TREMFYA®
JANSSEN CILAG
Anticuerpo monoclonal inhibidor de IL-23.
Composición.
Cada jeringa prellenada/autoinyector de 1 ml contiene: Guselkumab 100 mg. Excipientes: Sacarosa, L-Histidina, L-Histidina clorhidrato monohidrato, Polisorbato 80, Agua para inyectables. Cada autoinyector de 2 ml contiene: Guselkumab 200 mg. Excipientes: Sacarosa, L-Histidina, L-Histidina clorhidrato monohidrato, Polisorbato 80, Agua para inyectables. Cada vial de 20 ml contiene: Guselkumab 200 mg. Excipientes: Sacarosa, L-Histidina, L-Histidina clorhidrato monohidrato, Polisorbato 80, L-Metionina, EDTA disódico dihidrato, Agua para inyectables.
Indicaciones.
Psoriasis en placa: TREMFYA® está indicado para el tratamiento de pacientes adultos con psoriasis en placa moderada a severa que son candidatos para terapia sistémica o fototerapia. Artritis psoriásica: TREMFYA® está indicado para el tratamiento de pacientes adultos con artritis psoriásica activa. Colitis ulcerosa: TREMFYA® está indicado para el tratamiento de pacientes adultos con colitis ulcerosa activa de moderada a grave que hayan tenido una respuesta inadecuada, presenten pérdida de la respuesta o sean intolerantes al tratamiento convencional o a un tratamiento biológico. Enfermedad de Crohn: TREMFYA® está indicado para el tratamiento de pacientes adultos con enfermedad de Crohn activa de moderada a grave que hayan tenido una respuesta inadecuada, presenten pérdida de la respuesta o sean intolerantes al tratamiento convencional o a un tratamiento biológico.
Dosificación.
Evaluaciones e inmunizaciones recomendadas antes de iniciar el tratamiento: Evalúe a los pacientes para detectar infección por tuberculosis (TB) antes de iniciar el tratamiento con TREMFYA®. Para el tratamiento de la enfermedad de Crohn o la colitis ulcerosa, obtenga los niveles de enzimas hepáticas y bilirrubina antes de iniciar el tratamiento con TREMFYA®. Considere la posibilidad de aplicar todas las vacunas adecuadas para la edad de acuerdo con las pautas actuales de inmunización. Dosis recomendada para Psoriasis en placa: TREMFYA® se administra por inyección subcutánea. La dosis recomendada es de 100 mg en la Semana 0, Semana 4, y luego cada 8 semanas. Dosis recomendada para Artritis psoriásica: TREMFYA® se administra por inyección subcutánea. La dosis recomendada es de 100 mg en la Semana 0, Semana 4, y luego cada 8 semanas. TREMFYA® puede administrarse solo o en combinación con un fármaco antirreumático modificador de la enfermedad convencional (por ejemplo, metotrexato). Dosis recomendada para Colitis ulcerosa: Inducción: La dosis de inducción recomendada de TREMFYA® es de 200 mg administrados mediante infusión intravenosa durante un período de al menos una hora en la Semana 0, la Semana 4 y la Semana 8. Mantenimiento: La dosis recomendada de TREMFYA® es de: 100 mg administrados mediante inyección subcutánea en la semana 16 y luego cada 8 semanas, o 200 mg administrados mediante inyección subcutánea en la semana 12 y luego cada 4 semanas. Utilice la dosis recomendada efectiva más baja para mantener la respuesta terapéutica. Dosis recomendada para Enfermedad de Crohn: Inducción: La dosis de inducción recomendada de TREMFYA® es: 200 mg administrados mediante infusión intravenosa durante un período de al menos una hora en la Semana 0, Semana 4 y Semana 8 o 400 mg administrados mediante inyección subcutánea (administrados como dos inyecciones consecutivas de 200 mg cada una) en la Semana 0, Semana 4 y Semana 8. Mantenimiento: La dosis de mantenimiento recomendada de TREMFYA® es: 100 mg administrados por inyección subcutánea en la semana 16 y cada 8 semanas a partir de entonces, o 200 mg administrados mediante inyección subcutánea en la Semana 12 y cada 4 semanas a partir de entonces. Utilice la dosis mínima eficaz recomendada para mantener la respuesta terapéutica.
Contraindicaciones.
TREMFYA® está contraindicado en pacientes con antecedentes de reacciones de hipersensibilidad graves a guselkumab o a cualquiera de los excipientes.
Reacciones adversas.
Las siguientes reacciones adversas se tratan con mayor detalle en otras secciones del prospecto: Reacciones de hipersensibilidad (ver Contraindicaciones y Advertencias, Reacciones de hipersensibilidad). Infecciones (ver Advertencias, Infecciones).Tuberculosis (ver Advertencias, Tuberculosis). Hepatotoxicidad (ver Advertencias, Hepatotoxicidad). Experiencia en estudios clínicos: Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no pueden compararse directamente con las tasas de ensayos clínicos de otro medicamento y podrían no reflejar las tasas observadas en la práctica. Psoriasis en placa: En ensayos clínicos, un total de 1823 individuos con psoriasis en placa moderada a grave recibieron TREMFYA®. De estos, 1393 individuos fueron expuestos a TREMFYA® durante al menos 6 meses y 728 individuos fueron expuestos durante al menos 1 año. Los datos de dos ensayos controlados con placebo y con activo (PsO1 y PsO2) en 1441 individuos (edad media 44 años, 70% hombres, 82% blancos) se acumularon para evaluar la seguridad de TREMFYA® (100 mg administrados subcutáneamente en las Semanas 0 y 4, luego cada 8 semanas). Semanas 0 a 16: En el período de 16 semanas de los ensayos clínicos acumulados (PsO1 y PsO2) controlado con placebo, ocurrieron reacciones adversas en el 49% de los individuos del grupo TREMFYA® en comparación con el 47% de los individuos del grupo placebo y el 49% de los individuos del grupo de adalimumab aprobado en EE.UU. Se produjeron reacciones adversas graves en el 1,9% de los individuos del grupo TREMFYA® (6,3 eventos cada 100 pacientes-años de seguimiento) en comparación con el 1,4% de los pacientes del grupo placebo (4,7 eventos cada 100 pacientes-años de seguimiento) y en el 2,6% de los pacientes del grupo de adalimumab aprobado en EE.UU. (9,9 eventos cada 100 pacientes-años de seguimiento). La Tabla 1 resume las reacciones adversas que ocurrieron a una tasa de al menos el 1% y a una tasa mayor en el grupo de TREMFYA® que en el grupo de placebo durante el período de 16 semanas controlado con placebo.
Las reacciones adversas que ocurrieron en < 1%, pero > 0,1% de los individuos en el grupo de TREMFYA®, y con una tasa mayor que en el grupo de placebo hasta la semana 16 en PsO1 y PsO2 fueron migraña, infecciones por Candida y urticaria. Reacciones adversas específicas: Infecciones: Ocurrieron infecciones en el 23% del grupo TREMFYA® en comparación con el 21% del grupo placebo. Las infecciones más frecuentes (≥1%) fueron infecciones de las vías respiratorias superiores, gastroenteritis, infecciones por tinea e infecciones por herpes simple. Todos los casos fueron de intensidad leve a moderada y no llevaron a la suspensión de TREMFYA®. Enzimas hepáticas elevadas: Se informaron elevaciones de las enzimas hepáticas con más frecuencia en el grupo TREMFYA® (2,6%) que en el grupo placebo (1,9%). De los 21 individuos que informaron tener enzimas hepáticas elevadas en el grupo TREMFYA®, todos los eventos excepto uno fueron de intensidad leve a moderada y ninguno de los eventos llevo a la suspensión de TREMFYA®. Seguridad hasta la semana 48: No se identificaron nuevas reacciones adversas hasta la semana 48 con el uso de TREMFYA® y la frecuencia de las reacciones adversas fue similar al perfil de seguridad observado durante las primeras 16 semanas de tratamiento. Artritis psoriásica: TREMFYA® se estudió en dos ensayos controlados con placebo en individuos con artritis psoriásica (748 individuos tratados con TREMFYA® y 372 individuos tratados con placebo). De los 748 pacientes que recibieron TREMFYA®, 375 individuos recibieron TREMFYA® 100 mg en la Semana 0, Semana 4 y luego cada 8 semanas, y 373 individuos recibieron TREMFYA® 100 mg cada 4 semanas (c4s). El perfil de seguridad general observado en individuos con artritis psoriásica tratados con TREMFYA® generalmente es consistente con el perfil de seguridad en individuos con psoriasis en placas con la adición de bronquitis y disminución del recuento de neutrófilos. En el período de 24 semanas controlado con placebo, combinado en los dos estudios, se produjo bronquitis en el 1,6% de los individuos del grupo TREMFYA® cada 8 semanas y en el 2,9% de los individuos del grupo de TREMFYA® c4s en comparación con el 1,1% de los individuos del grupo de placebo. Se produjo una disminución del recuento de neutrófilos en el 0,3% de los individuos del grupo TREMFYA® c8s y en el 1,6% de los individuos del grupo TREMFYA® c4s en comparación con el 0% de los individuos del grupo placebo. La mayoría de los eventos de disminución del recuento de neutrófilos fueron leves, transitorios, no asociados con infección y no llevaron a la suspensión. Colitis ulcerosa: TREMFYA® se estudió durante un máximo de 12 semanas en pacientes con colitis ulcerosa activa de moderada a grave en un estudio de inducción aleatorizado, doble ciego, controlado con placebo (UC1) y en un estudio aleatorizado, doble ciego, controlado con placebo, de determinación de la dosis de inducción (UC3). La seguridad a largo plazo durante un máximo de 44 semanas se evaluó en pacientes que respondieron a la terapia de inducción en un estudio de mantenimiento (UC2), aleatorizado, doble ciego, controlado con placebo. En los estudios de inducción (UC1 y UC3), 522 pacientes recibieron al menos una dosis del régimen de inducción intravenosa de TREMFYA® (es decir 200 mg en la semana 0, la semana 4 y la semana 8). La respuesta clínica se definió como una disminución en la puntuación Mayo modificada (mMS) de ≥30 % y ≥2 puntos desde el inicio con una disminución ≥1 desde el inicio en la subpuntuación de sangrado rectal (RBS) o RBS de 0 o 1. En el estudio de mantenimiento (UC2), los sujetos que lograron una respuesta clínica después de 12 semanas de tratamiento de inducción intravenosa con TREMFYA® fueron aleatorizados y recibieron TREMFYA® 100 mg cada 8 semanas (con la primera dosis administrada en la semana 4 de UC2) o TREMFYA® 200 mg cada 4 semanas (con la primera dosis administrada en la semana 0 de UC2), mediante inyección subcutánea (s.c.) hasta 44 semanas adicionales. Se produjeron infecciones de las vías respiratorias en ≥2 % de los sujetos tratados con TREMFYA® y en una tasa mayor que con placebo (8,8 % de los sujetos tratados con TREMFYA® frente a 7,3 % de los sujetos tratados con placebo) hasta la semana 12 en los estudios de inducción (UC1 y UC3). Las infecciones de las vías respiratorias incluyeron COVID-19, gripe, nasofaringitis, infección de las vías respiratorias, infección de las vías respiratorias superiores e infección viral de las vías respiratorias. Las reacciones adversas que se produjeron en ≥3 % de los pacientes en el grupo de TREMFYA® y con una tasa más alta que el placebo hasta la semana 44 en el estudio de mantenimiento (UC2) se muestran en la Tabla 2.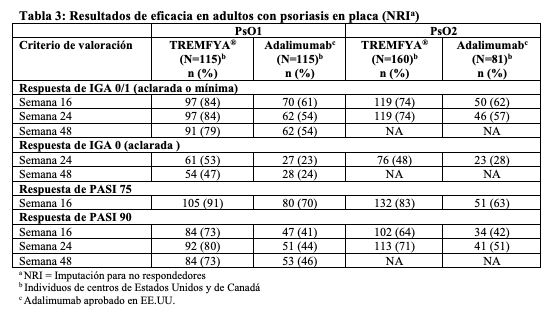
Enfermedad de Crohn: TREMFYA® se estudió en cuatro ensayos clínicos en sujetos con enfermedad de Crohn activa de moderada a grave. En dos ensayos aleatorizados, doble ciego y controlados con placebo (CD1, CD2) y un ensayo aleatorizado, doble ciego y de rango de dosis (CD4, NCT03466411), los sujetos recibieron TREMFYA® intravenoso en las semanas 0, 4 y 8, seguido de una de las dos pautas posológicas recomendadas de TREMFYA® subcutáneo, durante un máximo de 48 semanas. En un ensayo aleatorizado, doble ciego y controlado con placebo (CD3), los sujetos recibieron TREMFYA® subcutáneo en las semanas 0, 4 y 8, seguido de una de las dos pautas posológicas recomendadas de TREMFYA® subcutáneo, durante un máximo de 48 semanas. Ensayos CD1, CD2 y CD4: En los ensayos clínicos CD1, CD2 y CD4, se inscribieron 1349 pacientes, de los cuales 649 fueron aleatorizados para recibir TREMFYA® 200 mg mediante infusión intravenosa en la Semana 0, 4 y 8,seguido de TREMFYA® 100 mg subcutáneo cada 8 semanas (con la primera dosis administrada en la semana 16) durante hasta 32 semanas adicionales (desde la semana 16 hasta la semana 48) o TREMFYA® 200 mg subcutáneo cada 4 semanas (con la primera dosis administrada en la semana 12) durante hasta 36 semanas adicionales (desde la semana 12 hasta la semana 48); y 211 sujetos fueron aleatorizados para recibir placebo. Hasta la semana 12 en los estudios CD1, CD2 y CD4, se reportó cefalea (incluyendo cefalea, migraña y cefalea sinusal) en ≥3% de los sujetos tratados con TREMFYA® intravenoso, con una frecuencia mayor que en el grupo placebo (3,4% en los sujetos tratados con TREMFYA® frente a 1,9% en los sujetos tratados con placebo). En los sujetos tratados con TREMFYA® en el estudio CD2, se reportó un caso de tuberculosis extrapulmonar activa entre las semanas 12 y 48, y un caso de tuberculosis pulmonar activa después de la semana 48. Ambos sujetos presentaron resultados negativos en las pruebas de detección iniciales y residían en regiones endémicas de tuberculosis (ver Advertencias). En general, el perfil de seguridad en los sujetos tratados con TREMFYA® subcutáneo desde la semana 12 hasta la semana 48 en estos ensayos fue similar al reportado con TREMFYA® intravenoso hasta la semana 12. Ensayo CD3: En el estudio CD3, se inscribieron 347 sujetos, de los cuales 230 fueron aleatorizados para recibir TREMFYA®. La aleatorización fue 1:1:1 para TREMFYA® subcutáneo 400 mg en las semanas 0, 4 y 8, seguido de TREMFYA® subcutáneo 100 mg cada 8 semanas (con la primera dosis administrada en la semana 16) durante un máximo de 32 semanas adicionales (de la semana 16 a la semana 48); TREMFYA® subcutáneo 400 mg en las semanas 0, 4 y 8, seguido de TREMFYA® subcutáneo 200 mg cada 4 semanas (con la primera dosis administrada en la semana 12) durante un máximo de 36 semanas adicionales (de la semana 12 a la semana 48); o placebo. Las reacciones adversas notificadas en CD3 hasta la semana 48 en ≥3 % de los sujetos tratados con cualquiera de las pautas posológicas de TREMFYA® subcutáneo, con tasa más alta que con placebo, se muestran en la Tabla 3.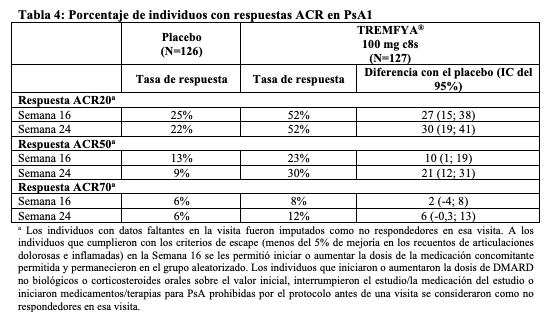
Reacciones adversas específicas: Enzimas hepáticas elevadas: Hasta la semana 12 en CD1, CD2 y CD4, se reportaron niveles de ALT ≥5 veces el LSN en 2/645 (0,3 %) sujetos tratados con TREMFYA® 200 mg intravenoso en las semanas 0, 4 y 8, y en 0/211 sujetos tratados con placebo. Estas elevaciones se produjeron sin aumentos concomitantes de la bilirrubina total. Hasta la semana 48 en CD3, se reportaron niveles de ALT ≥5 veces el LSN en 0/115 sujetos tratados con TREMFYA® 400 mg subcutáneo en las semanas 0, 4 y 8, seguido de TREMFYA® 100 mg subcutáneo cada 8 semanas; en 2/115 (1,7 %) sujetos tratados con TREMFYA® 400 mg subcutáneo en las semanas 0, 4 y 8, seguido de TREMFYA® 200 mg subcutáneo cada 4 semanas; y 0/117 sujetos tratados con placebo. Estas elevaciones se produjeron sin aumentos concomitantes de la bilirrubina total. Experiencia posterior a la comercialización: Las siguientes reacciones adversas se han informado después de la aprobación de TREMFYA®. Debido a que estas reacciones se informan voluntariamente a partir una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición a TREMFYA®. Trastornos del sistema inmunológico: Hipersensibilidad, incluyendo anafilaxis (ver Advertencias, Reacciones de hipersensibilidad) Trastornos de la piel y del tejido subcutáneo: Exantema (ver Advertencias, Reacciones de hipersensibilidad).
Advertencias.
Reacciones de hipersensibilidad: Se han informado reacciones de hipersensibilidad graves, incluyendo anafilaxis, con el uso de TREMFYA® posterior a la comercialización. Algunos casos requirieron hospitalización. Si se produce una reacción de hipersensibilidad grave, suspenda TREMFYA® e inicie la terapia adecuada. Infecciones: TREMFYA® puede aumentar el riesgo de infección. En ensayos clínicos realizados en individuos con psoriasis en placa, se produjeron infecciones en el 23% de los individuos del grupo TREMFYA® frente al 21% de los individuos en el grupo placebo durante las 16 semanas de tratamiento. Las infecciones de las vías respiratorias superiores, gastroenteritis, infecciones por tinea e infecciones por herpes simple ocurrieron con mayor frecuencia en el grupo TREMFYA® que en el grupo placebo (ver Reacciones adversas). La tasa de infecciones graves para el grupo TREMFYA® y el grupo placebo fue ≤0,2%. Se observó un riesgo similar de infección en ensayos controlados con placebo en individuos con artritis psoriásica, colitis ulcerosa y enfermedad de Crohn. El tratamiento con TREMFYA® no se debe iniciar en pacientes con cualquier infección activa clínicamente importante hasta que la infección se resuelva o sea tratada adecuadamente En pacientes con una infección crónica o antecedentes de infección recurrente, considere los riesgos y beneficios antes de prescribir TREMFYA®. Indicar a los pacientes que busquen ayuda médica si presentan signos o síntomas de infección crónica o aguda clínicamente importante. Si un paciente desarrolla una infección grave o clínicamente importante o no responde a la terapia estándar, se deberá monitorear de cerca al paciente y suspender TREMFYA® hasta que la infección se resuelva. Tuberculosis: Evaluar a los pacientes para detectar la infección de tuberculosis (TB) antes de iniciar el tratamiento con TREMFYA®. No administrar TREMFYA® a pacientes con infección de TB activa. Inicie el tratamiento para TB latente antes de administrar TREMFYA® Considere la terapia antituberculosa antes de iniciar TREMFYA® en pacientes con antecedentes de tuberculosis latente o activa en quienes no se pueda confirmar un tratamiento adecuado. Vigile a todos los pacientes para detectar signos y síntomas de tuberculosis activa durante y después del tratamiento con TREMFYA®. En ensayos clínicos, 105 individuos con psoriasis en placa, 71 individuos con artritis psoriásica, 31 pacientes con colitis ulcerosa y 36 pacientes con enfermedad de Crohn con TB latente, que fueron tratados simultáneamente con TREMFYA® y profilaxis apropiada para la TB, no desarrollaron TB activa. En ensayos clínicos de TREMFYA® en sujetos con enfermedad de Crohn, se informó tuberculosis activa en 2 sujetos durante el tratamiento con TREMFYA® (ver Reacciones adversas). Hepatotoxicidad: Se notificó una reacción adversa grave de lesión hepática inducida por fármacos en un sujeto de un ensayo clínico con enfermedad de Crohn, tras tres dosis de un régimen de inducción superior al recomendado. Este sujeto presentó un pico de alanina aminotransferasa (ALT) de 18 veces el límite superior de la normalidad (LSN), aspartato aminotransferasa (AST) de 11 veces el LSN y bilirrubina total de 2,4 veces el LSN. Posteriormente, se suspendió el tratamiento con TREMFYA® y las anomalías en las pruebas hepáticas se resolvieron tras la administración de corticosteroides. En pacientes con enfermedad de Crohn o colitis ulcerosa, se deben evaluar las enzimas hepáticas y la bilirrubina al inicio del tratamiento, durante al menos 16 semanas de tratamiento y, posteriormente, periódicamente, según el manejo habitual del paciente. Considere otras opciones de tratamiento en pacientes con evidencia de enfermedad hepática aguda o cirrosis. Se recomienda investigar rápidamente la causa del aumento de las enzimas hepáticas para identificar posibles casos de lesión hepática inducida por fármacos. Interrumpa el tratamiento si se sospecha lesión hepática inducida por fármacos hasta que se descarte este diagnóstico. Indique a los pacientes que busquen atención médica inmediata si experimentan síntomas que sugieran disfunción hepática. Vacunas: Evite el uso de vacunas de organismos vivos en pacientes tratados con TREMFYA®. Los medicamentos que interactúan con el sistema inmunitario pueden aumentar el riesgo de infección tras la administración de vacunas de organismos vivos. Antes de iniciar el tratamiento con TREMFYA®, complete todas las vacunas apropiadas para su edad de acuerdo con las directrices actuales de inmunización. No se dispone de datos sobre la respuesta a vacunas de organismos vivos o inactivos.
Interacciones.
Sustratos del CYP450: La formación de enzimas CYP450 puede ser alterada por niveles elevados de ciertas citokinas (por ejemplo, IL-1, IL-6, IL-10, TNFa, interferón) durante la inflamación crónica. Los resultados de un estudio exploratorio de interacción medicamentosa en individuos con psoriasis en placa moderada a grave sugirieron un bajo potencial de interacciones medicamentosas clínicamente relevantes para fármacos metabolizados por CYP3A4, CYP2C9, CYP2C19 y CYP1A2, pero no es posible descartar el potencial de interacción con fármacos metabolizados por CYP2D6. Sin embargo, los resultados fueron muy variables debido al bajo número de individuos en el estudio. Después del inicio de TREMFYA® en pacientes que reciben concomitantemente sustratos de CYP450, particularmente aquellos con un índice terapéutico estrecho, considerar el monitoreo del efecto terapéutico o de la concentración del fármaco, y considerar un ajuste de la dosis según fuera necesario. Uso en poblaciones específicas: Embarazo: Registro de exposición en el embarazo: Existe un registro de embarazo que monitorea los resultados del embarazo en mujeres expuestas a TREMFYA® durante el embarazo. Resumen de riesgos: Los datos disponibles de la literatura, los informes post-comercialización y el registro de embarazos en curso con el uso de TREMFYA® en mujeres embarazadas son insuficientes para establecer un riesgo asociado al fármaco de defectos congénitos mayores, aborto espontáneo u otros resultados adversos maternos o fetales. En un estudio combinado de desarrollo embriofetal y de desarrollo pre y postnatal, no se observaron efectos adversos en el desarrollo de crías nacidas de monas preñadas después de la administración subcutánea de guselkumab durante la organogénesis hasta el parto en dosis de hasta en dosis de hasta 18 veces la exposición (AUC) en seres humanos a los que se administran 200 mg por vía intravenosa y de 16 veces la exposición (AUC) a la dosis de 400 mg administrada por vía subcutánea. Se observaron muertes neonatales en los monos con 4 a 18 veces la exposición (AUC) en seres humanos a los que se administran 200 mg por vía intravenosa y con 4 a 16 veces la exposición (AUC) a la dosis de 400 mg administrada por vía subcutánea (ver Datos). Se desconoce la importancia clínica de estos hallazgos preclínicos. Todos los embarazos tienen un riesgo basal de defectos de nacimiento, abortos u otros desenlaces adversos. Se desconoce el riesgo basal de defectos significativos congénitos y de abortos espontáneos para la población indicada. En la población general de Estados Unidos, el riesgo basal de defectos significativos congénitos y de abortos espontáneos en embarazos clínicamente reconocidos es de 2% a 4% y de 15% a 20%, respectivamente. Consideraciones clínicas: Riesgo materno y embrionario/fetal asociado a la enfermedad: Los datos publicados sugieren que el riesgo de resultados adversos del embarazo en mujeres con enfermedad inflamatoria intestinal (EII) está asociado con una mayor actividad de la enfermedad. Los resultados adversos del embarazo incluyen parto prematuro (antes de las 37 semanas de gestación), bebés con bajo peso al nacer (menos de 2.500 g) y bebés pequeños para la edad gestacional al nacer. Reacciones adversas fetales/neonatales: El transporte de anticuerpos IgG endógenos a través de la placenta aumenta a medida que avanza el embarazo y alcanza su punto máximo durante el tercer trimestre. Por lo tanto, se espera que la administración de TREMFYA® pueda estar presente en bebés expuestos in utero. Se debe considerar el posible impacto clínico de la exposición a guselkumab en bebés expuestos in utero. Datos: Datos de animales: En un estudio combinado de desarrollo embriofetal y desarrollo prenatal y posnatal, monas preñadas recibieron dosis subcutáneas semanales de guselkumab desde el comienzo de la organogénesis hasta el parto en una dosis (50 mg/kg) que produjo exposiciones (AUC) 18 veces la exposición en seres humanos que recibieron 200 mg por vía intravenosa y 32 veces la exposición humana en 200 mg administrados por vía subcutánea. Se produjeron muertes neonatales entre las crías de las monas de control, tres monas que recibieron guselkumab en dosis de 10 mg/kg/semana (4 veces la exposición [AUC] en seres humanos que reciben 200 mg por vía intravenosao 400 mg por vía subcutánea) y tres monas que recibieron guselkumab en dosis de 50 mg/kg/semana (18 veces la exposición [AUC] en seres humanos que reciben 200 mg por vía intravenosa y 16 veces la exposición [AUC] después de una dosis subcutánea de 400 mg). Se desconoce la significancia clínica de estos hallazgos. No se observaron efectos relacionados con guselkumab sobre el desarrollo funcional o inmunológico en las crías desde su nacimiento hasta los 6 meses. Lactancia: Resumen de riesgos: No existen datos acerca de la presencia de guselkumab en la leche materna, los efectos sobre el lactante, ni de los efectos sobre la producción de leche. No se detectó guselkumab en la leche de monos cynomolgus lactantes. Las IgG maternas endógenas y los anticuerpos monoclonales se transfieren a la leche humana. Se desconocen los efectos de la exposición gastrointestinal local y el grado de exposición sistémica del lactante amamantado a guselkumab. Se deben considerar los beneficios de la lactancia para el desarrollo y la salud, junto con la necesidad clínica de TREMFYA® para la madre y cualquier potencial efecto adverso sobre el lactante causado por TREMFYA® o por la condición materna subyacente. Uso pediátrico: No se ha establecido la seguridad y la eficacia de TREMFYA® en pacientes pediátricos (menores de 18 años de edad). Uso en pacientes de edad avanzada: De los 5392 pacientes con psoriasis en placas, artritis psoriásica,colitis ulcerosa o enfermedad de Crohn expuestos a TREMFYA®, un total de 285 pacientes tenían 65 años o más y 28 pacientes tenían 75 años o más. Los estudios clínicos de TREMFYA®, dentro de cada indicación, no incluyeron un número suficiente de sujetos de 65 años o más para determinar si responden de forma diferente a los sujetos adultos más jóvenes. No se observaron diferencias clínicamente significativas en la farmacocinética de guselkumab en función de la edad.
Conservación.
Conservar en heladera (2°C - 8°C). No congelar. No agitar Mantener en el estuche para proteger de la luz.
Sobredosificación.
Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los Centros de Toxicología de: Hospital de Pediatría Dr. Ricardo Gutiérrez – Tel: (011) 4962-6666 / 2247 Hospital Profesor Alejandro Posadas – Tel: (011) 4654-6648 y 4658-7777.
Presentación.
TREMFYA® se presenta en una jeringa prellenada/autoinyector de dosis única de 100 mg/ml. TREMFYA® se presenta en uno o dos autoinyector/es de dosis única de 200 mg/ 2 ml. TREMFYA® I.V. se presenta en un vial de dosis única de 200 mg/ 20 ml.