Valixa®
BIOPAS
Valganciclovir.
Agente antiviral de uso sistémico.
Composición.
Cada comprimido recubierto contiene 450 mg de valganciclovir (en forma de clorhidrato), en un excipiente compuesto por povidona K30: 23,9 mg, crospovidona 23,9 mg, celulosa microcristalina 47,8 mg, ácido esteárico en polvo 6 mg y Opadry Pink YS-1-14551a: 18 mg (*). (*) Contiene hidroxipropilmetil celulosa, dióxido de titanio, polietilenglicol 400, óxido de hierro rojo y polisorbato 80. Cada mililitro de solución reconstituida contiene 50 mg de valganciclovir (en forma de clorhidrato), en un excipiente compuesto por manitol 5,78 g, ácido fumárico 0,20 g, povidona K30: 0,20 g, benzoato de sodio 0,10 g, sacarina sódica 0,03 g y sabor tutti-frutti 0,18 g.
Farmacología.
Código ATC: J05AB14. Grupo farmacoterapéutico: Agente antiviral de uso sistémico, nucleósidos y nucleótidos, excluidos inhibidores de la transcriptasa inversa. Propiedades farmacodinámicas: Mecanismo de acción: Valganciclovir es un éster L-valílico (profármaco) del ganciclovir. Después de su administración oral, valganciclovir se metaboliza de manera rápida y extensa a ganciclovir por las estearasas intestinales y hepáticas. Ganciclovir es un análogo sintético de la 2'-desoxiguanosina e inhibe la replicación de los virus herpéticos in vitro e in vivo. Los virus humanos sensibles a este medicamento son el citomegalovirus humano (CMV humano), los virus del herpes simple 1 y 2 (HSV-1 y HSV-2), los virus del herpes humano 6, 7 y 8 (HHV-6, HHV-7, HHV-8), el virus de Epstein-Barr (EBV), el virus de la varicela zóster (VZV) y el virus de la hepatitis B (VHB). En las células infectadas por CMV, ganciclovir se fosforila inicialmente a monofosfato de ganciclovir por la proteinquinasa vírica pUL97. La fosforilación posterior tiene lugar por quinasas celulares que producen trifosfato de ganciclovir, el cual se metaboliza lentamente dentro de la célula. Se ha demostrado que el metabolismo del trifosfato ocurre en células infectadas por HSV y por CMV humano, con vidas medias de 18 y 6-24 horas respectivamente, después de eliminar el ganciclovir extracelular. Como la fosforilación depende, fundamentalmente, de la quinasa vírica, el ganciclovir se fosforila preferentemente dentro de las células infectadas por el virus. La actividad virostática del ganciclovir se debe a la inhibición de la síntesis del ADN vírico a través de: (a) inhibición competitiva de la incorporación del trifosfato de desoxiguanosina al ADN a través de la ADN-polimerasa vírica, y (b) incorporación del trifosfato de ganciclovir al ADN vírico originando la terminación del ADN o limitando muchísimo la elongación posterior del ADN vírico. Actividad antivírica: La actividad in vitro antivírica, medida como CI50 del ganciclovir frente al CMV oscila en el intervalo de 0,08 mM (0,02 mg/ml) a 14 mM (3,5 mg/ml). El efecto clínico antiviral de Valixa se ha demostrado en el tratamiento de los pacientes de SIDA con retinitis por CMV recién diagnosticada. La eliminación de CMV disminuyó en orina desde el 46% (32/69) de los pacientes al comienzo del estudio hasta el 7% (4/55) de los pacientes después de cuatro semanas de tratamiento con Valixa. Eficacia clínica y seguridad: Pacientes adultos: Tratamiento de la retinitis por CMV: En un estudio se distribuyó aleatoriamente a pacientes recién diagnosticados de retinitis por CMV para recibir tratamiento de inducción con 900 mg de Valixa, dos veces por día, o con 5 mg/kg de ganciclovir intravenoso, dos veces por día. El porcentaje de pacientes con retinitis progresiva por CMV demostrada fotográficamente a las 4 semanas fue comparable en los dos grupos tratados, 7/70 y 7/71 pacientes progresaron en los grupos de ganciclovir intravenoso y valganciclovir respectivamente. Después del tratamiento de inducción, todos los pacientes de este estudio recibieron tratamiento de mantenimiento con Valixa en dosis de 900 mg una vez por día. La media del tiempo desde la aleatorización hasta la progresión de la retinitis por CMV del grupo que recibió tratamiento de inducción y mantenimiento con Valixa fue de 226 (160) días y la del grupo al que se administró terapia de inducción con ganciclovir por vía intravenosa y de mantenimiento con Valixa, de 219 (125) días. Prevención de la enfermedad por CMV en el trasplante: Se ha realizado un ensayo clínico doble-ciego, con doble enmascaramiento con comparador activo en pacientes con trasplante de corazón, hígado y riñón (no se incluyeron pacientes con trasplante pulmonar y gastrointestinal) con alto riesgo de enfermedad por CMV (D+/R-) que recibieron bien Valixa (900 mg una vez por día) o ganciclovir oral (1.000 mg tres veces por día), comenzando dentro de los 10 días del trasplante hasta el día 100 postrasplante. La incidencia de la enfermedad por CMV (síndrome por CMV más afección tisular invasiva) durante los primeros 6 meses postrasplante fue 12,1% en el grupo de Valixa (n = 239) comparado con 15,2% en el grupo de ganciclovir oral (n = 125). La gran mayoría de los casos se produjo tras del cese de la profilaxis (después del día 100) y los casos en el grupo de valganciclovir ocurrieron por término medio más tarde que los aparecidos en el de ganciclovir oral. La incidencia de rechazo agudo en los primeros 6 meses fue de 29,7% en pacientes randomizados a vanganciclovir comparado con 36,0% en el de ganciclovir oral, siendo la incidencia por pérdida de injerto equivalente, ya que se manifestó en cada grupo en un 0,8% de los pacientes. Se ha realizado un ensayo clínico doble-ciego, controlado con placebo en 326 pacientes con trasplante de riñón y alto riesgo de enfermedad por CMV (D+/R-), para evaluar la eficacia y la seguridad de Valixa prolongando la profilaxis de CMV de 100 a 200 días postrasplante. Los pacientes fueron aleatorizados (1:1) para recibir Valixa comprimidos (900 mg una vez por día) dentro de los 10 días de trasplante, un grupo hasta el día 200 postrasplante y el otro grupo hasta el día 100 postrasplante continuando otros 100 días con placebo. En la Tabla 1 se muestra la proporción de pacientes que desarrollaron la enfermedad por CMV durante los primeros 12 meses postrasplante.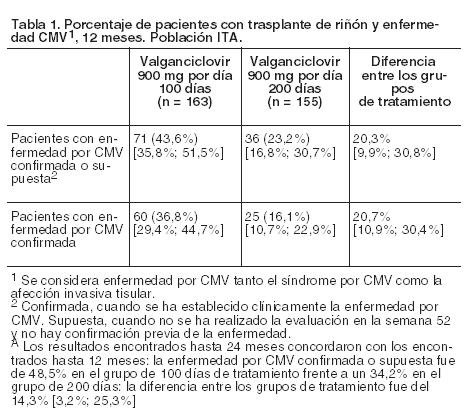
El desarrollo de la enfermedad por CMV fue significativamente menor en pacientes con trasplante de riñón de alto riesgo cuando la profilaxis de CMV con Valixa se extendió hasta el día 200 postrasplante comparado con aquéllos que recibieron Valixa como profilaxis de CMV hasta el día 100 postrasplante. La tasa de sobrevida del injerto, así como la incidencia de rechazo agudo comprobado por biopsia fueron similares en ambos grupos de tratamiento. La tasa de sobrevida del injerto a los 12 meses postrasplante fue del 98,2% (160/163) para el régimen de duración de 100 días y del 98,1% (152/155) para el de 200 días. Hasta los 24 meses postrasplante, se notificaron cuatro casos adicionales de pérdida de injerto, todos en el grupo de 100 días de dosificación. La incidencia de rechazo agudo comprobado por biopsia a los 12 meses postrasplante fue de 17,2% (28/163) para el régimen de 100 días y de 11,0% (17/155) para el de 200 días. Hasta los 24 meses postrasplante, se informó un caso adicional de pérdida del injerto en el grupo de duración de 200 días. Resistencia vírica: Después del tratamiento crónico con ganciclovir pueden surgir virus resistentes al valganciclovir por selección de mutaciones del gen de la quinasa vírica (UL97) responsable de la monofosforilación del ganciclovir, y/o del gen de la polimerasa vírica (UL54), o de ambos. Los virus con mutaciones del gen UL97 muestran resistencia al ganciclovir solo, mientras que aquellos con mutaciones del gen UL54 presentan resistencia a ganciclovir, pudiendo mostrar resistencia cruzada a otros antivirales cuyo mecanismo de acción sea la polimerasa viral. Tratamiento de la retinitis por CMV: En un estudio clínico el análisis genotípico de CMV en leucocitos polimorfonucleares (PMNL) aislados de 148 pacientes incorporados con retinitis por CMV mostró que el 2,2%, el 6,5%, el 12,8% y el 15,3% de aquellos contienen mutaciones de UL97 después del tratamiento con valganciclovir durante 3, 6, 12 y 18 meses, respectivamente. Prevención de la enfermedad por CMV en el trasplante: Ensayo con comparador activo: Se estudió la resistencia mediante el análisis genotípico de CMV en muestras de leucocitos polimorfonucleares (PMNL) recogidas (a) el día 100 (fin de la administración del fármaco en el estudio de profilaxis) y (b) en casos de sospecha de enfermedad por CMV hasta 6 meses después del trasplante. De los 245 pacientes randomizados que recibieron valganciclovir, se dispuso de 198 muestras del día 100 para examen y no se observaron mutaciones de resistencia al ganciclovir. Esto puede compararse con 2 mutaciones de resistencia a ganciclovir detectadas en 103 muestras examinadas de los pacientes en el grupo comparador de ganciclovir oral (1,9%). De los 245 pacientes randomizados que recibieron valganciclovir, se examinaron 50 muestras de pacientes con sospecha de enfermedad por CMV y no se observaron mutaciones de resistencia. De los 127 pacientes randomizados en el grupo comparador de ganciclovir, se examinaron muestras de 29 pacientes con sospecha de enfermedad por CMV, y se detectaron dos mutaciones de resistencia, lo que dio lugar a una incidencia de resistencia de 6,9%. Ensayo de extensión de la profilaxis de 100 a 200 días de tratamiento: El análisis genotípico se realizó en los genes UL54 y UL97 derivados del virus extraído a 72 pacientes que mostraron resistencia según el análisis de los siguientes criterios: los que tuvieron una carga viral positiva ( > 600 copias/ml) al final de la profilaxis y/o aquéllos en los que se confirmó la enfermedad por CMV hasta los 12 meses (52 semanas) postrasplante. Tres pacientes de cada grupo de tratamiento presentaron una mutación de resistencia a ganciclovir conocida. Población pediátrica: Tratamiento de la retinitis por CMV: La Agencia Europea de Medicamentos ha eximido de realizar estudios con Valixa en todos los subgrupos de población pediátrica en tratamiento por infección por CMV en pacientes inmunosuprimidos (véase la información sobre el uso en pediatría en Dosificación). Prevención de la enfermedad por CMV en el trasplante: En un ensayo Fase II de farmacocinética y seguridad en pacientes pediátricos (de 4 meses a 16 años de edad, n = 63) con un trasplante de órgano sólido, que fueron tratados con valganciclovir una vez por día continuando hasta los 100 días de acuerdo con el algoritmo de dosificación en pediatría (véase Dosificación), las exposiciones que se alcanzaron fueron similares a las de adultos (véase Farmacología, Propiedades farmacocinéticas). El seguimiento después del tratamiento fue de 12 semanas. La situación serológica por CMV D/R con respecto al inicio fue D+/R- en el 40%, D+/R+ en el 38%, D-/R+ en el 19% y D-/R- en el 3% de los casos. La presencia de virus CMV fue notificada en 7 pacientes. Las reacciones adversas observadas fueron de naturaleza similar a la de los adultos (véase Reacciones adversas). En un estudio Fase IV de tolerabilidad en pacientes pediátricos receptores de un trasplante renal (de 1 - 16 años de edad; n = 57) que recibieron valganciclovir una vez por día durante un período de hasta 200 días conforme al algoritmo de dosificación (véase Dosificación), la incidencia de infección por CMV fue baja. El período de seguimiento después del tratamiento fue de 24 semanas. La situación serológica por CMV D/R con respecto al inicio fue D+/R+ en el 45%, D+/R- en el 39%, D-/R+ en el 7%, D-/R- en el 7% y ND/R+ en el 2% de los casos. Se notificó la presencia de viremia por citomegalovirus en 3 pacientes y se sospechó un caso de síndrome por CMV en 1 paciente, que no fue confirmado mediante PCR del CMV en el laboratorio central. Las reacciones adversas observadas fueron de naturaleza similar a las de los adultos (véase Reacciones adversas). Estos datos respaldan la extrapolación a los niños de los datos sobre eficacia de los adultos y permiten dar recomendaciones posológicas para los pacientes pediátricos. En un estudio Fase I de farmacocinética y seguridad en pacientes con trasplante cardíaco (de 3 semanas a 125 días de edad; n = 14) que recibieron una dosis una vez por día de valganciclovir según el algoritmo de dosificación pediátrica (véase Dosificación) en 2 días consecutivos, las exposiciones fueron similares a las observadas en adultos (véase Farmacología, Propiedades farmacocinéticas). El seguimiento después del tratamiento se mantuvo durante 7 días. El perfil de seguridad concordó con el observado en otros estudios en pacientes pediátricos y adultos, aunque el número de pacientes y la exposición al valganciclovir en este estudio fueron limitados. Infección congénita por CMV: La eficacia y la seguridad del ganciclovir y el valganciclovir se investigaron en dos estudios en neonatos y lactantes con infección congénita sintomática por CMV. En el primer estudio, la farmacocinética y seguridad de una dosis única de valganciclovir (rango de dosis 14-162-20 mg/kg/dosis) fue estudiada en 24 neonatos (8 a 34 días de edad) con enfermedad congénita sintomática por CMV (véase Farmacología, Propiedades farmacocinéticas). Los neonatos recibieron tratamiento antiviral durante 6 semanas, en el que 19 de los 24 pacientes fueron tratados con valganciclovir oral hasta 4 semanas y las 2 semanas restantes con ganciclovir intravenoso. A los 5 pacientes restantes se administró ganciclovir intravenoso durante la mayoría del tiempo del estudio. En el segundo estudio, se evaluó la eficacia y la seguridad de 6 semanas versus 6 meses de tratamiento con valganciclovir en 109 lactantes de 2 a 30 días de edad con enfermedad congénita sintomática por CMV. Todos los lactantes recibieron valganciclovir por vía oral en dosis de 16 mg/kg dos veces por día durante 6 semanas. Después de 6 semanas de tratamiento, se asignó aleatoriamente a los lactantes, en una proporción 1:1, a continuar el tratamiento con valganciclovir con la misma dosis o a recibir el placebo correspondiente hasta completar 6 meses de tratamiento. Esta indicación de tratamiento no está actualmente recomendada para valganciclovir. El diseño de los estudios y los resultados obtenidos son demasiado escasos para sacar conclusiones precisas sobre la eficacia y seguridad de valganciclovir. Propiedades farmacocinéticas: Las propiedades farmacocinéticas del valganciclovir se han evaluado en pacientes que presentaban seropositividad al VIH y CMV, pacientes con SIDA y retinitis por CMV y en aquéllos con trasplante de órgano sólido. Absorción: Valganciclovir es un profármaco del ganciclovir. Se absorbe perfectamente en el tubo digestivo y se metaboliza en forma rápida y extensa en la pared intestinal y en el hígado a ganciclovir. La exposición sistémica a valganciclovir es transitoria y baja. La biodisponibilidad del ganciclovir, a partir de las dosis orales de valganciclovir, es aproximadamente del 60% en todas las poblaciones de pacientes estudiadas y el resultado de la exposición a ganciclovir es similar a la obtenida tras su aplicación intravenosa (véase la Tabla 2). Por comparación, la biodisponibilidad de ganciclovir después de la administración de 1.000 mg de ganciclovir oral (en cápsulas) es 6% - 8%. Valganciclovir en pacientes seropositivos para VIH y CMV: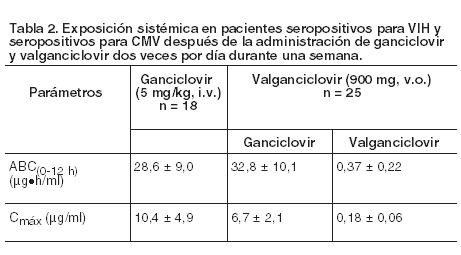
La eficacia de ganciclovir en el aumento del tiempo de progresión de la retinitis por CMV ha demostrado correlación con la exposición sistémica (ABC). Valganciclovir en pacientes con trasplante de órganos sólidos: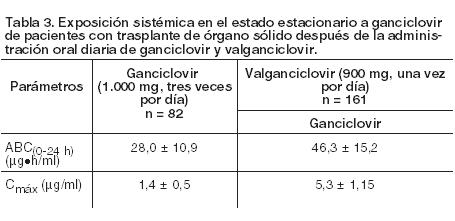
De acuerdo con el algoritmo de dosificación dependiendo de la función renal, la exposición sistémica de ganciclovir en los receptores de trasplante hepático, renal y cardíaco fue similar a la observada tras la administración oral de valganciclovir. Después de la administración de valganciclovir como solución oral, se han obtenido exposiciones sistémicas de ganciclovir equivalentes a las obtenidas con la formulación de comprimidos. Efecto de la comida: La relación de proporcionalidad entre el ABC de ganciclovir y la dosis de valganciclovir, tras la administración de éste último en un rango de dosis de 450 a 2.625 mg, sólo se ha demostrado después de la ingesta de alimentos. Cuando éstos se suministraron junto con valganciclovir a la dosis recomendada de 900 mg, se observaron valores mayores que en ayunas, tanto la media de ABC (aproximadamente 30%) como los valores medios de Cmáx (aproximadamente 14%) de ganciclovir. También, la variación entre individuos en la exposición a ganciclovir desciende cuando se toma Valixa con alimentos. En los estudios clínicos Valixa se ha administrado sólo con comida. Así pues, se recomienda administrar Valixa con las comidas (véase Dosificación). Distribución: Como el valganciclovir se convierte rápidamente en ganciclovir, no se ha determinado la unión de valganciclovir a las proteínas. La unión a proteínas plasmáticas de ganciclovir en concentraciones de 0,5 a 51 mg/ml fue de un 1% - 2%. El volumen de distribución del ganciclovir en el estado estacionario alcanza 0,680 ± 0,161 l/kg (n = 114) después de su administración intravenosa. Biotransformación: El valganciclovir se metaboliza de manera rápida y extensa a ganciclovir; no se conoce ningún otro metabolito. No existe ningún metabolito del ganciclovir radiactivo administrado por vía oral (en dosis única de 1.000 mg) que justifique más del 1% - 2% de la radiactividad recuperada en las heces o en la orina. Eliminación: Después de administrar Valixa, la vía principal de eliminación del valganciclovir consiste en la excreción renal de ganciclovir a través de filtración glomerular y secreción tubular activa. El clearance renal representa el 81,5% ± 22% (n = 70) del clearance sistémico del ganciclovir. En las estimaciones Bayesianas post-hoc para poblaciones, en pacientes con un CrCl > 60 ml/min, la media del clearance de ganciclovir es 14,05 ± 4,13 l/h. En pacientes con insuficiencia renal, la media del clearance de ganciclovir es 8,46 ± 1,67 l/h (CrCl 40 - 60 ml/min) y 7,00 ± 1,08 l/h (CrCl 25 - 40 ml/min). La vida media de ganciclovir a partir de valganciclovir es 4,1 ± 0,9 horas en pacientes seropositivos VIH y CMV. Farmacocinética en situaciones clínicas especiales: Pacientes con insuficiencia renal: La disminución de la función renal reduce el clearance de ganciclovir a partir de valganciclovir con el correspondiente aumento de la vida media terminal. Así pues, es necesario ajustar la dosis de los enfermos con insuficiencia renal (véanse Dosificación y Precauciones). Pacientes sometidos a hemodiálisis: No se puede dar la dosis recomendada de Valixa 450 mg comprimidos recubiertos en quienes estén recibiendo hemodiálisis, dado que la dosis individual de Valixa que precisan estos pacientes es menor que la contenida en los comprimidos recubiertos de 450 mg. Por lo tanto, no se debería usar Valixa comprimidos recubiertos en estos pacientes (véanse Dosificación y Precauciones). En pacientes sometidos a hemodiálisis, se recomienda administrar una dosis individualizada de Valixa polvo para solución oral (véanse Dosificación y Precauciones). Pacientes con alteraciones de la función hepática: La seguridad y la eficacia de Valixa no se han estudiado en pacientes con alteración hepática. Esta no debería afectar a la farmacocinética de ganciclovir, ya que éste se excreta por vía renal; por consiguiente, no se establecen recomendaciones posológicas específicas. Pacientes con fibrosis quística: En un estudio farmacocinético de Fase I en receptores de trasplante de pulmón con o sin fibrosis quística (FQ), 31 pacientes (16 FQ/15 no-FQ) recibieron profilaxis postrasplante con 900 mg/día de Valixa. El estudio indicó que, estadísticamente, la fibrosis quística no tenía una influencia significativa sobre la media total de exposición sistémica a ganciclovir en receptores de trasplante de pulmón. La exposición al ganciclovir en receptores de trasplante de pulmón fue comparable a la demostrada como eficaz para la prevención de citomegalovirus en otros receptores de trasplante de órgano sólido. Población pediátrica: En un ensayo Fase II de farmacocinética y seguridad en pacientes pediátricos (de 4 meses a 16 años de edad, n = 63) con un trasplante de órgano sólido, se administró valganciclovir una vez por día continuando hasta los 100 días. Los parámetros farmacocinéticos fueron similares entre los tipos de órgano y rango de edad y comparables a los de adultos. El modelo de población farmacocinético mostró que la biodisponibilidad fue aproximadamente del 60%. En el clearance influyó positivamente el área de superficie corporal y la función renal. En un estudio Fase I de farmacocinética y seguridad en pacientes pediátricos receptores de un trasplante cardíaco (de 3 semanas a 125 días de edad; n = 14), se administró valganciclovir una vez por día durante los 2 días del estudio. En base a la farmacocinética poblacional se estimó que la biodisponibilidad media era del 64%. Una comparación de los resultados de estos dos estudios y los hallazgos farmacocinéticos en la población de adultos muestra que los intervalos del ABC0-24 h fueron muy similares en todos los grupos de edad, incluidos los adultos. La media de los valores del ABC0-24 h y la Cmáx también fueron semejantes en los grupos pediátricos de edad < 12 años, aunque hubo una tendencia a la disminución de la media de los valores del ABC0-24 h y la Cmáx en todo el intervalo de edad pediátrica, que pareció correlacionarse con el aumento de la edad. Esta tendencia fue más evidente en lo que respecta a los valores medios del clearance y la vida media (t½); sin embargo, esto es previsible, dado que en el clearance influyen los cambios del peso, la altura y la función renal asociados con el crecimiento del paciente, tal como indica el modelo farmacocinético poblacional.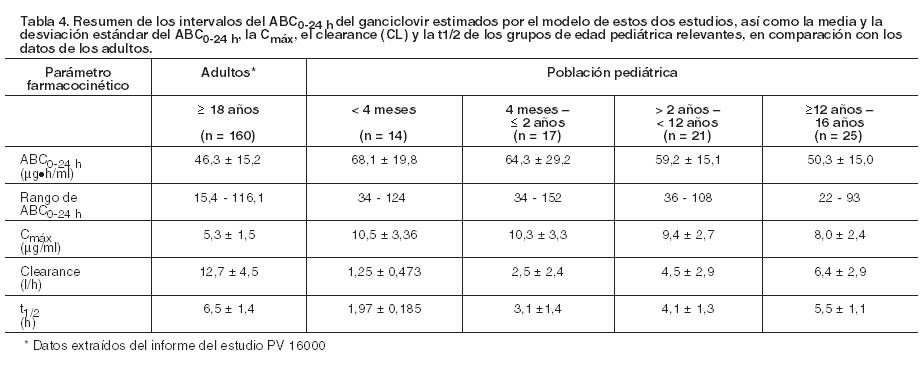
La dosis única diaria de Valixa se determinó en los dos estudios descriptos anteriormente en base al área de la superficie corporal (ASC) y al clearance de creatinina (CrCl) a partir de la fórmula de Schwartz modificada y se calculó usando el algoritmo de dosificación que se menciona en Dosificación. Los parámetros farmacocinéticos tras la administración de ganciclovir fueron también evaluados en dos estudios con neonatos y lactantes con enfermedad por CMV congénita. En el primer estudio, 24 neonatos de 8 a 34 días de edad, recibieron 6 mg/kg de ganciclovir intravenoso dos veces por día. Los pacientes fueron tratados con valganciclovir oral, donde el rango de la dosis de valganciclovir polvo para solución oral fue desde 14 mg/kg a 20 mg/kg dos veces por día, con una duración total de tratamiento de 6 semanas. Con una dosis de 16 mg/kg dos veces por día de valganciclovir polvo para solución oral se alcanzó una exposición de ganciclovir comparable con la de ganciclovir intravenoso 6 mg/kg dos veces por día en neonatos, y también se logró una exposición de ganciclovir similar a la dosis intravenosa de 5 mg/kg eficaz en un adulto. En el segundo estudio, 109 neonatos de 2 a 30 días de edad recibieron 16 mg/kg de valganciclovir polvo para solución oral dos veces por día durante 6 semanas, y posteriormente 96 de los 109 pacientes incorporados fueron asignados aleatoriamente a seguir recibiendo valganciclovir durante 6 meses o bien el placebo. Sin embargo, la media del ABC0-12 h fue menor en comparación con la media del ABC0-12 h del primer estudio.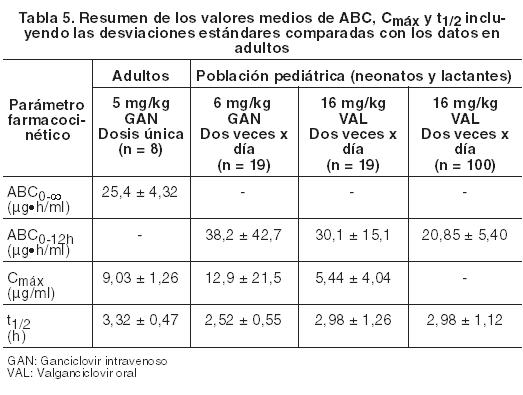
Estos datos son muy escasos para sacar conclusiones sobre las recomendaciones posológicas y la eficacia en la población pediátrica con infección congénita por CMV. Datos preclínicos sobre seguridad: Valganciclovir es un profármaco de ganciclovir y, por consiguiente, los efectos observados con ganciclovir son igualmente aplicables para valganciclovir. La toxicidad de valganciclovir en los estudios preclínicos de seguridad fue la misma que la registrada con ganciclovir y fue inducida con niveles de exposición a ganciclovir comparables, o más bajos que los alcanzados en seres humanos a los que se les administró la dosis de inducción. Estos hallazgos fueron gonadotoxicidad (pérdida de células testiculares) y nefrotoxicidad (uremia, degeneración celular) que fueron irreversibles, mielotoxicidad (anemia, neutropenia, linfocitopenia) y toxicidad gastrointestinal (necrosis de las células de la mucosa) que fueron reversibles. Estudios adicionales han demostrado que ganciclovir es mutagénico, carcinogénico, teratogénico, embriotóxico y espermatogénico (ejemplo, alteración de la fertilidad masculina) y suprime la fertilidad femenina.
Indicaciones.
Valixa está indicado para el tratamento de inducción y mantenimiento de la retinitis por citomegalovirus (CMV) en pacientes adultos con síndrome de inmunodeficiencia adquirida (SIDA). Valixa está indicado para la prevención de la enfermedad por CMV en adultos y niños (desde el nacimiento hasta los 18 años) seronegativos al CMV que han recibido un trasplante de órgano sólido de un donante seropositivo al CMV.
Dosificación.
Posología: Advertencia: se deben seguir estrictamente las recomendacones sobre la posología para evitar sobredosificación (véanse Precauciones y Sobredosificación). Después de su administración oral, el valganciclovir se metaboliza en forma rápida y extensa a ganciclovir; 900 mg de valganciclovir por vía oral, dos veces por día, es equivalente terapéuticamente a 5 mg/kg de ganciclovir administrado por vía intravenosa dos veces por día. La exposición sistémica a ganciclovir después de la administración oral de 900 mg de valganciclovir solución oral es equivalente a la de 900 mg de valganciclovir comprimidos recubiertos. Tratamiento de la retinitis por citomegalovirus (CMV): Pacientes adultos: Tratamiento de inducción de la retinitis por CMV: La dosis recomendada para los pacientes con retinitis activa por CMV es de 900 mg de valganciclovir, dos veces por día, durante 21 días y, siempre que sea posible, debe tomarse con alimentos. Un tratamiento prolongado de inducción puede incrementar el riesgo de toxicidad para la médula ósea (véase Precauciones). Tratamiento de mantenimiento de la retinitis por CMV: Después del tratamiento de inducción, o si se trata de pacientes con retinitis inactiva por CMV, se recomienda administrar una dosis de 900 mg de valganciclovir, una vez por día y, siempre que sea posible, debe tomarse con alimentos. Se puede repetir el tratamiento de inducción en aquellos pacientes en los que la retinitis empeore; sin embargo, se debe tener en cuenta la posibilidad de resistencia viral al fármaco. Pacientes pediátricos: La seguridad y eficacia de Valixa en el tratamiento de la retinitis por CMV en pacientes pediátricos no ha sido establecida en ensayos clínicos adecuados y bien controlados. Prevención de la enfermedad por CMV en el trasplante de órgano sólido. Pacientes adultos: La dosis recomendada en pacientes que han recibido un trasplante de riñón es de 900 mg, una vez por día, comenzando dentro de los 10 días postrasplante hasta los 100 días postrasplante. La profilaxis puede prolongarse hasta los 200 días postrasplante (véanse Precauciones, Reacciones adversas y Farmacología, Propiedades farmacodinámicas). La dosis recomendada en pacientes con trasplante de órgano sólido, distinto del de riñón, es de 900 mg, una vez por día, comenzando dentro de los 10 días postrasplante hasta los 100 días postrasplante. Siempre que sea posible, los comprimidos deben tomarse con alimentos. Población pediátrica: En pacientes pediátricos receptores de un trasplante de órgano sólido, edad contada desde el nacimiento, que están en riesgo de sufrir enfermedad por CMV, la dosis diaria recomendada de Valixa está basada en el área de superficie corporal (ASC) y el clearance de creatinina (CrCl) obtenido mediante la fórmula de Schwartz (CrClS), y se calcula mediante la siguiente ecuación: Dosis pediátrica (mg) = 7 x ASC x CrClS (véase a continuación la fórmula de Mosteller para el cálculo del ASC y la fórmula de Schwartz para el cálculo del CrCl). Si el CrCl calculado mediante la fórmula de Schwartz excede de 150 ml/min/1,73m2, se usará en la ecuación el valor máximo de 150 ml/min/1,73m2:
En pacientes pediátricos receptores de un trasplante renal, la dosis recomendada en mg una vez por día (7 x ASC x CrClS) comenzará a administrarse en los 10 días postrasplante y se mantendrá hasta los 200 días postrasplante. En los pacientes pediátricos que han recibido un trasplante de órgano sólido que no sea de riñón, la dosis recomendada en mg una vez por día (7 x ASC x CrClS) comenzará a administrarse en los 10 días postrasplante y se mantendrá hasta 100 días postrasplante. Todas las dosis calculadas deben redondearse hasta el incremento de 25 mg más próximo para obtener la dosis que se debe administrar. Si la dosis calculada excede de 900 mg, se administrará una dosis máxima de 900 mg. La solución oral es la formulación preferible, dado que permite administrar la dosis calculada conforme a la fórmula anterior; no obstante, pueden usarse los comprimidos recubiertos de Valixa si las dosis calculadas se encuentran dentro del margen del 10% de las dosis de los comprimidos disponibles, y si el paciente puede tragar los comprimidos. Por ejemplo, si la dosis calculada está entre 405 mg y 495 mg, se puede administrar un comprimido de 450 mg. Se recomienda monitorizar la concentración de creatinina sérica regularmente, y considerar los cambios de la altura y el peso, y adaptar la dosis convenientemente durante el período de profilaxis. Pautas posológicas especiales: Pacientes con insuficiencia renal: Los niveles séricos de creatinina o el clearance de creatinina se deben vigilar cuidadosamente. Se debe ajustar la posología según el clearance de creatinina, tal y como se indica en la Tabla 6 (tanto para comprimidos recubiertos como para polvo para solución oral) (véanse Precauciones y Farmacología, Propiedades farmacocinéticas). El clearance de creatinina estimado (ml/min) se puede calcular según la creatinina sérica mediante las siguientes fórmulas:
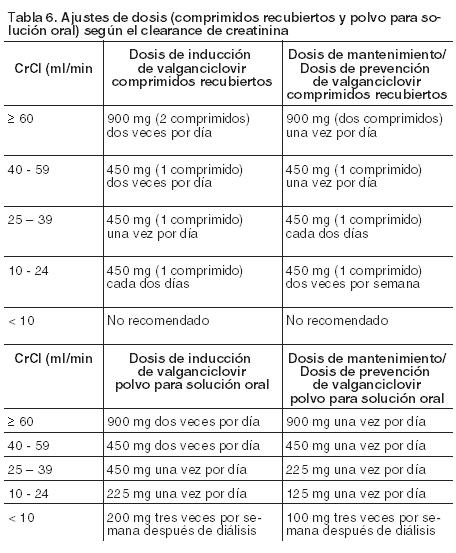
Pacientes sometidos a hemodiálisis: Para pacientes en hemodiálisis (CrCl < 10 ml/min) no se puede dar una recomendación de dosis si se les administra comprimidos recubiertos. Por consiguiente, Valixa comprimidos recubiertos no se debe emplear en estos pacientes (véanse Precauciones y Farmacología, Propiedades farmacocinéticas). Sin embargo, es necesario ajustar la dosis del polvo para solución oral en los pacientes en hemodiálisis (CrCl < 10 ml/min) (véanse Precauciones y Farmacología, Propiedades farmacocinéticas) y en la Tabla 6 se da una recomendación de dosis. Pacientes con insuficiencia hepática: La seguridad y eficacia de Valixa no han sido estudiadas en pacientes con insuficiencia hepática (véase Farmacología, Propiedades farmacocinéticas). Población pediátrica: La posología en pacientes pediátricos receptores de trasplantes de órganos sólidos se individualizará según la función renal, junto con la altura y el peso del paciente. Pacientes de edad avanzada: La seguridad y eficacia de Valixa se desconocen en esta población. Pacientes con leucopenia, neutropenia, anemia, trombocitopenia y pancitopenia graves: Antes de comenzar el tratamiento, véase Precauciones. Si se produce un deterioro significativo del recuento de células sanguíneas durante el tratamiento con Valixa, se deberá considerar el empleo de factores de crecimiento hematopoyético y/o una suspensión de la medicación (véase Precauciones). Formas de administración: Valixa (en sus dos formas farmacéuticas) se administra por vía oral, y siempre que sea posible, debe tomarse con alimentos (véase Farmacología, Propiedades, Propiedades farmacocinéticas). Para pacientes pediátricos que no puedan tragar los comprimidos recubiertos de Valixa, se puede administrar Valixa polvo para suspensión oral. Precauciones que deben tomarse antes de manipular o administrar Valixa comprimidos recubiertos: Los comprimidos no se deben romper ni triturar. Valixa se considera potencialmente teratógeno y carcinógeno para los seres humanos, por lo que se recomienda precaución cuando se manipulen comprimidos rotos (véase Precauciones). Evite el contacto directo de los comprimidos rotos o triturados con la piel o las mucosas. En caso de que ocurra el contacto, lave cuidadosamente la zona con agua y jabón; lave los ojos con agua estéril, o a falta de ésta con abundante agua. Precauciones que deben tomarse antes de manipular o administrar Valixa polvo para suspensión oral: Valixa polvo para suspensión oral requiere ser reconstituido antes de su administración oral. Preparación de Valixa polvo para solución oral: Antes de suministrar el medicamento al paciente, el profesional de la salud deberá preparar la solución oral de Valixa como se indica a continuación (véase Precauciones; y Prospecto Información para el Paciente). Medir 91 ml de agua en una probeta graduada. Quitar el tapón a prueba de niños, añadir el agua en el frasco y cerrar el frasco con el tapón a prueba de niños. Agitar el frasco cerrado hasta que se disuelva todo el polvo formando una solución clara, de incolora a parda. Quitar el tapón a prueba de niños y colocar el adaptador en el cuello del frasco. Cerrar bien fuerte el frasco con el tapón a prueba de niños. Esto asegurará el asentamiento apropiado del adaptador al frasco y la función del tapón a prueba de niños. Almacenar la solución oral reconstituida a una temperatura de 2° C a 8° C por un período de tiempo que no exceda los 49 días. No congelar. Escribir la fecha de vencimiento de la solución oral reconstituida en la etiqueta del frasco. El paciente deberá recibir el Prospecto Información para el Paciente, que incluye las instrucciones de administración para pacientes, junto con 2 dispensadores orales con graduación desde 25 mg hasta 500 mg. Se recomienda que el paciente use el dispensador (véase Prospecto Información para el Paciente).
Contraindicaciones.
Valixa está contraindicado en pacientes con hipersensibilidad a valganciclovir, ganciclovir o a alguno de sus excipientes. Debido a la semejanza en la estructura química de valganciclovir (principio activo de Valixa) y de aciclovir y valaciclovir, es posible que ocurra una reacción de hipersensibilidad cruzada entre estos medicamentos. Por lo tanto, Valixa está contraindicado en pacientes con hipersensibilidad a aciclovir y valaciclovir. Valixa está contraindicado durante la lactancia (véase Precauciones).
Reacciones adversas.
Resumen del perfil de seguridad: El valganciclovir es un profármaco del ganciclovir, que se metaboliza de manera rápida y extensa a ganciclovir después de su administración oral. Puede esperarse que los efectos adversos conocidos asociados con la utilización de ganciclovir se manifiesten también con valganciclovir. Todas las reacciones adversas observadas en los estudios clínicos con valganciclovir se habían observado antes con ganciclovir. Las reacciones adversas más comunes comunicadas después de la administración de valganciclovir en adultos son neutropenia, anemia y diarrea. Valganciclovir, se asocia con un mayor riesgo de diarrea comparado con ganciclovir intravenoso. Además, valganciclovir se vincula con un riesgo más alto de neutropenia y leucopenia comparado con ganciclovir oral. Se observa con más frecuencia neutropenia grave (RAN < 500 células/ml) en pacientes con SIDA y con retinitis por CMV en tratamiento con valganciclovir que en aquéllos con trasplante de órgano sólido que reciben valganciclovir (véase Precauciones). En la Tabla 7 se detalla la frecuencia de las reacciones adversas notificadas en los ensayos clínicos con valganciclovir, ganciclovir oral, o ganciclovir intravenoso. Las reacciones adversas reflejadas en la tabla se comunicaron en ensayos clínicos para el tratamiento de inducción y mantenimiento de la retinitis por CMV en pacientes con SIDA, o para la profilaxis de la enfermedad por CMV en aquéllos con trasplante de corazón, riñón o hígado. El término (grave) que aparece en paréntesis en la tabla indica que la reacción adversa se ha comunicado en pacientes tanto de intensidad leve/moderada como intensidad grave/que supone amenaza para la vida en esa frecuencia específica. El perfil de seguridad de Valixa no cambió al ampliar la profilaxis hasta 200 días en pacientes adultos receptores de un trasplante renal con riesgo elevado de padecer una enfermedad por CMV (D+/R-). La incidencia notificada de leucopenia fue ligeramente mayor en el grupo de tratamiento durante 200 días, mientras que la de neutropenia, anemia y trombocitopenia fue similar en ambos grupos. Lista tabulada de las reacciones adversas: Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Las frecuencias se definen como muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1.000 a < 1/100) y raras (≥ 1/10.000 a < 1/1.000).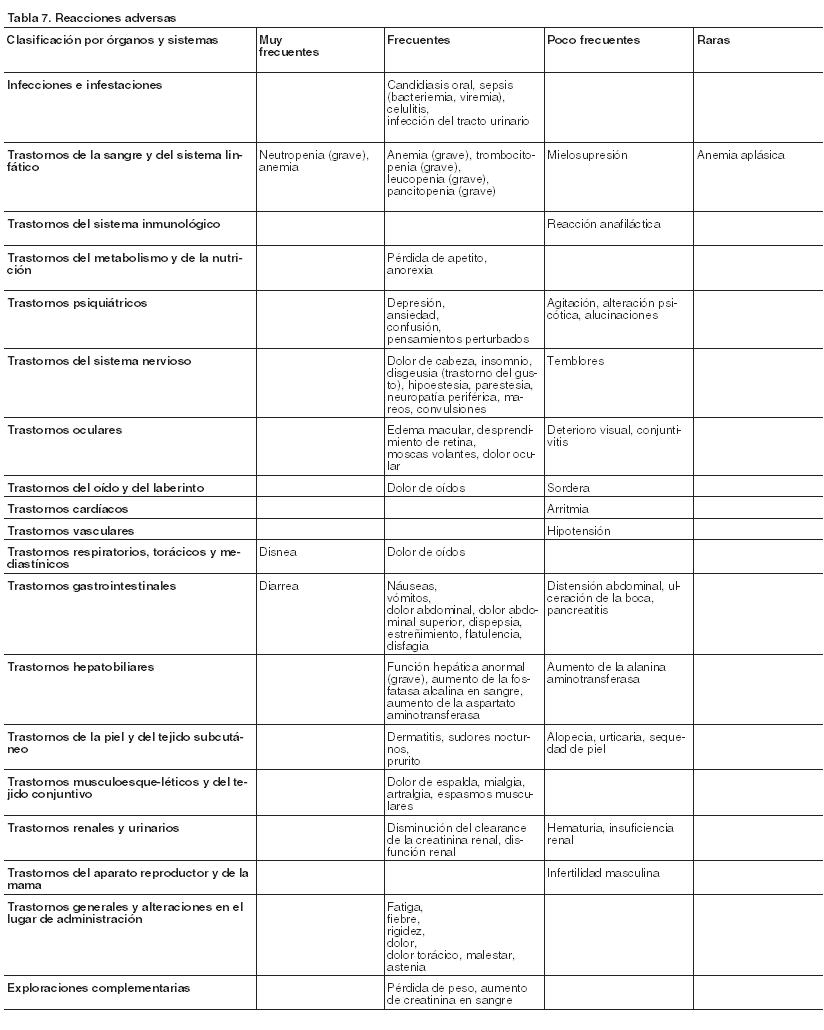
Se puede asociar la trombocitopenia grave con amenaza de la vida por una hemorragia. El desprendimiento de retina sólo se ha notificado en pacientes con SIDA tratados con Valixa para la retinitis por CMV. Población pediátrica: Valixa se ha estudiado en 179 pacientes pediátricos (de 3 semanas a 16 años de edad) receptores de un trasplante de órgano sólido con riesgo de desarrollar enfermedad por CMV y en 133 neonatos (de 2 a 31 días de edad) con enfermedad congénita por CMV sintomática; la duración de la exposición al ganciclovir fue de 2 a 200 días. Las reacciones adversas notificadas más frecuentemente en los tratamientos en ensayos clínicos pediátricos fueron diarrea, náuseas, neutropenia, leucopenia y anemia. En pacientes sometidos a trasplante de órgano sólido, el perfil de seguridad general fue similar en los pacientes pediátricos y en los adultos. Sin embargo, la incidencia de ciertos acontecimientos adversos, como infección de las vías respiratorias altas, pirexia, dolor abdominal y disuria, que pueden ser característicos de los niños, fue mayor en esta población que en los adultos. También se informó neutropenia con una incidencia ligeramente mayor en los dos estudios con pacientes pediátricos que recibieron un trasplante de órgano sólido cuando se comparó con adultos, si bien no existió ninguna correlación entre la neutropenia y los acontecimientos adversos infecciosos en la población pediátrica. En pacientes pediátricos receptores de un trasplante renal, la prolongación de la exposición al valganciclovir hasta 200 días no se asoció con un aumento general de la incidencia de eventos adversos. La incidencia de neutropenia grave (recuento absoluto de neutrófilos < 500/ml) fue mayor en los pacientes pediátricos receptores de un trasplante renal tratados hasta el día 200 comparado con los tratados hasta el día 100 y si se compara con pacientes adultos receptores d