TRUVADA®
GADOR
Grupo farmacoterapéutico: antiviral para uso sistémico; antivirales para el tratamiento de infecciones por el VIH, combinaciones. Código ATC: J05AR03
Composición.
Cada comprimido recubierto de TRUVADA® contiene: Emtricitabina 200 mg. Tenofovir disoproxil fumarato (equivalente a 245 mg de disoproxilo de tenofovir) 300 mg. Excipientes: Croscaramelosa sódica, Lactosa monohidrato, Estearato de magnesio, Almidón pregelatinizado, Celulosa microcristalina, Opadry II azul Y-30-10701* c.s. * Laca alumínica FD&C azul N°2, hidroxipropilmetilcelulosa 2910, lactosa monohidrato, dióxido de titanio y triacetina.
Farmacología.
Descripción: Los comprimidos de TRUVADA® son comprimidos de asociación en dosis fijas, que contienen emtricitabina (FTC) y tenofovir disoproxil fumarato (TDF). La FTC es un análogo nucleosídico sintético de la citidina. El TDF se convierte in vivo en tenofovir, un análogo (nucleótido) fosfonato nucleósido acíclico de 5'-monofosfato de adenosina. La FTC y el tenofovir presentan actividad inhibitoria contra la retrotranscriptasa del VIH-1. Emtricitabina: El nombre químico de la FTC es 5-fluoro-1-(2R,5S)-[2-(hidroximetil)-1,3-oxatiolan-5-il]citosina. La FTC es el enantiómero (-) de un análogo tio de la citidina, el cual difiere de otros análogos de la citidina porque tiene un flúor en la posición 5. Su fórmula molecular es C8H10FN3O3S, y su peso molecular, 247,24. Tiene la siguiente fórmula estructural: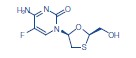
La FTC es un polvo cristalino, de color blanco a blanquecino, con una solubilidad de aproximadamente 112 mg/ml en agua a 25°C. El coeficiente de partición (log p) de la emtricitabina es -0,43, y el pKa, 2,65. Tenofovir Disoproxil Fumarato: El TDF es una sal de ácido fumárico del éster bis-isopropoxicarboniloximetil derivado del tenofovir. El nombre químico del tenofovir DF es fumarato de 9-[(R)-2-[[bis[[(isopropoxicarbonil)oxi]-metoxi]fosfinil]metoxi]propil]adenina (1:1). Su fórmula molecular es C19H30N5O10P·C4H4O4, y su peso molecular, 635,52. Tiene la siguiente fórmula estructural: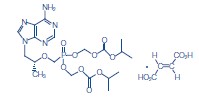
El tenofovir disoproxil fumarato es un polvo cristalino, de color blanco a blanquecino, con una solubilidad de aproximadamente 13,4 mg/ml en agua a 25 °C. El coeficiente de partición (log p) del disoproxilo de tenofovir es 1,25 y el pKa, 3,75. Todas las dosificaciones se expresan en términos de TDF, excepto cuando se indique lo contrario. Los comprimidos de TRUVADA® se administran por vía oral. Cada comprimido recubierto contiene 200 mg de emtricitabina y 300 mg de tenofovir DF (que equivalen a 245 mg de disoproxilo de tenofovir) como principios activos. Los comprimidos también incluyen los siguientes excipientes: croscaramelosa sódica, lactosa monohidrato, estearato de magnesio, celulosa microcristalina y almidón pregelatinizado (sin gluten). Los comprimidos están recubiertos con Opadry II azul Y-30-10701, que contiene laca alumínica FD&C azul N°2, hidroxipropilmetilcelulosa 2910, lactosa monohidrato, dióxido de titanio y triacetina. Farmacología clínica: Mecanismo de acción: TRUVADA® es una asociación en dosis fijas de los antivirales FTC y TDF [véase Microbiología]. Farmacocinética: TRUVADA®: Un comprimido de TRUVADA® fue comparable a una cápsula de FTC (200 mg de emtricitabina) más un comprimido de TDF (300 mg de tenofovir DF), después de su administración como dosis única a sujetos sanos en ayunas (N = 39). Emtricitabina: Las propiedades farmacocinéticas de la FTC se resumen en la tabla 1. Después de la administración oral de FTC, FTC se absorbe rápidamente y alcanza concentraciones plasmáticas máximas al cabo de una a dos horas después de la administración de la dosis. Menos del 4 % de la FTC se une in vitro a las proteínas plasmáticas de seres humanos, y la unión es independiente de la concentración, dentro del rango de 0,02 a 200 mg/ml. Después de la administración de FTC radiomarcada, se recupera aproximadamente el 86 % en la orina y el 13 % como metabolitos. Los metabolitos de la FTC son 3'-sulfóxido diastereómeros y su conjugado con ácido glucurónico. La emtricitabina se elimina mediante una combinación de filtración glomerular y secreción tubular activa. Después de una dosis oral única de FTC, la vida media plasmática de la FTC es de aproximadamente 10 horas. Tenofovir Disoproxil Fumarato: Las propiedades farmacocinéticas del TDF se resumen en la tabla 1. Después de la administración oral de TDF, las concentraciones máximas de tenofovir en el suero se alcanzaron en 1,0 ± 0,4 horas. Menos del 0,7 % del tenofovir se une in vitro a las proteínas plasmáticas de seres humanos, y la unión es independiente de la concentración, dentro del rango de 0,01 a 25 mg/ml. Aproximadamente entre el 70 y el 80 % de la dosis intravenosa de tenofovir se recupera como fármaco inalterado en la orina. El tenofovir se elimina mediante una combinación de filtración glomerular y secreción tubular activa. Después de una dosis única por vía oral de TDF, la vida media de eliminación terminal del tenofovir es de aproximadamente 17 horas.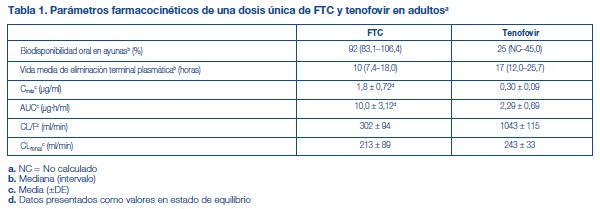
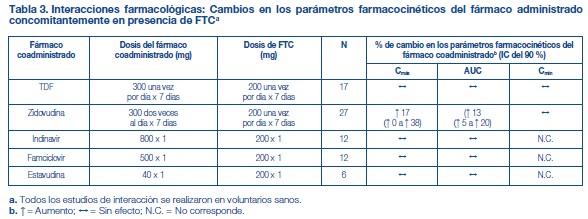
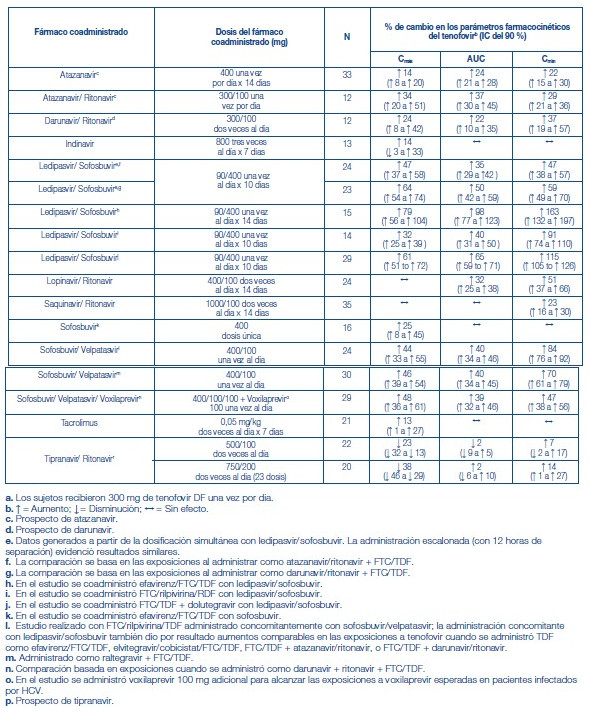
Efectos de los alimentos en la absorción oral: Se puede administrar TRUVADA® con o sin alimentos. La administración de TRUVADA® después de una comida con alto contenido de grasas (784 kcal; 49 gramos de grasa) o de una comida ligera (373 kcal; 8 gramos de grasa) retrasó el tiempo hasta la Cmáx del tenofovir en aproximadamente 0,75 horas. Los aumentos medios del AUC y de la Cmáx del tenofovir fueron de aproximadamente el 35 % y el 15 %, respectivamente, cuando se administró con una comida con alto contenido de grasas o con una comida ligera, en comparación con su administración en ayunas. En estudios de inocuidad y eficacia anteriores, el TDF (tenofovir) se administró con alimentos. Las exposiciones sistémicas de la FTC (AUC y Cmáx) no se vieron afectadas cuando se administró TRUVADA® con comidas con un alto contenido de grasas o con comidas ligeras. Poblaciones específicas: Raza: Emtricitabina: No se identificaron diferencias farmacocinéticas atribuibles a la raza después de la administración de FTC. Tenofovir Disoproxil Fumarato: No hubo un número suficiente de sujetos de grupos raciales y étnicos, aparte de la raza blanca, para poder determinar adecuadamente las posibles diferencias farmacocinéticas entre estas poblaciones después de la administración de TDF. Sexo: Emtricitabina y tenofovir Disoproxil Fumarato: Las propiedades farmacocinéticas de la FTC y del tenofovir son similares en los pacientes de ambos sexos. Pacientes pediátricos: TRUVADA® no debe administrarse a pacientes pediátricos infectados por el VIH-1 que sean menores de 12 años o que tengan un peso inferior a 35 kg. Emtricitabina: Las propiedades farmacocinéticas de la emtricitabina en estado de equilibrio se determinaron en 27 sujetos pediátricos infectados por el VIH-1 de entre 13 y 17 años de edad que recibieron una dosis diaria de 6 mg/kg hasta una dosis máxima de 240 mg en solución oral o una cápsula de 200 mg; 26 de los 27 pacientes de este grupo etario recibieron la cápsula de 200 mg de emtricitabina. Los valores medios (±DE) de la Cmáx y el AUC fueron de 2,7 ± 0,9 mg/ml y 12,6 ± 5,4 mg•h/ml, respectivamente. Las exposiciones alcanzadas en los sujetos pediátricos de 12 a menos de 18 años de edad fueron similares a las alcanzadas en los adultos que recibieron dosis de 200 mg una vez por día. Tenofovir DF: Las propiedades farmacocinéticas del tenofovir en estado de equilibrio se evaluaron en 8 sujetos pediátricos infectados por el VIH-1 (de 12 a menos de 18 años de edad). Los valores medios (±DE) de la Cmáx y el AUCtau son 0,38 ± 0,13 mg/ml y 3,39 ± 1,22 mg•h/ml, respectivamente. La exposición al tenofovir alcanzada en estos sujetos pediátricos que recibieron dosis diarias de 300 mg de tenofovir DF por vía oral fue similar a las exposiciones alcanzadas en los adultos que recibieron 300 mg de tenofovir DF una vez por día. Pacientes geriátricos: No se han evaluado totalmente las propiedades farmacocinéticas de la FTC y del tenofovir en personas de edad avanzada (65 años o más). Pacientes con disfunción renal: Las propiedades farmacocinéticas de la FTC y del tenofovir están alteradas en los pacientes con disfunción renal [véase Advertencias]. En los pacientes adultos con depuración de creatinina inferior a 50 ml/min, aumentaron la Cmáx y el AUC0-inf de la FTC y del tenofovir. No hay datos disponibles que permitan hacer recomendaciones sobre la dosis a utilizar en pacientes pediátricos con disfunción renal. Pacientes con disfunción hepática: Se han estudiado las propiedades farmacocinéticas del tenofovir después de una dosis de 300 mg de TDF en pacientes no infectados por el VIH y con disfunción hepática de moderada a grave. No hubo alteraciones importantes en las propiedades farmacocinéticas del tenofovir en los pacientes con disfunción hepática, en comparación con los pacientes con función hepática normal. No se han estudiado las propiedades farmacocinéticas de TRUVADA® ni de la FTC en los pacientes con disfunción hepática; sin embargo, las enzimas hepáticas no metabolizan significativamente la FTC, por lo que la repercusión de la disfunción hepática debería ser limitada. Evaluación de las interacciones farmacológicas: Las propiedades farmacocinéticas de la FTC y del tenofovir en estado de equilibrio no se vieron afectadas cuando se administraron FTC y TDF juntos, en comparación con la administración de cada fármaco por separado. Los estudios in vitro y los estudios clínicos farmacocinéticos sobre interacciones farmacológicas han demostrado que la posibilidad de interacciones mediadas por el CYP que afectan a la FTC y al tenofovir con otros medicamentos es baja. El TDF es un sustrato de los transportadores de la glucoproteína P (P-gp) y de la proteína de resistencia de cáncer de mama (BCRP). Cuando el TDF se administra concomitantemente con un inhibidor de estos transportadores, puede observarse un aumento de la absorción. No se observaron interacciones farmacológicas clínicamente significativas entre FTC y famciclovir, indinavir, estavudina, TDF y zidovudina (véanse las tablas 2 y 3). De manera similar, no se observaron interacciones farmacológicas clínicamente significativas entre TDF y efavirenz, metadona, nelfinavir, anticonceptivos orales, la ribavirina o el sofosbuvir en estudios realizados en voluntarios sanos (véanse las tablas 4 y 5).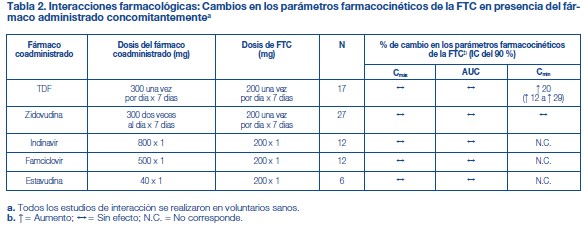
No se observó ningún efecto en los parámetros farmacocinéticos de los siguientes fármacos al coadministrarlos con TRUVADA®: abacavir, didanosina (comprimidos amortiguados), FTC, entecavir y lamivudina.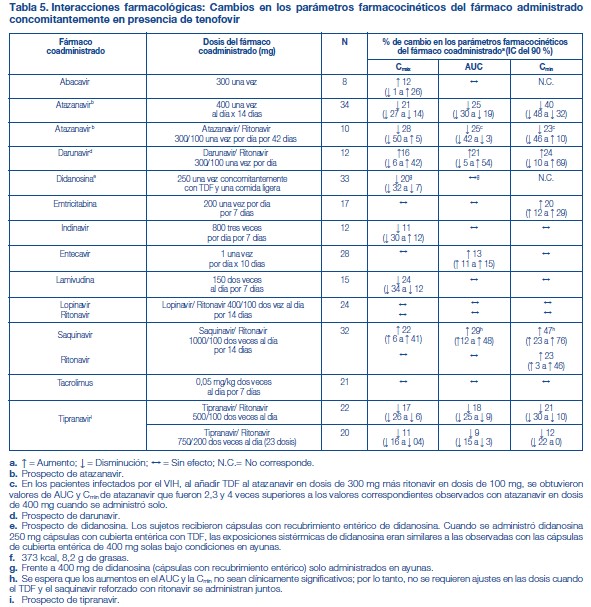
Microbiología: Mecanismo de acción: Emtricitabina: La FTC, un análogo nucleosídico sintético de la citidina, es fosforilado por enzimas celulares para formar 5´-trifosfato de emtricitabina. El 5´-trifosfato de emtricitabina (FTC-TP), que inhibe la actividad de la retrotranscriptasa (RT) del VIH-1, al competir con el sustrato natural 5´-trifosfato de desoxicitidina e incorporarse al ADN viral incipiente, lo que produce la terminación de la cadena. El FTC-TP es un inhibidor débil de las polimerasas a, b y e del ADN de mamíferos, y de la polimerasa c del ADN mitocondrial. Tenofovir Disoproxil Fumarato: El TDF es un diéster de fosfonato nucleosídico acíclico, análogo del monofosfato de adenosina. El TDF requiere la hidrólisis inicial del diéster para su conversión a tenofovir y fosforilaciones subsiguientes por medio de enzimas celulares, para formar el difosfato de tenofovir. El TDF-DP,que inhibe la actividad de la RT del VIH-1, al competir con el sustrato natural 5'-trifosfato de desoxiadenosina y, después de su incorporación al ADN, por la terminación de la cadena de ADN. El TDF-DP es un inhibidor débil de las polimerasas a y b del ADN de mamíferos, y de la polimerasa c del ADN mitocondrial. Actividad antiviral: Emtricitabina y Tenofovir Disoproxil Fumarato: no se observó ningún antagonismo en estudios de asociación que evaluaron la actividad antiviral de la FTC y del tenofovir juntos en cultivo celular. Emtricitabina: Se evaluó la actividad antiviral de la FTC contra cepas aisladas clínicas y de laboratorio del VIH-1 en líneas celulares linfoblastoides, línea celular MAGI-CCR5 y células mononucleares de sangre periférica. Los valores de concentración eficaz al 50 % (CE50) de la FTC fueron de entre 0,0013 y 0,64 mM (0,0003 a 0,158 mg/ml). No se observó ningún antagonismo en los estudios de asociación de fármacos de FTC con inhibidores nucleosídicos de la RT (abacavir, lamivudina, estavudina, zidovudina), inhibidores no nucleosídicos de la RT (delavirdina, efavirenz, nevirapina) e inhibidores de la proteasa (amprenavir, nelfinavir, ritonavir, saquinavir). La emtricitabina presentó actividad antiviral en cultivo celular contra los subtipos A, B, C, D, E, F y G del VIH-1 (los valores de la CE50 variaron entre 0,007 y 0,075 mM) y actividad específica de cepa contra el VIH-2 (los valores de CE50 variaron entre 0,007 y 1,5 mM). Tenofovir Disoproxil Fumarato: Se evaluó la actividad antiviral del tenofovir contra cepas aisladas clínicas y de laboratorio del VIH-1 en líneas celulares linfoblastoides, células primarias monocitomacrofágicas y linfocitos de sangre periférica. Los valores de CE50 correspondientes al tenofovir estuvieron dentro de los límites de 0,04 y 8,5 mM. No se observó ningún antagonismo en los estudios de asociación de fármacos de tenofovir con inhibidores nucleosídicos de la RT (abacavir, didanosina, lamivudina, estavudina, zidovudina), inhibidores no nucleosídicos de la RT (delavirdina, efavirenz, nevirapina) e inhibidores de la proteasa (amprenavir, indinavir, nelfinavir, ritonavir, saquinavir). El tenofovir presentó actividad antiviral en cultivo celular contra los subtipos A, B, C, D, E, F, G y O del VIH-1 (los valores de CE50 variaron entre 0,5 y 2,2 mM), y actividad específica de cepa contra el VIH-2 (los valores de CE50 variaron entre 1,6 mM y 5,5 mM). Resistencia: Emtricitabina y Tenofovir Disoproxil Fumarato: Se han seleccionado en un cultivo celular cepas aisladas del VIH-1 con una disminución de la sensibilidad a la asociación de FTC y tenofovir. El análisis genotípico de estas cepas aisladas identificó las sustituciones de aminoácidos M184V/I y/o K65R en la RT viral. Además, con el uso de tenofovir se ha seleccionado una sustitución K70E en la RT del VIH-1, lo cual redujo la susceptibilidad al tenofovir. En el Estudio 934, un estudio clínico con pacientes sin tratamiento antirretroviral previo [véase Estudios clínicos] se realizó un análisis de resistencia en cepas aisladas del VIH-1 de todos los pacientes con fracaso virológico confirmado con más de 400 copias/ml de ARN del VIH-1 en la semana 144 o que habían abandonado el estudio en forma temprana. La aparición de sustituciones asociadas con la resistencia al efavirenz se produjo con mayor frecuencia y fue similar en todos los grupos de tratamiento. Se observó la sustitución de aminoácidos M184V, asociada con resistencia a la FTC y la lamivudina, en 2/19 de las cepas aisladas de pacientes analizados en el grupo tratado con FTC + TDF, y en 10/29 de las cepas aisladas de pacientes analizados en el grupo tratado con zidovudina y lamivudina. En las 144 semanas del estudio 934, ningún paciente había presentado una sustitución K65R o K70E detectable en su VIH-1, según los análisis genotípicos habituales. Emtricitabina: Se han seleccionado cepas aisladas del VIH-1 resistentes a la FTC en cultivo celular e in vivo. El análisis genotípico de estas cepas aisladas demostró que la disminución de la sensibilidad a la FTC estaba asociada a una sustitución del gen de la retrotranscriptasa del VIH-1 en el codón 184, lo que produjo una sustitución del aminoácido metionina por valina o isoleucina (M184V/I). Tenofovir Disoproxil Fumarato: Se han seleccionado en un cultivo celular cepas aisladas del VIH-1 con disminución de la sensibilidad al tenofovir. Estos virus expresaron una sustitución K65R en la retrotranscriptasa y mostraron una disminución de 2 a 4 veces en la sensibilidad al tenofovir. En los pacientes sin tratamiento previo con antirretrovirales, las cepas aisladas de 8/47 (17 %) pacientes analizados presentaron la sustitución K65R en el grupo tratado con TDF después de 144 semanas; siete casos se produjeron en las primeras 48 semanas de tratamiento, y uno en la semana 96. En los pacientes con tratamiento previo con antirretrovirales, 14/304 (5 %) cepas aisladas de pacientes que no respondieron al TDF hasta la semana 96 mostraron una disminución mayor a 1,4 veces (mediana de 2,7) en la sensibilidad al tenofovir. El análisis genotípico de las cepas aisladas resistentes demostró una sustitución del aminoácido K65R en la TR del VIH-1. Resistencia cruzada: Emtricitabina y Tenofovir Disoproxil Fumarato: Se ha reconocido la resistencia cruzada entre ciertos inhibidores nucleosídicos de la retrotranscriptasa (INRT). Las sustituciones M184V/I o K65R seleccionadas en cultivo celular por la asociación de FTC y tenofovir también se observan en algunas cepas aisladas del VIH-1 de pacientes que no respondieron al tratamiento con tenofovir en asociación con FTC o lamivudina y abacavir o didanosina. Por lo tanto, la resistencia cruzada entre estos fármacos puede presentarse en los pacientes cuyos virus hospedan alguna de estas sustituciones de aminoácidos o ambas. Emtricitabina: Las cepas aisladas resistentes a la FTC (M184V/I) presentaron resistencia cruzada a la lamivudina pero mantuvieron la sensibilidad en cultivo celular a los NRTIs didanosina, estavudina, tenofovir y zidovudina y a los NNRTIs (delavirdina, efavirenz y nevirapina). Las cepas aisladas del VIH-1 que contienen la sustitución K65R, seleccionadas in vivo por abacavir, didanosina, y tenofovir presentaron una disminución de la sensibilidad a la inhibición por la FTC. Los virus que hospedan sustituciones que confieren una disminución de la sensibilidad a la estavudina y la zidovudina (M41L, D67N, K70R, L210W, T215Y/F, K219- Q/E), o a la didanosina (L74V) se mantuvieron sensibles a la FTC. El VIH-1 que contiene la sustitución K103N asociada con la resistencia a los NNRTIs fue sensible a la FTC. Tenofovir Disoproxil Fumarato: Las sustituciones K65R y K70E seleccionadas por el tenofovir también se seleccionan en algunos pacientes infectados por el VIH-1 que reciben tratamiento con abacavir o didanosina. Los inóculos del VIH-1 con las sustituciones K65R y K70E también evidenciaron una disminución de la susceptibilidad a la emtricitabina y la lamivudina. Por lo tanto, puede producirse resistencia cruzada entre estos NRTI en los pacientes cuyos virus albergan sustituciones K65R o K70E. Las cepas aisladas del VIH-1 de sujetos (N = 20) cuyo VIH-1 expresó una media de tres sustituciones de aminoácidos de la RT asociadas a la zidovudina (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) presentaron una disminución de 3,1 veces en la sensibilidad al tenofovir. Los pacientes cuyos virus expresaban una sustitución L74V sin sustituciones asociadas con la resistencia a la zidovudina (N = 8) presentaron una disminución de la respuesta al TDF. Se dispone de datos limitados en el caso de pacientes cuyos virus expresaban una sustitución Y115F (N = 3), una sustitución Q151M (N = 2) o una inserción T69 (N = 4), todos los cuales presentaban una disminución de la respuesta.
Indicaciones.
TRUVADA® está indicado en asociación con otros antirretrovirales para el tratamiento de la infección causada por el VIH-1 en pacientes adultos y pediátricos de 12 años o más [consulte Estudios clínicos].
Dosificación.
Dosis recomendada para el tratamiento de la infección por HIV-1 en pacientes adultos y pediátricos con un peso corporal de al menos 35 kg: TRUVADA® es un producto de asociación de dosis fijas de dos fármacos que contiene emtricitabina (FTC) y tenofovir disoproxil fumarato (TDF). La dosis recomendada de TRUVADA® en pacientes adultos y pediátricos de 12 años o más, con un peso corporal de al menos 35 kg, es de un comprimido (que contiene 200 mg de FTC y 300 mg de TDF) una vez al día, administrado por vía oral con o sin alimentos [véase Farmacología Clínica]. Ajuste de la dosis en pacientes con disfunción renal: La Tabla 6 presenta un ajuste del intervalo de administración para pacientes con disfunción renal. No es necesario ajustar la dosis en los pacientes con disfunción renal leve (depuración de creatinina de 50 a 80 ml/min). No se han evaluado clínicamente la inocuidad ni la eficacia de estas recomendaciones de ajuste del intervalo de administración en los pacientes con disfunción renal moderada (depuración de creatinina de 30 a 49 ml/min); por lo tanto, se debe controlar rigurosamente la respuesta clínica al tratamiento y la función renal en estos pacientes [véase Advertencias]. No hay datos disponibles que permitan hacer recomendaciones sobre la dosis a utilizar en pacientes pediátricos con disfunción renal.
Debe vigilarse sistemáticamente la depuración de creatinina estimada, el fósforo sérico, la glucosa en orina y la proteinuria en todos los pacientes con disfunción renal leve [véase Advertencias]. Formas farmacéuticas y concentraciones: TRUVADA® se comercializa en comprimidos recubiertos. Cada comprimido contiene 200 mg de emtricitabina y 300 mg de tenofovir DF (que equivale a 245 mg de disoproxilo de tenofovir). Los comprimidos son de color azul, llevan la inscripción "GILEAD" en un lado y "701" en el otro.
Contraindicaciones.
TRUVADA® está contraindicado en los pacientes con hipersensibilidad previamente comprobada a cualquiera de los componentes del producto. TRUVADA® debe utilizarse únicamente en asociación con otros antirretrovirales.
Reacciones adversas.
En otros apartados del prospecto se tratan las siguientes reacciones adversas: Exacerbaciones graves agudas de la hepatitis B en pacientes con infección por HBV [véase Advertencias]. Nueva aparición o empeoramiento de la disfunción renal [véase Advertencias]. Síndrome de reconstitución inmune [véase Advertencias]. Pérdida ósea y defectos de mineralización [véase Advertencias]. Acidosis láctica/Severa hepatomegalia con esteatosis [consulte Advertencias]. Experiencia en estudios clínicos: Debido a que los estudios clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los estudios clínicos de un fármaco no pueden compararse directamente con las tasas de los estudios clínicos de otro fármaco, y puede que no reflejen las tasas observadas en la práctica. Estudios clínicos en sujetos adultos: En el Estudio 934, 511 sujetos sin terapia antirretroviral previa recibieron efavirenz (EFV) administrado en asociación con FTC+TDF (N=257) o zidovudina (AZT)/lamivudina (3TC) (N=254) durante 144 semanas. Las reacciones adversas más frecuentes (incidencia superior o igual al 10 %, todos los grados), incluían diarrea, náuseas, fatiga, cefalea, mareos, depresión, insomnio, anomalías del sueño y erupción cutánea. La tabla 10 presenta las reacciones adversas aparecidas con el tratamiento (grados 2 a 4) que se produjeron en el 5 % o más de los pacientes tratados en cualquiera de los grupos de tratamiento en este estudio. La alteración del color de la piel, manifestada por hiperpigmentación se observó en el 3% de los sujetos que tomaban FTC + TDF y generalmente fue leve y asintomática. Se desconocen su mecanismo y su importancia clínica.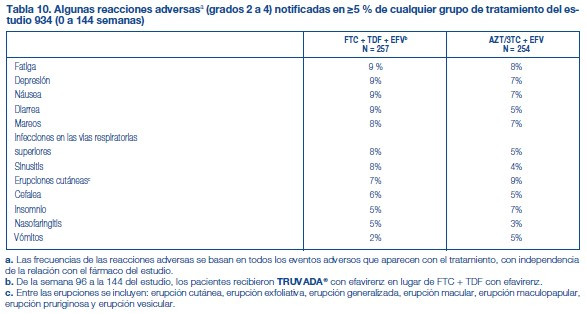
Anomalías de laboratorio: Por lo general, las anomalías de laboratorio observadas en este estudio fueron coherentes con las observadas en otros estudios de TDF y/o FTC (tabla 11).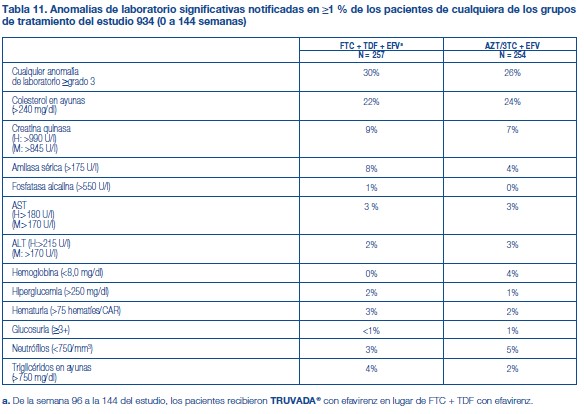
Estudios clínicos en sujetos pediátricos de 12 años o más: Emtricitabina: Además de las reacciones adversas notificadas en los adultos, se observaron anemia e hiperpigmentación en el 7% y 32 %, respectivamente, de los sujetos pediátricos (edades de entre 3 meses y menos de 18 años) que recibieron tratamiento con FTC en el más grande de los dos estudios pediátricos abiertos no controlados (N = 116). Para obtener información adicional, consulte el prospecto de emtricitabina. Tenofovir Disoproxil Fumarato: En un estudio clínico pediátrico realizado en sujetos de 12 a menos de 18 años de edad, las reacciones adversas observadas en los sujetos pediátricos que recibieron tratamiento con TDF coincidieron con las reacciones observadas en los estudios clínicos realizados con TDF en adultos [véase Advertencias]. Experiencia posterior a la comercialización: Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de TDF. No se han identificado otras reacciones adversas durante el uso de FTC después de su aprobación. Debido a que las reacciones posteriores a la comercialización se notifican voluntariamente a partir de una población de tamaño indeterminado, no siempre es posible calcular de manera fiable su frecuencia ni establecer una relación causal con la exposición al fármaco. Trastornos del sistema inmunitario: Reacción alérgica, incluso angioedema. Trastornos de la nutrición y el metabolismo: Acidosis láctica, hipopotasemia, hipofosfatemia. Trastornos respiratorios, torácicos y mediastínicos: Disnea. Trastornos digestivos: Pancreatitis, aumento de la amilasa, dolor abdominal. Trastornos hepatobiliares: Esteatosis hepática, hepatitis, aumento de las enzimas hepáticas (con mayor frecuencia la AST, la ALT, la gamma GT). Trastornos de la piel y del tejido subcutáneo: Erupción cutánea. Trastornos musculoesqueléticos y del tejido conjuntivo: Rabdomiólisis, osteomalacia (manifestada como dolor óseo y que puede contribuir a las fracturas), debilidad muscular, miopatía. Trastornos renales y urinarios: Insuficiencia renal aguda, insuficiencia renal, necrosis tubular aguda, síndrome de Fanconi, tubulopatía renal proximal, nefritis intersticial. (incluso casos agudos), diabetes insípida nefrógena, disfunción renal, aumento de la creatinina, proteinuria, poliuria. Trastornos generales y alteraciones en el lugar de la administración: Astenia. Las siguientes reacciones adversas, enumeradas bajo los encabezados de sistemas y órganos anteriores, pueden producirse a consecuencia de la tubulopatía renal proximal: rabdomiólisis, osteomalacia, hipopotasemia, debilidad muscular, miopatía, hipofosfatemia.
Advertencias.
Generales: Exacerbaciones agudas graves de la hepatitis B en pacientes con Infección por el VHB: A todos los pacientes se les debería realizar la prueba para detectar la presencia crónica del virus de la hepatitis B (VHB) antes, o al iniciar el tratamiento con TRUVADA® [véase Posología y forma de administración (4)]. Se han notificado casos de exacerbaciones graves agudas de la hepatitis B (descompesación hepática e insuficiencia hepática) en los pacientes con infección por el VHB que han interrumpido la administración de TRUVADA®. Se debe controlar rigurosamente a los pacientes que padecen una infección por el VHB con seguimiento clínico y de laboratorio durante, por lo menos, varios meses después de interrumpir el tratamiento con TRUVADA®. Si fuese conveniente, puede estar justificado el inicio del tratamiento contra la hepatitis B, especialmente en pacientes con enfermedad hepática avanzada o cirrosis, ya que la exacerbación de la hepatitis después del tratamiento puede llevar a una descompensación hepática y a insuficiencia hepática. A las personas que no estén infectadas por el VHB se les debe ofrecer la vacunación. Nueva aparición o empeoramiento de la disfunción renal: La emtricitabina y el tenofovir se eliminan principalmente por los riñones. Se han notificado casos de disfunción renal, entre ellos casos de insuficiencia renal aguda y síndrome de Fanconi (lesión tubular renal con hipofosfatemia grave), asociados con el uso de TDF, un componente de TRUVADA® [véase Reacciones adversas]. Antes de iniciar el tratamiento y durante el uso de TRUVADA®, según se requiera clínicamente, evaluar la creatinina sérica, la depuración de creatinina estimada, la glucosa en orina y la proteinuria en todos los pacientes. En los pacientes con enfermedad renal crónica evaluar también el fósforo sérico. Debe evitarse el uso de TRUVADA® con el uso concomitante o reciente de un fármaco nefrotóxico (p. ej., múltiples antinflamatorios no esteroideos o a dosis altas [AINE]) [véase Interacciones farmacológicas]. Se han notificado casos de disfunción renal grave tras la administración de múltiples AINE o a dosis altas en pacientes infectados por el VIH con factores de riesgo de disfunción renal que parecían estables con TDF. Algunos pacientes precisaron hospitalización y un tratamiento renal sustitutivo. En caso necesario, deben considerarse alternativas terapéuticas a los AINE en los pacientes con riesgo de disfunción renal. El dolor óseo persistente o empeoramiento del mismo, el dolor en las extremidades, las fracturas y/o el dolor o la debilidad muscular pueden ser manifestaciones de tubulopatía renal proximal y requerirán una evaluación de la función renal de los pacientes con riesgo de disfunción renal. Se recomienda el ajuste del intervalo de administración de TRUVADA® y el control estricto de la función renal en todos los pacientes con una depuración de creatinina estimada de 30 a 49 ml/min [véase Dosificación]. No se dispone de información sobre la inocuidad ni sobre la eficacia en los pacientes con disfunción renal que hayan recibido TRUVADA® siguiendo estas pautas posológicas, por lo que el posible beneficio del tratamiento con TRUVADA® debe evaluarse en relación con el posible riesgo de toxicidad renal. No se recomienda administrar TRUVADA® a pacientes con una depuración de creatinina estimada inferior a 30 ml/min o a pacientes que necesiten hemodiálisis. Síndrome de reconstitución inmune: Se han notificado casos de síndrome de reconstitución inmune en pacientes que recibieron tratamiento antirretroviral combinado, incluso con TRUVADA®. Durante la fase inicial del tratamiento antirretroviral combinado, los pacientes cuyo sistema inmunitario responde pueden presentar una respuesta inflamatoria ante infecciones oportunistas residuales o indolentes (por ejemplo, infección por Mycobacterium avium, citomegalovirus, neumonía por Pneumocystis jirovecii o tuberculosis), que pueden requerir evaluación y tratamiento adicionales. También se han notificado casos de trastornos autoinmunitarios (como enfermedad de Graves, polimiositis y síndrome de Guillain Barré), que se produjeron en el contexto de reconstitución inmune; sin embargo, el momento de la aparición de estos trastornos es más variable, y pueden aparecer muchos meses después del inicio del tratamiento. Pérdida ósea y defectos de mineralización: Densidad mineral ósea. En los estudios clínicos con adultos infectados por el VIH-1 y en un estudio con personas no infectadas por el VIH-1, el TDF, (un componente de TRUVADA®) se asoció a disminuciones ligeramente superiores de la densidad mineral ósea (DMO) y a aumentos en los marcadores bioquímicos del metabolismo óseo, lo que sugiere un mayor recambio óseo frente a los comparadores [véanse Reacciones adversas]. Las concentraciones de la hormona paratiroidea en el suero y de 1,25-vitamina D también fueron más altas en los pacientes tratados con TDF. Se realizaron estudios clínicos para evaluar el TDF en sujetos pediátricos y adolescentes. En circunstancias normales, la DMO aumenta rápidamente en los pacientes pediátricos. En los sujetos de 2 a menos de 18 años de edad que estaban infectados por el VIH-1, los efectos óseos fueron similares a los observados en los sujetos adultos, lo que sugiere un mayor recambio óseo. La ganancia total de DMO corporal fue menor en el grupo de sujetos pediátricos infectados por el VIH-1 tratados con TDF que en los grupos de control. Se observaron tendencias similares en los sujetos adolescentes de 12 a menos de 18 años de edad tratados por la hepatitis B crónica. En todos los estudios pediátricos, el crecimiento del esqueleto (estatura) no se vio afectado. Para obtener información adicional, consulte el prospecto de tenofovir DF. Se desconocen los efectos de los cambios asociados al TDF en la DMO, y de los marcadores bioquímicos en la salud ósea a largo plazo y en el riesgo futuro de fracturas. Debe considerarse la evaluación de la DMO en los pacientes adultos y pediátricos que tengan antecedentes de fracturas óseas patológicas u otros factores de riesgo de osteoporosis o pérdida de masa ósea. Si bien no se ha estudiado el efecto de los suplementos de calcio y vitamina D, dichos suplementos pueden ser beneficiosos. Se debe obtener asesoramiento adecuado si se sospecha la presencia de anomalías óseas. Defectos de mineralización. Se han notificado casos de osteomalacia asociada a tubulopatía renal proximal, que se manifiesta como dolor óseo o dolor en las extremidades y que puede contribuir a las fracturas, en relación con el uso de TDF [véase Reacciones adversas]. Asimismo, se han notificado artralgia y dolor o debilidad muscular en casos de tubulopatía renal proximal. Deben tenerse en cuenta la hipofosfatemia y la osteomalacia secundaria a tubulopatía renal proximal en los pacientes con riesgo de disfunción renal que presentan síntomas musculares u óseos persistentes o un empeoramientos de los mismos mientras reciben tratamiento con fármacos que contienen TDF [véase Advertencias]. Acidosis láctica/Hepatomegalia severa con esteatosis: Se han informado casos de acidosis láctica y hepatomegalia grave con esteatosis, incluso casos mortales, con el uso de análogos de nucleósidos, incluido el TDF y la FTC, componentes de TRUVADA®, solos o en combinación con otros antirretrovirales. El tratamiento con TRUVADA® deberá ser suspendido en caso de que algún paciente desarrolle manifestaciones clínicas y/o parámetros de laboratorio sugestivos de acidosis láctica o hepatotoxicidad pronunciada (que pueden incluir hepatomegalia y esteatosis incluso en ausencia de elevación de transaminasas). Riesgo de reacciones adversas debido a interacciones con otros fármacos: El uso concomitante de TRUVADA® y otros fármacos puede resultar en interacciones con otros fármacos conocidas o potencialmente significativas, algunas de las cuales pueden llevar a posibles reacciones adversas clínicamente significativas por mayores exposiciones de fármacos concomitantes [véase Interacciones farmacológicas]. Ver Tabla 7 para los pasos para prevenir o tratar estas posibles y conocidas interacciones farmacológicas significativas, incluyendo las recomendaciones de administración. Considerar el potencial para interacciones farmacológicas antes y durante el tratamiento con TRUVADA®; revisar las medicaciones concomitantes durante la terapia con TRUVADA®; y monitorear con respecto a reacciones adversas asociadas con los fármacos concomitantes.
Interacciones.
Fármacos que afectan la función renal: La FTC y el tenofovir se excretan principalmente por los riñones mediante una combinación de filtración glomerular y secreción tubular activa [véase Farmacología clínica]. No se observaron interacciones farmacológicas debido a la competencia por la excreción renal. Sin embargo, la administración de TRUVADA® junto con medicamentos que se eliminan por secreción tubular activa puede aumentar las concentraciones de FTC, tenofovir y/o el fármaco coadministrado. Algunos ejemplos son, entre otros, aciclovir, dipivoxilo de adefovir, cidofovir, ganciclovir, valaciclovir, valganciclovir, aminoglucósidos (p. ej., gentamicina) y AINE múltiples o a dosis altas [véase Advertencias]. Los fármacos que disminuyen la función renal también pueden aumentar las concentraciones de FTC y/o tenofovir. Interacciones establecidas y significativas: La Tabla 7 presenta una lista de las interacciones farmacológicas establecidas o clínicamente significativas. Las interacciones farmacológicas descritas se basan en estudios realizados con TRUVADA®, los componentes de TRUVADA® (FTC y TDF) como agentes individuales y/o en combinación, o son interacciones farmacológicas predichas que pueden ocurrir con TRUVADA® [véase Farmacología Clínica].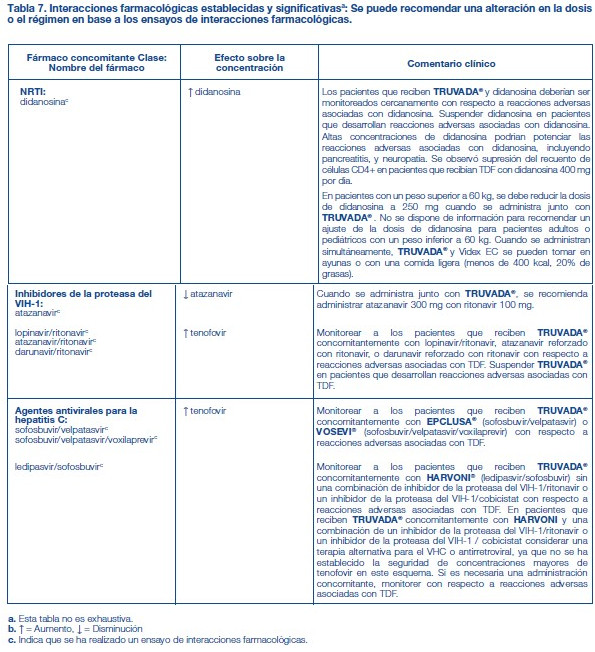
Toxicología preclínica: Carcinogénesis, mutagénesis, alteración de la fertilidad Emtricitabina: En los estudios de carcinogénesis oral a largo plazo de la FTC, no se observaron aumentos relacionados con el fármaco en la incidencia de tumores en ratones, en dosis de hasta 750 mg/kg/día (26 veces la exposición sistémica en los seres humanos con la dosis terapéutica de 200 mg/día), ni en ratas, en dosis de hasta 600 mg/kg/día (31 veces la exposición sistémica en los seres humanos con la dosis terapéutica). La FTC no fue genotóxica en la prueba bacteriana de mutación inversa (prueba de Ames), la prueba de linfoma en ratones ni en los estudios de micronúcleos en ratones. La FTC no afectó la fertilidad en ratas macho, con exposiciones aproximadamente 140 veces superiores, ni en ratones macho y hembra, con exposiciones aproximadamente 60 veces superiores a las exposiciones (AUC) en los seres humanos, con la dosis diaria recomendada de 200 mg. La fertilidad fue normal en las crías de los ratones expuestos diariamente desde antes del nacimiento (exposición intrauterina) hasta la maduración sexual, con exposiciones diarias (AUC) aproximadamente 60 veces superiores a las exposiciones en los seres humanos, con la dosis diaria recomendada de 200 mg. Tenofovir Disoproxil Fumarato: Los estudios a largo plazo de la carcinogénesis oral del TDF en ratas y ratones se llevaron a cabo con exposiciones de hasta aproximadamente 16 veces (ratones) y 5 veces (ratas) superiores a las observadas en los seres humanos con la dosis terapéutica para la infección por el VIH-1. Con las dosis altas en ratones hembra, aumentaron los adenomas hepáticos con exposiciones 16 veces superiores a las de los seres humanos. En las ratas, el estudio arrojó resultados carcinogénicos negativos con exposiciones de hasta 5 veces las observadas en los seres humanos con la dosis terapéutica. El TDF fue mutagénico en la prueba de linfoma en ratones in vitro y dio un resultado negativo en la prueba de mutagénesis bacteriana in vitro (prueba de Ames). En un estudio de micronúcleos en ratones in vivo, el TDF resultó negativo cuando se administró a ratones macho