tasa de filtración glomerular estimada (TFGe) ≥30 ml/min han recibido remdesivir sin realizar un ajuste de la dosis para el tratamiento de la COVID-19. No se debe utilizar remdesivir en pacientes con una TFGe < 30 ml/min Insuficiencia hepática: No se ha evaluado la farmacocinética de remdesivir en pacientes con insuficiencia hepática. Se desconoce si el ajuste de la dosis es adecuado en pacientes con insuficiencia hepática. Población pediátrica: No se ha establecido todavía la seguridad y la eficacia de remdesivir en niños menores de 12 años de edad y que pesen menos de 40 kg. No se dispone de datos. Forma de administración: Para vía intravenosa. Remdesivir se administra mediante perfusión intravenosa tras su reconstitución y dilución posterior. No se debe administrar como inyección intramuscular (IM). Para consultar las instrucciones sobre la reconstitución y la dilución del medicamento antes de la administración, (ver precauciones especiales de eliminación y otras manipulaciones).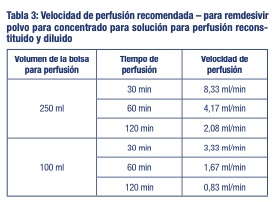
Preparación de remdesivir solución para perfusión Reconstitución Extraiga el número necesario de viales de un solo uso de su lugar de almacenamiento. Para cada vial: Reconstituya asépticamente remdesivir polvo para concentrado para solución para perfusión añadiendo 19 ml de agua estéril para preparaciones inyectables utilizando una jeringa y aguja de tamaño adecuado por vial. Deseche el vial si el vacío no arrastra el agua estéril para preparaciones inyectables hacia el interior del vial. Agite inmediatamente el vial durante 30 segundos. Deje que el contenido del vial se asiente durante 2 a 3 minutos. Se debe formar una solución transparente. Si el contenido del vial no se ha disuelto completamente, agite el vial de nuevo durante 30 segundos y deje que el contenido se asiente durante 2 a 3 minutos. Repita este procedimiento según sea necesario hasta que el contenido del vial se disuelva completamente. Inspeccione el vial para asegurarse de que el cierre del envase no tiene defectos y que la solución no tiene partículas. Diluya inmediatamente después de la reconstitución. Dilución: Se debe tener cuidado para evitar la contaminación microbiana accidental. Dado que este producto no contiene ningún conservante ni agente bacteriostático, se debe utilizar una técnica aséptica para preparar la solución parenteral final. Siempre se recomienda administrar los medicamentos IV inmediatamente después de la preparación, cuando sea posible. Utilizando la Tabla 4, determine el volumen de solución inyectable de cloruro de sodio 9 mg/ml (0,9 %) a extraer de la bolsa para perfusión.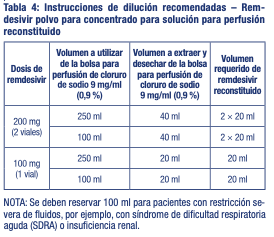
Extraiga y deseche el volumen requerido de cloruro de sodio 9 mg/ml de la bolsa utilizando una jeringa y aguja de tamaño adecuado según la Tabla 4. Extraiga el volumen requerido de remdesivir polvo para concentrado para solución para perfusión reconstituido utilizando una jeringa de tamaño adecuado según la Tabla 4. Deseche cualquier parte no utilizada que quede en el vial de remdesivir. Transfiera el volumen requerido de remdesivir polvo para concentrado para solución para perfusión reconstituido a la bolsa para perfusión seleccionada. Invierta suavemente la bolsa 20 veces para mezclar la solución en la bolsa. No la agite. La solución preparada es estable durante 4 horas a temperatura ambiente (entre 20 °C y 25 °C) o durante 24 horas en heladera (entre 2 °C y 8 °C) (incluido cualquier momento antes de la dilución en líquidos para perfusión intravenosa). Una vez finalizada la perfusión, enjuague con al menos 30 ml de cloruro de sodio 9 mg/ml. Eliminación: La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Contraindicaciones.
Hipersensibilidad al (a los) principio(s) activo(s) o a alguno de los excipientes.
Reacciones adversas.
Resumen del perfil de seguridad: La reacción adversa más frecuente en voluntarios sanos es la elevación de transaminasas (14 %). La reacción adversa más frecuente en pacientes con COVID-19 son náuseas (4 %). Tabla de reacciones adversas: Las reacciones adversas en la Tabla 5 se enumeran a continuación según la clasificación de órganos del sistema y la frecuencia. Las frecuencias se definen del siguiente modo: Muy frecuentes (≥1/10); frecuentes (≥1/100 a < 1/10); poco frecuentes (≥1/1.000 a < 1/100); raras (≥1/10.000 a < 1/1.000).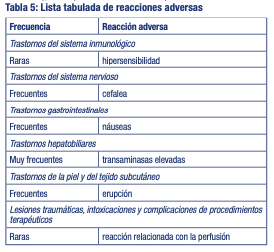
Descripción de las reacciones adversas seleccionadas: Transaminasas elevadas: En estudios en voluntarios sanos, los aumentos en la ALT, la aspartato aminotransferasa (AST) o ambos en sujetos que recibieron remdesivir fueron de grado 1 (10 %) o de grado 2 (4 %). En un estudio clínico aleatorizado, doble ciego, controlado con placebo en pacientes con COVID-19 (Estudio ACTT-1 del NIAID), la incidencia de reacciones adversas no graves de grado ≥3 de niveles elevados de aminotransferasa, incluida la ALT, la AST o ambas fue del 4 % de los pacientes que recibieron remdesivir en comparación con el 6 % de los que recibieron placebo. En un ensayo clínico aleatorizado, abierto y multicéntrico (Estudio GS-US-540-5773) en pacientes hospitalizados con COVID-19 grave que recibieron remdesivir durante 5 (n = 200) o 10 días (n = 197), se notificaron alteraciones en las pruebas analíticas de cualquier grado [≥1,25 veces el límite superior de la normalidad (LSN)] de AST elevada y ALT elevada en un 40 % y 42 % de los pacientes, respectivamente, que recibieron remdesivir. Se notificaron alteraciones en las pruebas analíticas de grado ≥3 (≥5,0 veces el LSN) de AST elevada y ALT elevada en un 7 % de los pacientes que recibieron remdesivir. En un ensayo clínico aleatorizado, abierto y multicéntrico (Estudio GS-US-540-5774) en pacientes hospitalizados con COVID-19 moderada que recibieron remdesivir durante 5 (n = 191) o 10 días (n = 193) en comparación con el tratamiento de referencia (n = 200), se notificaron alteraciones en las pruebas analíticas de cualquier grado de AST elevada y ALT elevada en un 32 % y 33 % de los pacientes, respectivamente, que recibieron remdesivir, y en un 33 % y 39 % de los pacientes, respectivamente, que recibieron el tratamiento de referencia. Se notificaron alteraciones en las pruebas analíticas de grado ≥3 de AST elevada y ALT elevada en un 2 % y 3 % de los pacientes, respectivamente, que recibieron remdesivir y en un 6 % y 7 %, respectivamente, que recibieron el tratamiento de referencia. Notificación de sospechas de reacciones adversas Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia al siguiente link: https://www.argentina.gob.ar/anmat/farmacovigilancia/notificanos/eventosadversos y/o al Departamento de Farmacovigilancia de GADOR S.A. vía email a farmacovigilancia@gador.com o telefónicamente al 0800-220-2273 (CARE).
Advertencias.
Hipersensibilidad, incluidas las reacciones anafilácticas y las relacionadas con la perfusión Se han observado reacciones de hipersensibilidad que incluyen reacciones anafilácticas y relacionadas con la perfusión durante y después de la administración de remdesivir. Los signos y los síntomas pueden incluir hipotensión, hipertensión, taquicardia, bradicardia, hipoxia, pirexia, disnea, sibilancia, angioedema, erupción, náuseas, vómitos, diaforesis y escalofrío. Se pueden considerar velocidades de perfusión más lentas, con un tiempo de perfusión máximo de hasta 120 minutos, para prevenir potencialmente estos signos y síntomas. Si se presentan signos y síntomas de una reacción de hipersensibilidad clínicamente significativa, suspenda inmediatamente la administración de remdesivir e inicie el tratamiento adecuado. Elevaciones de las transaminasas Se han observado elevaciones de las transaminasas en los ensayos clínicos con remdesivir, que incluyó a voluntarios sanos y pacientes con COVID-19. Se debe determinar la función hepática en todos los pacientes antes de comenzar el tratamiento con remdesivir y se debe controlar mientras se les administre cuando sea clínicamente adecuado. No se han realizado estudios clínicos con remdesivir en pacientes con insuficiencia hepática. Solo se debe utilizar remdesivir en pacientes con insuficiencia hepática si el posible beneficio supera el posible riesgo. El tratamiento con remdesivir no se debe iniciar en pacientes con alanina aminotransferasa (ALT) ≥5 veces el límite superior de la normalidad al inicio. Se debe interrumpir el tratamiento con remdesivir en pacientes que presenten: ALT ≥5 veces el límite superior de la normalidad durante el tratamiento con remdesivir. Se puede reanudar el tratamiento con remdesivir cuando la ALT sea < 5 veces el límite superior de la normalidad. O Elevación de la ALT junto con signos o síntomas de inflamación hepática o aumento de la bilirrubina conjugada, la fosfatasa alcalina o el índice internacional normalizado (IIN) Insuficiencia renal: En los estudios en animales, en ratas y monos, se observó toxicidad renal grave. El mecanismo de esta toxicidad renal no se conoce totalmente. No se puede descartar su relevancia en los seres humanos. Se debe determinar la TFGe en todos los pacientes antes de comenzar el tratamiento con remdesivir y mientras se les administre cuando sea clínicamente adecuado. No se debe utilizar remdesivir en pacientes con una TFGe < 30 ml/min. Excipientes: Remdesivir contiene éter sulfobutilico de beta ciclodextrina sódica, que se elimina por vía renal y se acumula en pacientes con función renal disminuida, lo que puede posiblemente afectar negativamente a la función renal. Por lo tanto, no se debe utilizar remdesivir en pacientes con una TFGe < 30 ml/min. Riesgo de disminución de la actividad antiviral cuando se administra de forma concomitante con cloroquina o hidroxicloroquina No se recomienda la administración concomitante de remdesivir y fosfato de cloroquina o sulfato de hidroxicloroquina en base a los datos in vitro que demuestran un efecto antagonista de la cloroquina sobre la activación metabólica intracelular y la actividad antiviral de remdesivir. Interacción con otros medicamentos y otras formas de interacción No se han realizado estudios clínicos de interacciones con remdesivir. Actualmente se desconoce el potencial general de interacciones; los pacientes deben permanecer bajo una estrecha observación durante los días de la administración de remdesivir. Debido al antagonismo observado in vitro, no se recomienda el uso concomitante de remdesivir con fosfato de cloroquina o sulfato de hidroxicloroquina. Efectos de otros medicamentos sobre remdesivir: in vitro, remdesivir es un sustrato para las esterasas en plasma y tejido, las enzimas metabolizadoras de fármacos CYP2C8, CYP2D6 y CYP3A4, y es un sustrato para los polipéptidos transportadores de aniones orgánicos 1B1 (OATP1B1) y los transportadores de la glucoproteína P (gp-P). No se ha estudiado el potencial de interacción de remdesivir con inhibidores/inductores de la vía hidrolítica (esterasa) o CYP2C8, 2D6 o 3A4. Se desconoce el riesgo de interacción clínicamente relevante. Los inhibidores potentes pueden provocar un aumento de la exposición a remdesivir. El uso de inductores potentes (p. ej., rifampicina) puede reducir las concentraciones plasmáticas de remdesivir y no se recomienda. Se ha descrito que la dexametasona es un inductor moderado de CYP3A y gp-P. La inducción depende de la dosis y se produce después de varias dosis. Es poco probable que la dexametasona tenga un efecto clínicamente significativo sobre el remdesivir, ya que el remdesivir tiene una tasa de extracción hepática moderada-alta y se usa durante un periodo corto en el tratamiento con COVID-19. Efectos de remdesivir sobre otros medicamentos in vitro, remdesivir es un inhibidor de CYP3A4, OATP1B1 y OATP1B3. No se ha establecido la relevancia clínica de estas interacciones farmacológicas in vitro. Remdesivir puede aumentar de forma transitoria las concentraciones plasmáticas de medicamentos que son sustratos de CYP3A o de OATP 1B1/1B3. No hay datos disponibles, sin embargo, parece indicar que los medicamentos que son sustratos de CYP3A4 o sustratos de OATP 1B1/1B3 se deben administrar al menos 2 horas después de remdesivir. Remdesivir indujo CYP1A2 y potencialmente CYP3A in vitro. La administración concomitante de remdesivir con sustratos de CYP1A2 o CYP3A4 con un índice terapéutico estrecho puede dar lugar a la pérdida de su eficacia. La dexametasona es un sustrato de CYP3A4 y, aunque remdesivir inhibe CYP3A4, debido a la rápida eliminación de remdesivir después de la administración IV, es poco probable que remdesivir tenga un efecto significativo sobre la exposición a dexametasona. Fertilidad, embarazo y lactancia Embarazo: No hay datos o éstos son limitados relativos al uso de remdesivir en mujeres embarazadas. Los estudios en animales son insuficientes con respecto a la toxicidad para la reproducción. No se debe utilizar remdesivir durante el embarazo a menos que el estado clínico de las mujeres requiera tratamiento con este. Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces durante el tratamiento. Lactancia: Se desconoce si remdesivir se excreta en la leche materna o los efectos sobre el lactante o sobre la producción de leche. En los estudios en animales, el metabolito análogo de nucleósido GS-441524 se ha detectado en la sangre de crías de ratas lactantes de madres que recibieron remedsivir. Por lo tanto, se puede suponer la excreción de remdesivir y/o los metabolitos en la leche de los animales lactantes. Debido al potencial de transmisión viral a los lactantes negativos al SARS-CoV-2 y a las reacciones adversas del medicamento en los lactantes, se debe tomar la decisión de suspender la lactancia o suspender/abstenerse de administrar el tratamiento con remdesivir teniendo en cuenta el beneficio de la lactancia materna para el niño y el beneficio del tratamiento para la mujer. Fertilidad: No hay datos en humanos disponibles sobre el efecto de remdesivir en la fertilidad. En ratas macho, no se observó ningún efecto sobre el apareamiento o la fertilidad con el tratamiento con remdesivir. En ratas hembras, sin embargo, se observó un deterioro de la fertilidad. Se desconoce la relevancia en los seres humanos. Efectos sobre la capacidad para conducir y utilizar máquinas La influencia de remdesivir sobre estas capacidades se prevé que sea nula o insignificante.
Conservación.
Mantener en su envase original a temperatura ambiente hasta 30°C. Solución reconstituida y diluida para perfusión: Una vez reconstituido, VEKLURY® se debe diluir inmediatamente. Conservar la solución diluida de remdesivir para perfusión hasta 4 horas a una temperatura ambiente (entre 20-25°C) o 24 horas en heladera (entre 2 °C y 8 °C). No deje pasar más de 24 horas entre la dilución y la administración. Precauciones especiales de eliminación y otras manipulaciones Preparar la solución para perfusión en condiciones asépticas y en el mismo día de la administración. Remdesivir se debe inspeccionar visualmente para detectar partículas y cambios de color antes de la administración, siempre que la solución y el envase lo permitan. De observarse lo anterior, la solución se debe desechar y preparar una solución nueva. Remdesivir se debe reconstituir con 19 ml de agua estéril para preparaciones inyectables y diluir en solución inyectable de cloruro de sodio 9 mg/ml (0,9 %) antes de ser administrado mediante perfusión intravenosa durante 30 a 120 minutos.
Sobredosificación.
El tratamiento de la sobredosis con remdesivir debe consistir en medidas generales de apoyo, incluida la vigilancia de las constantes vitales, así como la observación del estado clínico del paciente. No existe ningún antídoto específico para tratar la sobredosis con remdesivir.
Presentación.
Vial de vidrio transparente de tipo I con cierre elastomérico y un precinto de aluminio con tapón desprendible. Tamaño de envase: 1 vial.
Revisión.
07/2021. G00222800-01.