TRUXIMA®
ADIUM
Agente antineoplásico, anticuerpo monoclonal.
Composición.
Cada vial de 50 mL contiene: Rituximab 500 mg. Excipientes: Cloruro sódico, citrato de trisodio dihidratado, polisorbato 80, agua para preparaciones inyectables. Cada vial de 10 mL contiene: Rituximab 100 mg. Excipientes: Cloruro sódico, citrato de trisodio dihidratado, polisorbato 80, agua para preparaciones inyectables.
Propiedades.
Mecanismo de acción: Truxima es un medicamento biosimilar. Rituximab se une específicamente al antígeno CD20, una fosfoproteína transmembrana no-glucosilada, expresada en los linfocitos pre-B y B maduros. El antígeno se expresa en más del 95 % de todos los linfomas no-Hodgkin de células B. CD20 se expresa tanto en células B normales como en tumorales, pero no en células madre hematopoyéticas, células pro-B, células plasmáticas normales ni en otros tejidos normales. Este antígeno no se internaliza tras la unión del anticuerpo ni se elimina de la superficie celular. CD20 no circula en plasma como antígeno libre, y, por esta razón, no compite por la unión con los anticuerpos. El dominio Fab de rituximab se une al antígeno CD20 en la superficie de los linfocitos B, mientras que el dominio Fc puede reclutar efectores de la respuesta inmune para mediar la lisis de las células B. Los mecanismos posibles de la lisis celular mediada por efector incluyen citotoxicidad dependiente del complemento (CDC) como resultado de la unión de C1q, y la citotoxicidad celular dependiente de anticuerpos (ADCC) mediada por uno o más receptores Fcc de la superficie de los granulocitos, macrófagos y células NK (natural killer). También se ha demostrado que la unión del rituximab al antígeno CD20 de los linfocitos B induce la muerte celular por apoptosis. Tras completarse la administración de la primera dosis de rituximab, los recuentos de células B periféricas disminuyeron por debajo de lo normal. En los pacientes tratados de neoplasias hematológicas, la recuperación de células B comenzó a los 6 meses de tratamiento y en general se recuperaron los niveles normales en 12 meses después de finalizado el tratamiento aunque en algunos pacientes puede tardar más (hasta un tiempo medio de recuperación de 23 meses después de la terapia de inducción). En pacientes con artritis reumatoide se observa una depleción inmediata de células B en sangre periférica después de las dos perfusiones de 1.000 mg de rituximab separadas por un intervalo de 14 días. El recuento de células B en sangre periférica empieza a aumentar desde la semana 24 y la evidencia de repoblación se observa, en la mayoría de los pacientes, en la semana 40, independientemente de que rituximab se administre en monoterapia o en combinación con metotrexato. En una pequeña proporción de pacientes la depleción de células B periféricas se ha prolongado durante 2 años o más tras la última dosis con rituximab. En pacientes con granulomatosis con poliangeítis o poliangeítis microscópica el número de células B en sangre periférica disminuyó a < 10 células/ml después de 2 perfusiones semanales de rituximab 375 mg/m2, y ese nivel se mantuvo en muchos pacientes a los 6 meses. La mayoría de los pacientes (81 %) mostraron signos de recuperación de células B, con recuentos de > 10 células/ml a los 12 meses, aumentando al 87 % de los pacientes a los 18 meses. Farmacocinética: En pacientes con LNH que recibieron perfusiones únicas o múltiples de rituximab bien solo bien en combinación con terapia CHOP, los parámetros poblacionales típicos de aclaramiento no específico (CL1), aclaramiento específico (CL2) a los que probablemente contribuyeron las células B o la carga tumoral, y el volumen de distribución en el compartimento central (V1), que varió en función del área de la superficie corporal (ASC) y la terapia CHOP, se estimaron en 0,14 l/día, 0,59 l/día y 2,7 litros, respectivamente. La mediana de la semivida de eliminación terminal estimada de rituximab fue 22 días (intervalo 6,1 a 52 días). La Cmax media luego de 4 perfusiones es de 486 mg/ml (intervalo 77,5 a 996,6 mg/ml). Y de hasta 550 mg/ml (intervalo 171-1177 mg/ml) tras la octava perfusión. Se detectó rituximab en el plasma de los pacientes a los 3-6 meses de finalizar el último tratamiento. En pacientes con LLC, el resultado de Cmax fue 408 mg/ml (rango, 97-764 mg/ml) después de la quinta perfusión y el resultado de semivida fue 32 días (rango, 14-62 días). En artritis reumatoide la media de la semivida de eliminación terminal fue de 20,8 días (rango 8,58 a 35,9 días); el aclaramiento sistémico medio 0,23 l/día (rango 0,091 a 0,67 l/día); y el volumen de distribución medio en el estado estacionario 4,6 l (rango: 1,7 a 7,51 l). Un análisis farmacocinético poblacional reveló que la superficie corporal y el género eran las covariables más importantes que justificaban la variabilidad interindividual de los parámetros farmacocinéticos. Después de ajustar por la superficie corporal, los varones mostraron un volumen de distribución mayor y un aclaramiento más rápido que las mujeres. Las diferencias de género en la farmacocinética no se consideran clínicamente relevantes y no exigen ningún ajuste posológico. No se dispone de datos farmacocinéticos en pacientes con insuficiencia hepática o renal. En base al análisis farmacocinético poblacional de los datos en pacientes con granulomatosis con poliangeítis y poliangeítis microscópica, la mediana de la semivida de eliminación terminal fue de 23 días (intervalo, 9 a 49 días). El aclaramiento medio y el volumen de distribución de rituximab fueron 0,313 l/día (intervalo, 0,116 a 0,726 l/día) y 4,50 l (intervalo 2,25 a 7,39 l) respectivamente. Los parámetros farmacocinéticos de rituximab en estos pacientes parecen similares a los observados en pacientes con artritis reumatoide.
Indicaciones.
Linfoma no-Hodgkin (LNH): Truxima está indicado en combinación con quimioterapia en el tratamiento de pacientes con linfoma no-Hodgkin folicular estadio III-IV que no hayan sido tratados previamente. Truxima está indicado para el tratamiento de mantenimiento en pacientes con linfoma folicular que hayan respondido al tratamiento de inducción. Truxima en monoterapia está indicado en el tratamiento de pacientes con linfoma no-Hodgkin folicular estadio III-IV que son quimiorresistentes o están en su segunda o posterior recidiva tras la quimioterapia. Truxima está indicado en combinación con quimioterapia CHOP (ciclofosfamida, doxorubicina, vincristina, prednisolona) en el tratamiento de pacientes con linfoma no-Hodgkin difuso de células B grandes CD20 positivas. Leucemia linfática crónica (LLC): Truxima está indicado en combinación con quimioterapia en el tratamiento de pacientes con LLC que no hayan sido tratados previamente o que estén en recidiva o refractarios a un tratamiento previo Hay datos limitados sobre la eficacia y el perfil de seguridad en pacientes previamente tratados con anticuerpos monoclonales, incluido Truxima o en pacientes refractarios a un tratamiento previo con Truxima y quimioterapia. Artritis reumatoide: Truxima, en combinación con metotrexato, está indicado en pacientes adultos para el tratamiento de artritis reumatoide activa grave en pacientes que hayan presentado una respuesta inadecuada o intolerancia otros fármacos antirreumáticos modificadores de la enfermedad (FAME), incluyendo uno o más tratamientos con inhibidores del factor de necrosis tumoral (TNF) Truxima ha demostrado reducir la tasa de progresión del daño articular medido con rayos-x y mejorar la función física, cuando se administra en combinación con metotrexato. Granulomatosis con poliangeítis y poliangeítis microscópica: Truxima, en combinación con glucocorticoides, está indicado para la inducción de la remisión en pacientes adultos con granulomatosis con poliangeítis (Wegener) (GPA) y con poliangeítis microscópica (PAM), activa y grave.
Dosificación.
Truxima se debe administrar bajo la estrecha supervisión de un profesional sanitario con experiencia, en un entorno que disponga de forma inmediata de un equipo completo de reanimación. Siempre se debe administrar premedicación consistente en un antipirético y un antihistamínico, por ejemplo paracetamol y difenhidramina, antes de cada administración de Truxima. En pacientes con Linfoma no-Hodgkin y leucemia linfática crónica se debe considerar la premedicación con glucocorticoides si Truxima no se va a administrar en combinación con quimioterapia que incluya glucocorticoides. En pacientes con artritis reumatoide se debe administrar 100 mg de metilprednisolona intravenosa 30 minutos antes de la perfusión de Truxima para reducir la incidencia y la gravedad de las reacciones relacionadas con la perfusión (RRI). En pacientes con granulomatosis con poliangeítis (Wegener) o con poliangeítis microscópica antes de la primera perfusión de Truxima, se recomienda administrar metilprednisolona por vía intravenosa de 1 a 3 días, a una dosis de 1.000 mg al día (la última dosis de metilprednisolona se puede administrar el mismo día que la primera perfusión de Truxima). Esto se debe continuar con prednisona por vía oral a una dosis de 1 mg/kg/día (sin exceder los 80 mg/día, y reducir la dosis tan rápido como sea posible, basándose en la necesidad clínica) durante y después del tratamiento con Truxima. Posología Linfoma no-Hodgkin folicular: Terapia combinada: Para el tratamiento de inducción en pacientes con linfoma folicular en recidiva o refractario o que no hayan sido previamente tratados, la posología recomendada de Truxima en combinación con quimioterapia es de 375 mg/m2 de superficie corporal por ciclo, hasta 8 ciclos. Truxima debe ser administrado el día 1 de cada ciclo de quimioterapia, después de la administración intravenosa del componente glucocorticoide de la quimioterapia, si procede. Terapia de mantenimiento: Linfoma folicular previamente no tratado: La posología recomendada de Truxima, cuando se utiliza para el tratamiento de mantenimiento en pacientes con linfoma folicular no previamente tratados que han respondido a la terapia de inducción es de 375 mg/m2 de superficie corporal una vez cada 2 meses (empezando dos meses después de la última dosis de la terapia de inducción) hasta progresión de la enfermedad o hasta un periodo máximo de dos años. Linfoma folicular en recaída o refractario: La posología recomendada de Truxima, cuando se utiliza para el tratamiento de mantenimiento en pacientes con linfoma folicular que están en recaída o son refractarios que han respondido a la terapia de inducción es de 375 mg/m2 de superficie corporal una vez cada 3 meses (empezando 3 meses después de la última dosis de la terapia de inducción) hasta progresión de la enfermedad o hasta un periodo máximo de dos años. Monoterapia: Linfoma folicular en recaída o refractario: La posología recomendada de Truxima en monoterapia usado como tratamiento de inducción en pacientes adultos con linfoma folicular estadio III-IV que sean quimiorresistentes o estén en su segunda o subsiguientes recidivas tras quimioterapia es de 375 mg/m2 de superficie corporal administrada en forma de perfusión intravenosa una vez por semana durante cuatro semanas. La posología recomendada para repetir el tratamiento con Truxima en monoterapia en pacientes con linfoma no-Hodgkin folicular en recidiva o refractario que ya habían respondido a un tratamiento previo con Truxima en monoterapia es de 375 mg/m2 de superficie corporal administrada en forma de perfusión intravenosa una vez por semana durante cuatro semanas. Linfoma no-Hodgkin difuso de células B grandes Truxima debe usarse en combinación con quimioterapia CHOP. La posología recomendada es de 375 mg/m2 de superficie corporal el primer día de cada ciclo de quimioterapia, durante 8 ciclos, tras la perfusión intravenosa del componente glucocorticoide del CHOP. No se han establecido la seguridad y eficacia de la combinación de Truxima con otras quimioterapias en linfoma no Hodgkin difuso de células B grandes Ajustes de dosis durante el tratamiento: No están recomendadas las reducciones de dosis de Truxima. Cuando Truxima se administre en combinación con quimioterapia, se deben aplicar las reducciones de dosis estándar a la quimioterapia. Leucemia linfática crónica: En pacientes con LLC se recomienda una profilaxis con una adecuada hidratación y administración de uricostáticos 48 horas antes de comenzar la terapia para disminuir el riesgo del síndrome de lisis tumoral. Para todos los pacientes con LLC cuyo recuento de linfocitos sea > 25 x 109/l se recomienda administrar 100 mg de prednisona/prednisolona intravenosa poco antes de la perfusión con Truxima para disminuir el riesgo y la gravedad de las reacciones agudas de la perfusión y/o el síndrome de liberación de citoquinas. La dosis recomendada de Truxima en combinación con quimioterapia para pacientes no tratados previamente o que estén en recidiva o refractarios a un tratamiento previo es 375 mg/m2 de superficie corporal administrada el día 0 del primer ciclo de tratamiento seguido de 500 mg/m2 de superficie corporal administrada el día 1 de los siguientes ciclos hasta llegar a 6 ciclos en total. La quimioterapia debe ser administrada después de la perfusión de Truxima. Artritis reumatoide: Cada ciclo de Truxima se compone de dos perfusiones intravenosas de 1.000 mg. La dosis recomendada de Truxima es de 1.000 mg en perfusión intravenosa, seguida, dos semanas más tarde, de una segunda perfusión intravenosa de 1.000 mg. La necesidad de más ciclos debe evaluarse a las 24 semanas del ciclo anterior. Repetir el tratamiento si queda actividad residual de la enfermedad si no se debe retrasar el retratamiento hasta que se reactive la enfermedad. Los datos disponibles indican que la respuesta clínica normalmente se alcanza entre la semana 16-24 después del ciclo de tratamiento inicial. La terapia continua debe evaluarse cuidadosamente en pacientes que no han mostrado evidencia de los beneficios terapéuticos durante este periodo de tiempo. Granulomatosis con poliangeítis y poliangeítis microscópica La dosis recomendada de Truxima para el tratamiento para la inducción de la remisión de granulomatosis con poliangeítis y poliangeítis microscópica es de 375 mg/m2 de superficie corporal, administrada en forma de perfusión intravenosa una vez por semana durante 4 semanas (cuatro perfusiones en total). Se recomienda profilaxis para la neumonía por Pneumocystis jiroveci en pacientes con granulomatosis con poliangeítis o con poliangeítis microscópica durante y tras el tratamiento con Truxima, según sea apropiado. Poblaciones especiales: Pacientes de edad avanzada: No se requiere ajustar la dosis en los pacientes de edad avanzada ( > 65 años). Población pediátrica: No se ha establecido todavía la seguridad y eficacia de Truxima en niños. No se dispone de datos. Forma de administración: La solución preparada de Truxima se debe administrar como perfusión intravenosa empleando una vía específica. Las soluciones preparadas no se deben administrar en perfusión rápida o en bolo intravenoso. Los pacientes deben ser estrechamente monitorizados para detectar el inicio de un síndrome de liberación de citoquinas (ver sección advertencias). Se debe interrumpir inmediatamente la perfusión en aquellos pacientes que muestren evidencia de reacciones graves, especialmente disnea grave, broncoespasmo o hipoxia. En los pacientes con linfoma no-Hodgkin se debe evaluar posteriormente la evidencia de síndrome de lisis tumoral incluyendo pruebas de laboratorio adecuadas, y la evidencia de infiltración pulmonar por radiología torácica. En ningún paciente se debe reiniciar la perfusión hasta la remisión completa de todos los síntomas, y normalización de los valores de laboratorio y de los resultados de la radiología torácica. A partir de ese momento, la perfusión se puede reiniciar inicialmente como máximo a la mitad de la velocidad de la perfusión previa. Si se presentasen por segunda vez las mismas reacciones adversas graves, se debe considerar seriamente, y caso por caso, la decisión de interrumpir el tratamiento. Las reacciones relacionadas con la perfusión (RRP) de grado leve o moderado se resuelven generalmente reduciendo la velocidad de perfusión. La velocidad de perfusión se puede incrementar cuando mejoren los síntomas. Primera perfusión: La velocidad inicial recomendada de la perfusión es de 50 mg/h, y después de los primeros 30 minutos se puede aumentar, en incrementos de 50 mg/h cada 30 minutos, hasta un máximo de 400 mg/h. Perfusiones posteriores: Todas las indicaciones: Las perfusiones posteriores de Truxima se pueden comenzar con una velocidad de 100 mg/h, y aumentar, en incrementos de 100 mg/h cada 30 minutos, hasta un máximo de 400 mg/h. Sólo en Artritis Reumatoide: Posología en perfusiones posteriores alternativas más rápida: Si los pacientes no experimentan una reacción adversa grave a la perfusión con la primera o posteriores perfusiones de una dosis de Truxima de 1.000 mg administrada durante los tiempos estándar de perfusión, se puede administrar una perfusión más rápida en la segunda o posteriores perfusiones usando la misma concentración que en perfusiones anteriores (4 mg/ml en un volumen de 250 ml). Iniciar a una velocidad de 250 mg/hora durante los primeros 30 minutos y después 600 mg/hora durante los siguientes 90 minutos. Si se tolera la perfusión más rápida, este mismo régimen se puede utilizar cuando se administren perfusiones posteriores. En pacientes con enfermedades cardiovasculares clínicamente significativas, incluyendo arritmias, o reacciones graves a la perfusión previas a cualquier terapia biológica anterior o a rituximab, no se debe administrar la perfusión más rápida.
Contraindicaciones.
Hipersensibilidad al principio activo o a proteínas murinas o a alguno de los excipientes. • Infecciones graves y activas Para el uso en Linfoma no-Hodgkin y Leucemia linfática crónica: Pacientes en un estado inmunocomprometido grave. Para el uso en artritis reumatoide, granulomatosis con poliangeítis y poliangeítis microscópica: Insuficiencia cardiaca grave (clase IV de la New York Heart Association) o enfermedades cardiacas graves no controladas.
Reacciones adversas.
En pacientes que recibieron rituximab, las reacciones adversas al fármaco (RAFs) observadas con mayor frecuencia fueron las RRP y en la mayoría de los pacientes ocurrieron durante la primera perfusión. La incidencia de los síntomas relacionados con la perfusión disminuyó sustancialmente con las posteriores perfusiones y fue menor del 1 % después de ocho dosis de rituximab. Un alto porcentaje de pacientes (~30-55 %) experimentaron reacciones infecciosas (en su mayoría bacterianas y virales). En la tabla 1 están incluidas las frecuencias de las RAFs notificadas con rituximab tanto sólo como en combinación con quimioterapia. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Las frecuencias se definen como muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1.000 a < 1/100); raras (≥ 1/10.000 a < 1/1.000) y muy raras ( < 1/10.000); y no conocidas (no puede estimarse a partir de los datos disponibles).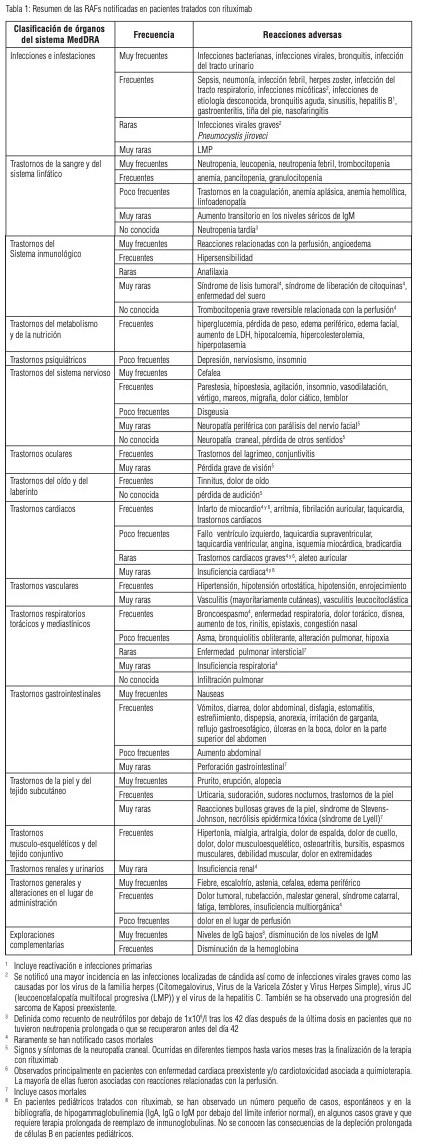
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Departamento de Farmacovigilancia del laboratorio vía email: fvigilancia@raffo.com.ar o a través de los teléfonos (011) 4509-7100/7127, y/o del Sistema Nacional de Farmacovigilancia al siguiente link: http://sistemas.anmat.gov.ar/aplicaciones_net/applications/fvg_eventos_adversos_nuevo/index.html
Precauciones.
Para mejorar la trazabilidad de los medicamentos biológicos, debe consignarse (o indicarse) claramente el nombre y número de lote del medicamento administrado en la historia clínica del paciente. Interacciones: Actualmente existen datos limitados sobre las posibles interacciones medicamentosas con Truxima. En pacientes con LLC la administración concomitante de rituximab y fludarabina o ciclofosfamida, no parece tener efectos sobre la farmacocinética de éstos. Además, no hay un efecto aparente de la fludarabina y ciclofosfamida sobre la farmacocinética del rituximab. La coadministración con metotrexato no modifica la farmacocinética de rituximab en los pacientes con artritis reumatoide. Los pacientes con títulos de anticuerpos humanos antimurinos o antiquiméricos (HAMA/HACA) pueden sufrir reacciones alérgicas o de hipersensibilidad al ser tratados con otros anticuerpos monoclonales terapéuticos o de diagnóstico. En pacientes con artritis reumatoide, que recibieron un tratamiento secuencial con un FAME biológico después de rituximab, la incidencia de infecciones clínicamente relevantes durante el tratamiento con rituximab fue de 6,01 por cien pacientes año, comparado con 4,97 por cien pacientes año tras el tratamiento con el FAME biológico. Anticoncepción en hombres y mujeres: Debido al largo tiempo de permanencia de rituximab en el organismo en pacientes con depleción de células B, las mujeres en edad fértil deben usar métodos anticonceptivos eficaces durante el tratamiento y hasta 12 meses después del tratamiento con Truxima. Embarazo: Se sabe que las inmunoglobulinas IgG atraviesan la barrera placentaria. No se han determinado los niveles de linfocitos B en recién nacidos de madres expuestas a Truxima en ensayos clínicos. No existen datos suficientes ni controlados en mujeres embarazadas, sin embargo se han notificado depleción transitoria de células B y linfocitopenia en algunos niños nacidos de madres expuestas a rituximab durante el embarazo. Se han observado efectos similares en los estudios realizados en animales. Por estos motivos Truxima no debe administrarse a una mujer embarazada a menos que el beneficio esperado supere el riesgo potencial. Lactancia: Se desconoce si rituximab se excreta en la leche materna. Sin embargo, teniendo en cuenta que la IgG materna se excreta en la leche y que se ha detectado rituximab en la leche de monas en periodo de lactancia, las mujeres no deben dar el pecho a sus hijos durante el tratamiento con Truxima ni durante los 12 meses siguientes. Fertilidad: Los estudios realizados en animales no muestran efectos perjudiciales de rituximab en los órganos reproductores. Efectos sobre la capacidad para conducir y utilizar máquinas: No se han realizado estudios de los efectos de Truxima sobre la capacidad para conducir y utilizar máquinas, aunque la actividad farmacológica y las reacciones adversas notificadas hasta la fecha sugieren que la influencia de rituximab sobre la capacidad para conducir y utilizar máquinas sería nula o insignificante.
Advertencias.
Leucoencefalopatía Multifocal Progresiva (LMP): Se han notificado casos muy raros de muerte por LMP tras el uso de rituximab. Los pacientes deben ser monitorizados a intervalos regulares para detectar cualquier nuevo signo o síntoma neurológico así como cualquier empeoramiento que pueda indicar LMP. Si se sospechase que el paciente sufre LMP, debe suspenderse la administración de rituximab hasta que se haya descartado dicha posibilidad. El médico debe evaluar a los pacientes para determinar si los síntomas son indicativos de alteración neurológica, y si es así, si estos síntomas son indicativos de LMP. Se debe considerar si esta clínicamente indicada la consulta con un neurólogo. Si existe alguna duda, además de la evaluación, deberá considerarse un estudio de imagen de resonancia magnética preferiblemente con contraste, un análisis del líquido cefalorraquídeo (LCR) para detectar ADN del virus JC y repetir las evaluaciones neurológicas. El médico debe estar especialmente alerta a los síntomas indicativos de LMP, que el paciente pueda no advertir (p.ej. síntomas cognitivos, neurológicos o psiquiátricos). Se le debe aconsejar al paciente que informe a su pareja o a la persona que le cuide, acerca de su tratamiento, ya que ellos puedan detectar síntomas de los que el paciente no es consciente. Si el paciente desarrolla LMP, se debe suspender el tratamiento con Truxima permanentemente. En pacientes inmunodeprimidos con LMP, se ha observado la estabilización o mejora del desenlace clínico tras la reconstitución del sistema inmune. Se desconoce si la detección precoz de LMP y la suspensión del tratamiento con Truxima pueden llevar a una estabilización similar o a una mejoría del desenlace clínico. Reacciones relacionadas con la perfusión: Truxima está asociado con reacciones relacionadas con la perfusión (RRP), que pueden estar mediadas por la liberación de citoquinas y/u otros mediadores químicos. En el caso de las enfermedades reumatológicas, siempre antes de cada perfusión de Truxima se debe administrar premedicación consistente en un medicamento analgésico/antipirético y un medicamento antihistamínico. En artritis reumatoide, también se debe administrar premedicación con glucocorticoides antes de cada perfusión de Truxima para reducir la frecuencia y gravedad de las RRP. El síndrome de liberación de citoquinas puede ser clínicamente indistinguible de las reacciones de hipersensibilidad aguda. Este conjunto de reacciones descritas a continuación incluyen el síndrome de liberación de citoquinas, el síndrome lisis tumoral y reacciones anafilácticas y de hipersensibilidad. Se han notificado casos de reacciones graves relacionadas con la perfusión con resultado de muerte, durante el uso post-comercialización de la formulación de rituximab intravenoso, con un inicio entre los 30 minutos y 2 horas después del comienzo de la primera perfusión de rituximab intravenoso. En pacientes con Linfoma no-Hodgkin y leucemia linfática crónica se caracterizaron por acontecimientos pulmonares y en algunos casos incluyeron lisis tumoral rápida y características del síndrome de lisis tumoral además de fiebre, escalofríos, rigidez, hipotensión, urticaria, angioedema y otros síntomas. En pacientes con artritis reumatoide, la mayoría de los eventos notificados relacionados con la perfusión en los ensayos clínicos fueron de leves a moderados en cuanto a gravedad. Los síntomas más frecuentes fueron reacciones alérgicas como cefalea, prurito, irritación de garganta, enrojecimiento, erupciones, urticaria, hipertensión y fiebre. En general, el porcentaje de pacientes que experimenta alguna reacción a la perfusión es más alto después de la primera perfusión que tras la segunda en cualquier ciclo de tratamiento. La incidencia de RRP disminuye con las sucesivas perfusiones. Las reacciones notificadas revirtieron, por lo general, tras disminuir la velocidad de perfusión de rituximab o suspender la perfusión y administrar un antipirético, un antihistamínico y, en ocasiones, oxígeno, una solución salina intravenosa o broncodilatadores, y, en caso de necesidad, glucocorticoides. Los pacientes con afecciones cardiacas pre-existentes o que han tenido una reacción cardiopulmonar adversa previa se deben vigilar estrechamente. Dependiendo de la gravedad de la RRP y de las intervenciones necesarias se suspenderá el tratamiento con Truxima de forma temporal o permanente. En la mayoría de los casos, la perfusión se pudo reanudar al 50 % de la velocidad anterior (p. ej., de 100 mg/h a 50 mg/h), una vez resueltos completamente todos los síntomas. Las RRP en pacientes con granulomatosis con poliangeítis y poliangeítis microscópica fueron similares a las observadas en ensayos clínicos en pacientes con artritis reumatoide. El síndrome de liberación de citoquinas grave se caracteriza por disnea grave, frecuentemente acompañada de broncoespasmo e hipoxia, además de fiebre, escalofríos, rigidez, urticaria y angioedema. Este síndrome puede estar asociado con algunas características del síndrome de lisis tumoral tales como hiperuricemia, hiperpotasemia, hipocalcemia, hiperfosfatemia, fallo renal agudo, elevación de la lactato dehidrogenasa (LDH) y puede estar asociado con fallo respiratorio agudo y muerte. El fallo respiratorio agudo puede estar acompañado de infiltración intersticial o edema pulmonar, visibles a la exploración radiológica torácica. El síndrome se manifiesta frecuentemente dentro de la primera o segunda hora después de iniciar la primera perfusión. Los pacientes con historial de insuficiencia pulmonar o con infiltración tumoral pulmonar, pueden tener un riesgo mayor de mal pronóstico y deben aumentarse las precauciones durante su tratamiento. En aquellos pacientes que desarrollen síndrome de liberación de citoquinas grave se debe interrumpir la perfusión inmediatamente y deben recibir tratamiento sintomático de choque. Dado que a la mejoría inicial de los síntomas clínicos puede seguir una recidiva, se debe monitorizar estrechamente a estos pacientes hasta que el síndrome de lisis tumoral y la infiltración pulmonar se hayan resuelto o hayan sido descartados. Una vez resueltos completamente los signos y síntomas, raramente se repite el síndrome de liberación de citoquinas en tratamientos posteriores. Los pacientes con gran masa tumoral o con un elevado número de células tumorales circulantes (≥ 25 x 109/l) como los pacientes con leucemia linfática crónica (LLC), que pueden tener un riesgo mayor de desarrollar un síndrome de liberación de citoquinas especialmente grave, solo se deben tratar extremando las precauciones. Estos pacientes se deben monitorizar muy estrechamente durante la primera perfusión. En estos pacientes se debe considerar reducir la velocidad de la primera perfusión o un fraccionamiento de la dosis durante más de dos días durante el primer ciclo y algún ciclo posterior si el recuento de linfocitos es aún > 25 x 109/l. En el 77 % de los pacientes tratados con rituximab se han observado todo tipo de reacciones adversas relacionadas con la perfusión (incluyendo síndrome de liberación de citoquinas acompañado de hipotensión y broncoespasmo en el 10 % de los pacientes). Generalmente, estos síntomas son reversibles tras la interrupción de la perfusión de rituximab y la administración de un antipirético, un antihistamínico, y ocasionalmente, oxígeno, solución salina intravenosa o broncodilatadores, y, en caso de necesidad, glucocorticoides. Para reacciones graves, ver síndrome de liberación de citoquinas. Se han notificado casos de reacciones de hipersensibilidad, incluyendo anafilácticas, después de la administración intravenosa de proteínas. A diferencia del síndrome de liberación de citoquinas, las reacciones de hipersensibilidad verdaderas se presentan típicamente durante los primeros minutos de la perfusión. Conviene disponer para uso inmediato de medicamentos utilizados para combatir las reacciones de hipersensibilidad, es decir, adrenalina, antihistamínicos y glucocorticoides, por si ocurriera una reacción alérgica durante la administración de Truxima. Las manifestaciones clínicas de anafilaxia pueden parecerse a las del síndrome de liberación de citoquinas (anteriormente descrito). Las reacciones atribuibles a la hipersensibilidad se han notificado menos frecuentemente que las atribuidas a la liberación de citoquinas. Además de las reacciones notificadas en algunos hubo casos de infarto de miocardio, fibrilación auricular, edema pulmonar y trombocitopenia reversible aguda. Dado que se puede producir hipotensión durante la administración de Truxima, se debe considerar interrumpir los tratamientos antihipertensivos 12 horas antes de dicha perfusión. Trastornos cardiacos: Se han notificado casos de angina de pecho, arritmias cardiacas tales como flutter y fibrilación auricular, fallo cardiaco y/o infarto de miocardio en pacientes tratados con rituximab. Por lo tanto, se deben monitorizar cuidadosamente los pacientes con historial de enfermedad cardiaca y/o cardiotoxicidad asociada a la quimioterapia. No existen datos sobre la seguridad de Truxima en pacientes con artritis reumatoide, granulomatosis o poliangeítis o poliangeítis microscópica e insuficiencia cardiaca moderada (clase III de la NYHA) o enfermedad cardiovascular grave no controlada. En pacientes con isquemia miocárdica preexistente se ha notificado con rituximab su exacerbación sintomática, resultando en angina de pecho, así como fibrilación auricular y flutter. Por lo tanto, si el paciente refiere antecedentes de cardiopatía, y en los que han experimentado previamente reacciones adversas cardiopulmonares, se sopesará el riesgo de complicaciones cardiovasculares derivadas de las reacciones a la perfusión antes de administrar Truxima y se monitorizará rigurosamente a los pacientes durante el tratamiento. Dado que se puede producir hipotensión durante la perfusión de rituximab, se evaluará la necesidad de interrumpir temporalmente cualquier medicamento antihipertensivo 12 horas antes de la perfusión de Truxima. Toxicidad hematológica: Aunque Truxima en monoterapia no tiene efecto mielosupresor, se recomienda prudencia antes de aplicar el tratamiento a pacientes con un recuento de neutrófilos < 1,5 × 109/l y/o plaquetas < 75 × 109/l, puesto que la experiencia clínica en esta población es limitada. Rituximab se ha utilizado en 21 pacientes sometidos a trasplante autólogo de médula ósea y en otros grupos de riesgo con una función de la médula ósea presumiblemente reducida, sin que haya inducido mielotoxicidad. Se deben realizar recuentos de sangre total de forma regular, incluyendo recuentos de neutrófilos y de plaquetas, durante el tratamiento con Truxima. Infecciones: Basado en el mecanismo de acción de Truxima y en el conocimiento de que las células B desempeñan un papel importante en el mantenimiento de la respuesta inmune, los pacientes tratados con Truxima pueden tener un mayor riesgo de infección. Durante el tratamiento con Truxima pueden producirse infecciones graves e incluso mortales. Truxima no debe ser administrado a pacientes con infecciones graves activas (ej. tuberculosis, sepsis e infecciones oportunistas) ni a aquéllos con enfermedades reumáticas e inmunodeficiencia grave (p. ej., a los que tengan niveles de CD4 o CD8 muy bajos). El médico debe tener especial precaución cuando considere el uso de Truxima en pacientes con historial de infecciones crónicas o recurrentes o en unas condiciones subyacentes que puedan provocar una mayor predisposición a infecciones graves p.ej., hipogammaglobulinemia. En el caso de las enfermedades reumáticas, e recomienda que los niveles de inmunoglobulina se determinen antes de iniciar el tratamiento con Truxima. Se evaluará de inmediato y se tratará convenientemente a todo paciente que manifieste signos y síntomas de infección después del tratamiento con Truxima. Antes de administrar los ciclos siguientes del tratamiento con Truxima, en estos pacientes debe ser re-evaluado el riesgo potencial de infecciones. Se han notificado casos de reactivación de hepatitis B en pacientes tratados con rituximab que incluyeron hepatitis fulminante con fallecimiento. La mayoría de estos pacientes habían estado expuestos también a quimioterapia citotóxica. Información limitada de un estudio en pacientes con LLC en recaída o refractarios, sugiere que el tratamiento con rituximab puede empeorar el resultado de una infección primaria por hepatitis B. En todos los pacientes se debe llevar a cabo la detección del virus de la Hepatitis B (VHB) antes de iniciar el tratamiento con Truxima. Al menos debe incluir HBsAg y HBcAc. Esto puede ser complementado con otros marcadores apropiados de acuerdo a las guías locales. Los pacientes con hepatitis B activa no deben ser tratados con Truxima. En pacientes con serología positiva de hepatitis B (bien HBsAg o HBcAc), se debe consultar con un especialista en enfermedades hepáticas antes de iniciar el tratamiento y deben ser monitorizados y tratados siguiendo los estándares médicos locales para prevenir la reactivación de la hepatitis B. Se han notificado casos muy raros de leucoencefalopatía multifocal progresiva (LMP), durante el uso post-comercialización de rituximab. En el caso de LNH y LLC, La mayoría de los pacientes habían recibido rituximab en combinación con quimioterapia o como parte de un transplante de células madre hematopoyéticas. Inmunizaciones: En pacientes con LNH y LLC no se ha estudiado la seguridad de la inmunización con vacunas de virus vivos después de recibir tratamiento con Truxima, por tanto, no se recomienda la vacunación con virus vivos. Los pacientes tratados con Truxima pueden recibir vacunas inactivadas, sin embargo, con las vacunas inactivadas los porcentajes de respuesta puede ser menores. En un estudio no aleatorizado de pacientes con LNH de bajo grado con recidivas, que recibieron rituximab en monoterapia cuando se comparó con el grupo control no tratado sano, el porcentaje de respuesta a la vacunación fue menor con el antígeno de recuerdo del Tétanos (16 % vs 81 %), y con neoantígeno Keyhole Limpet Haemocyanin (KLH) (4 % vs 76 % cuando se determinó un incremento > de 2 en el título de anticuerpo) En pacientes con LLC se esperan resultados similares ya que ambas enfermedades tienen muchas similitudes, aunque no se han estudiado en ensayos clínicos. Esto implicó que los títulos de anticuerpos medios antes del tratamiento frente a antígenos como Streptococcus pneumoniae, gripe A, paperas, rubéola, y varicela, se mantuvieron hasta al menos 6 meses después del tratamiento con rituximab. Reacciones de la piel: Se han notificado casos de reacciones cutáneas graves tales como necrólisis epidérmica tóxica (Síndrome de Lyell) y Síndrome de Stevens-Johnson, algunas con desenlace mortal. En caso de que aparezcan tales reacciones con una sospecha de relación con Truxima, el tratamiento debe suspenderse permanentemente. Población con artritis reumatoide Metotrexato (MTX) naïve: El uso de Truxima no está recomendado en pacientes que no han sido tratados previamente con MTX ya que no se ha establecido una relación beneficio-riesgo favorable. Uso concomitante/secuencial con otros fármacos antirreumáticos modificadores de la enfermedad (FAMEs) en artritis reumatoide: No está recomendado el uso concomitante de Truxima y otros tratamientos antirreumáticos distintos a los incluidos en la indicación y la posología de artritis reumatoide. Existen datos limitados en los ensayos clínicos para evaluar totalmente la seguridad del uso secuencial de otros FAMEs (incluidos los inhibidores del TNF y otros biológicos tras la terapia con Truxima. Los datos disponibles indican que la incidencia de infección clínicamente relevante no cambia cuando estas terapias se utilizan en pacientes previamente tratados con rituximab, sin embargo los pacientes deben de ser estrechamente monitorizados para ver signos de infección si se utilizan agentes biológicos o FAMEs después del tratamiento con Truxima. Neutropenia tardía: En el tratamiento de las enfermedades reumatológicas, se deben medir los neutrófilos en sangre antes de cada ciclo con Truxima y regularmente hasta 6 meses tras la finalización del tratamiento, y si hay signos o síntomas de infección. Neoplasias malignas: Los medicamentos inmunomoduladores pueden aumentar el riesgo de neoplasias malignas. En base a la limitada experiencia con rituximab en pacientes con artritis reumatoide los datos existentes no parecen sugerir un aumento del riesgo de neoplasias malignas. Sin embargo, no se puede excluir un posible riesgo de desarrollo de tumores sólidos en este momento. Síndrome de