Tecentriq®
ROCHE
Atezolizumab.
Agente antineoplásico, anticuerpo monoclonal.
Composición.
Cada vial con un concentrado de 20 ml contiene 1.200 mg de atezolizumab, que corresponde a una concentración previa a la dilución de 60 mg/ml, en un excipiente compuesto por: L-histidina 62,0 mg, ácido acético glacial 16,5 mg, sacarosa 821,6 mg, polisorbato 20: 8,0 mg y agua para inyectables c.s.p. 20 ml.
Farmacología.
Código ATC: (Pendiente de asignación). Grupo farmacoterapéutico: Agentes antineoplásicos, anticuerpos monoclonales. Propiedades farmacodinámicas: Mecanismo de acción: PD-L1 puede expresarse en las células tumorales y/o en las células inmunes infiltrantes de tumor, y puede contribuir a la inhibición de la respuesta antitumoral del sistema inmune en el microambiente tumoral. La unión de PD-L1 a los receptores PD-1 y B7.1 que se encuentran en las células T y en las células presentadoras de antígenos, suprime la actividad de las células T citotóxicas, la proliferación de células T y la producción de citocinas. Atezolizumab es un anticuerpo monoclonal que se une a PD-L1 y bloquea las interacciones con los receptores PD-1 y B7.1. Esto libera la inhibición mediada por PD-L1/PD-1 de la respuesta inmune, incluida la activación de la respuesta antitumoral del sistema inmune sin inducir la citotoxicidad celular dependiente de anticuerpo. En los modelos tumorales de ratones singénicos, el bloqueo de la actividad de PD-L1 redujo el crecimiento del tumor. Eficacia clínica y seguridad: Carcinoma urotelial: Tecentriq fue investigado en el Estudio 1, de dos cohortes, multicéntrico, abierto, que incluyó a pacientes con carcinoma urotelial metastásico o localmente avanzado. En la cohorte 2 del Estudio 1, Tecentriq fue administrado a 310 pacientes con carcinoma urotelial localmente avanzado o metastásico que presentaron progresión de la enfermedad durante o después de un régimen de quimioterapia basado en platino o progresión de la enfermedad dentro de los 12 meses de tratamiento con un régimen de quimioterapia neoadyuvante o adyuvante basado en platino. Este estudio excluyó a pacientes con: antecedentes de enfermedad autoinmune, metástasis cerebrales dependientes de corticosteroides o activas, administración de una vacuna atenuada con agentes vivos dentro de los 28 días previos a la incorporación, o administración de agentes inmunoestimuladores sistémicos o medicación inmunosupresora sistémica. Los pacientes recibieron una infusión intravenosa de 1.200 mg de Tecentriq cada 3 semanas hasta toxicidad inaceptable o progresión radiográfica o clínica. Las evaluaciones de la respuesta tumoral se realizaron cada 9 semanas durante las primeras 54 semanas y cada 12 semanas en adelante. Los principales criterios de valoración de eficacia incluyeron la tasa de respuesta objetiva (ORR, por sus siglas en inglés) confirmada, según la estimación de un Comité de revisión independiente (IRF, por sus siglas en inglés), utilizando los criterios de evaluación de respuesta en tumores sólidos v1.1 (RECIST, por sus siglas en inglés) y la duración de la respuesta (DOR, por sus siglas en inglés). En esta cohorte, la mediana de edad fue 66 años, 78% eran hombres, 91% eran pacientes caucásicos. El 26% de los pacientes tenía carcinoma urotelial que no se había originado en la vejiga y el 78% metástasis viscerales. El 62% presentó un puntaje de ECOG de 1 y el 35% una depuración de creatinina < 60 ml/min. El 19 % experimentó progresión de la enfermedad luego de la quimioterapia neoadyuvante o adyuvante basada en platino. El 41% había recibido ≥2 regímenes sistémicos previos en el contexto metastásico. Al 73% se administró cisplatino previo, al 26% carboplatino y al 1% otros regímenes basados en platino. Las muestras de tumor fueron evaluadas en forma prospectiva utilizando el ensayo VENTANA PD-L1 (SP142) en un laboratorio central y los resultados fueron utilizados para definir los subgrupos de los análisis preespecificados. De 310 pacientes, el 32% evidenciaba una expresión de PD-L1 ≥ 5% (definida como tinción de PD-L1 en las células inmunes (CI) infiltrantes de tumor en ≥ 5% del área del tumor). El 68% restante presentaba una expresión de PD-L1 < 5% (tinción de PD-L1 en las CI infiltrantes de tumor en < 5% del área del tumor). La Tabla 1 detalla la ORR confirmada en todos los pacientes y los dos subgrupos de PD-L1. La mediana de seguimiento de esta cohorte fue 14,4 meses. En 59 pacientes con progresión de la enfermedad luego de la terapia adyuvante o neoadyuvante, la ORR fue de 22,0% (IC 95%: 12,3%, 34,7%).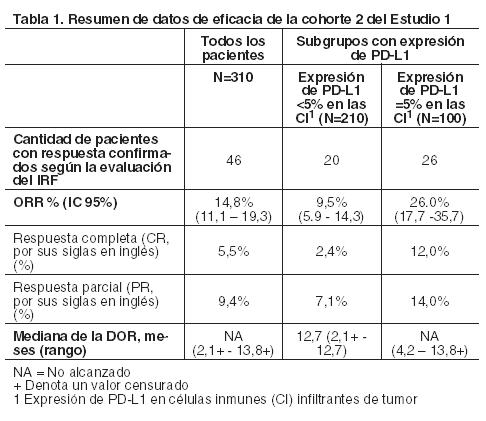
Cáncer de pulmón de células no pequeñas metastásico: CPCNP metastásico previamente tratado: La eficacia de Tecentriq fue investigada en dos estudios multicéntricos, internacionales, aleatorizados, abiertos, en pacientes con CPCNP metastásico que progresaron durante o después de un régimen que contenía platino. El Estudio 2 fue realizado en 1225 pacientes en el que la población del análisis primario incluía a los primeros 850 pacientes aleatorizados. El Estudio 3 fue incorporó 287 pacientes. En ambos estudios los pacientes elegibles fueron estratificados según el estado de la expresión de PD-L1 en células inmunes (CI) infiltrantes de tumor según la cantidad de regímenes quimioterápicos previos, y según la histología. Los pacientes fueron aleatorizados (1:1) para recibir Tecentriq por vía intravenosa con una dosis de 1.200 mg cada 3 semanas hasta toxicidad inaceptable o progresión radiográfica o clínica, o docetaxel por vía intravenosa a 75 mg/m2 cada 3 semanas hasta toxicidad inaceptable o progresión de la enfermedad. Estos estudios excluyeron a pacientes con: antecedentes de enfermedad autoinmune, metástasis cerebrales dependientes de corticosteroides o activas, administración de una vacuna viva atenuada dentro de los 28 días previos a la incorporación, administración de inmunoestimuladores sistémicos dentro de las 4 semanas o medicación inmunosupresora sistémica dentro de las 2 semanas previas a la inclusión. Las evaluaciones tumorales se realizaron cada 6 semanas en las primeras 36 semanas, y cada 9 semanas en adelante. En el Estudio 2 las muestras de tumor fueron evaluadas en forma prospectiva para determinar la expresión de PD-L1 en las células tumorales (CT) y las CI con el ensayo VENTANA PD-L1 (SP142) y los resultados fueron utilizados para definir los subgrupos de expresión de PD-L1 para los análisis que se presentan a continuación. En el Estudio 2, entre los pacientes de la población de análisis primario, la mediana de la edad fue de 64 años (rango: 33 a 85) y el 61% de los pacientes eran hombres. La mayoría era de raza blanca (70%). Aproximadamente 3/4 tenía enfermedad no escamosa (74%), el 10% tenía una mutación conocida en EGFR, el 0,2% tenía reordenamientos conocidos de ALK, y la mayoría fumaba o había fumado anteriormente (82%). El estado funcional del Eastern Cooperative Group (ECOG) al inicio fue de 0 (37%) o 1 (63%). El 75% recibió sólo un régimen previo basado en platino. En el Estudio 3 la mediana de la edad fue de 62 años (rango: 36 a 84), y el 59% de los pacientes eran hombres. La mayoría era de raza blanca (79%). Aproximadamente 2/3 tenían enfermedad no escamosa (66%), el 7% presentaba mutación conocida en EGFR, el 1% reordenamientos de ALK y la mayoría fumaba o había fumado anteriormente (80%). El estado funcional de ECOG al inicio fue 0 (33%) o 1 (67%). Aproximadamente 2/3 de los pacientes recibieron sólo un esquema de tratamiento previo basado en platino. EI principal criterio de valoración de eficacia en el Estudio 2 fue la sobrevida global (SG) en la población del análisis primario (los primeros 850 pacientes aleatorizados). El principal criterio de valoración de eficacia en el Estudio 3 fue la sobrevida global (SG). Otros criterios de valoración de eficacia en el Estudio 3 incluyeron las tasas de respuesta objetiva evaluadas por el investigador y la duración de la respuesta según RECIST v1.1. En la Tabla 2 y la Figura 1 se analizan los resultados del Estudio 2 con una mediana de seguimiento de 21 meses.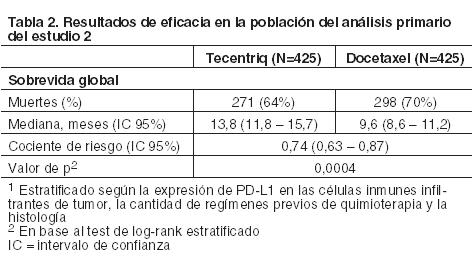
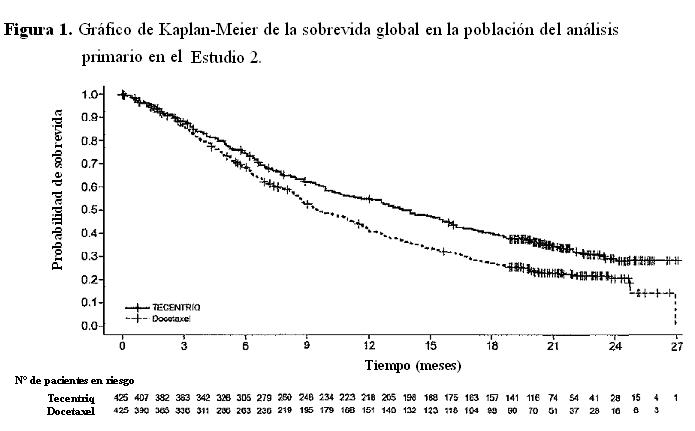
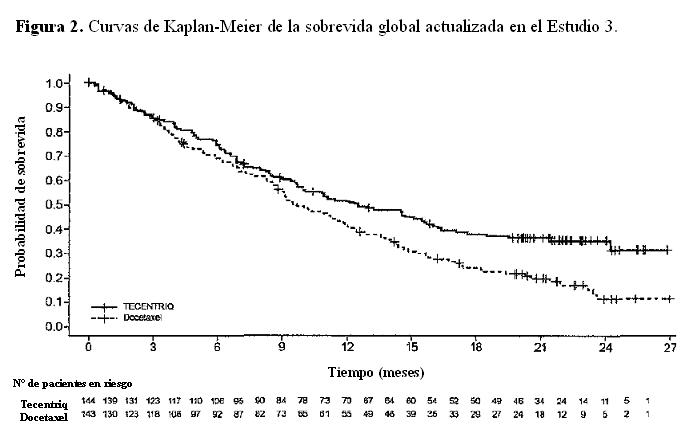
Las muestras de tumor fueron evaluadas en forma prospectiva con el ensayo VENTANA PD-L1 (SP142) en un laboratorio central y los resultados fueron utilizados para definir los subgrupos de expresión de PD-L1 para los análisis preespecificados. De los 850 pacientes, el 16% tenía una expresión de PD-L1 elevada definida como ≥50% de las CT o ≥10% de las CI. En un análisis exploratorio de eficacia de la SG en los subgrupos, realizado en base a la expresión de PD-L1, el cociente de riesgo fue 0,41 (IC 95%: 0,27, 0,64) en el subgrupo con expresión elevada de PD-L1 y 0,82 (IC 95%: 0,68 - 0,98) en los pacientes cuya expresión de PD-L1 era baja. Se presentan los resultados del análisis actualizado de la sobrevida en el Estudio 3 con una mediana de seguimiento de 22 meses para todos los pacientes aleatorizados (Tabla 3 y Figura 2).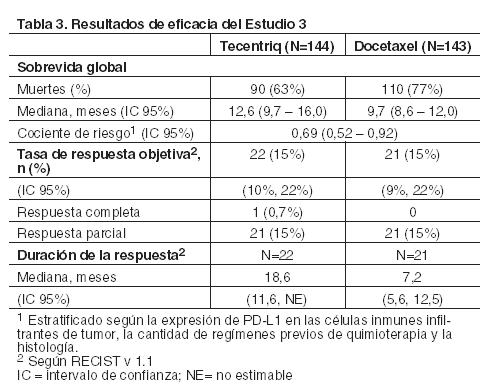
Inmunogenicidad: Al igual que con todas las proteínas terapéuticas, la inmunogenicidad es un riesgo posible. De las 275 pacientes en el Estudio 1, 114 (41,5%) presentaron anticuerpos antiterapéuticos (ATA) emergentes del tratamiento (inducidos o aumentados por el mismo) en uno o más de los intervalos posteriores a la dosis. De las 135 pacientes en el Estudio 3, 73 (54,1%) tenían ATA emergentes del tratamiento (inducidos o aumentados por el mismo) en uno o más intervalos posteriores a la dosis. En el Estudio 1 y el Estudio 3, la presencia de ATA no pareció causar un impacto clínicamente significativo en la farmacocinética, la seguridad o la eficacia. Los resultados de los ensayos de inmunogenicidad dependen en gran parte de varios factores, entre ellos, la sensibilidad y especificidad de los ensayos, la metodología utilizada, la manipulación de las muestras, el momento en que se recolectaron las muestras, los medicamentos concomitantes y la enfermedad de base. Por estos motivos, la comparación de la incidencia de ATA contra Tecentriq y la de anticuerpos contra otros productos podría resultar confusa. Propiedades farmacocinéticas: Las exposiciones de los pacientes a atezolizumab se incrementaron en forma proporcional a la dosis en todo el rango de dosis, de 1 mg/kg a 20 mg/kg incluyendo la dosis fija de 1.200 mg administrada cada 3 semanas. En base a un análisis de población que incluyó a 472 pacientes en el rango de dosis, la depuración en la población típica fue 0,20 l/día, el volumen de distribución en estado de equilibrio de 6,9 litros y la vida media terminal de 27 días. El análisis PK de la población sugiere que el estado de equilibrio se alcanza después de 6 a 9 semanas (2 a 3 ciclos) de dosis repetidas. La acumulación sistémica en el área bajo la curva (ABC), la concentración máxima (Cmáx) y la concentración valle (Cmin) fueron de 1,91, 1,46 y 2,75 veces, respectivamente. En un análisis post-hoc, se observó que la depuración de atezolizumab se reducía con el tiempo, con una disminución máxima media (% coeficiente de variación [CV%]) desde el valor inicial de aproximadamente 17,1% (40,6%). No obstante, el descenso en la depuración no fue considerado clínicamente relevante. Poblaciones especiales: La edad (21-89 años), el peso corporal, el sexo, los anticuerpos antiterapéuticos (ATA) positivos, los niveles de albumina, la carga tumoral, la región o la etnia, la insuficiencia renal leve o moderada (tasa de filtración glomerular estimada (eGFR) de 30 a 89 ml/min/1,73 m2), la insuficiencia hepática leve (bilirrubina ≤ ULN y AST > ULN o bilirrubina < 1,0 a 1,5 x ULN y cualquier AST), el nivel de expresión de PD-L1 o el estado de ECOG no causaron un efecto clínicamente significativo en la exposición sistémica de atezolizumab. Se desconoce el efecto de la insuficiencia renal severa (eGFR 15 a 29 ml/min/1,73 m2) o la insuficiencia hepática moderada o severa (bilirrubina > ULN y AST > ULN o bilirrubina ≥ 1,0 a 1,5 x ULN y cualquier AST) en la farmacocinética de atezolizumab. Población pediátrica: No se ha establecido la seguridad y eficacia de Tecentriq en pacientes pediátricos. Pacientes de edad avanzada: De los 310 pacientes con carcinoma urotelial tratados con Tecentriq en el Estudio l, el 59% tenía 65 años o más. De los 142 pacientes con CPCNP tratados con Tecentriq en el Estudio 3, el 39% tenía 65 años o más. No se registraron diferencias globales en la seguridad o la eficacia entre pacientes ≥65 años y más jóvenes. Pacientes con insuficiencia renal: En base a un análisis farmacocinético de la población no se recomienda ajustar la dosis de Tecentriq en pacientes con insuficiencia renal. Pacientes con insuficiencia hepática: Según un análisis farmacocinético de la población, no se recomienda ajustar la dosis de Tecentriq en pacientes con insuficiencia hepática leve. Tecentriq no ha sido estudiado en pacientes con insuficiencia hepática moderada o severa. Datos preclínicos de seguridad. Carcinogenicidad, mutagenicidad y deterioro de la fertilidad: No se han realizado estudios para evaluar el potencial carcinogénico o genotóxico de atezolizumab. Tampoco se han llevado a cabo ensayos de fertilidad con atezolizumab en animales; no obstante, en un estudio de toxicidad de 26 semanas con dosis repetida en monos cynomolgus se incluyó una evaluación de los órganos reproductores en machos y hembras. La administración semanal de atezolizumab a monos hembras con la dosis máxima evaluada causó un ciclo menstrual irregular y falta de cuerpos lúteos nuevos en los ovarios. Este efecto ocurrió con un ABC estimada de aproximadamente 6 veces el ABC en pacientes que recibieron la dosis recomendada y fue reversible. No hubo efecto alguno en los órganos reproductores de monos machos. Toxicología y farmacología en animales: En modelos animales, la inhibición de la señalización de PD-L1/PD-1 potenció las respuestas inflamatorias y la severidad de algunas infecciones. Los ratones transgénicos sin actividad PD-1 (knock-out) infectados por Mycobacterium tuberculosis tuvieron una sobrevida considerablemente menor en comparación con los controles wild type, lo que se correlacionó con un aumento en la proliferación bacteriana y las respuestas inflamatorias en estos animales. Los ratones transgénicos sin actividad PD-L1 y PD-1 (knock-out) y los ratones que recibieron anticuerpos inhibidores de PD-L1 también presentaron una sobrevida menor luego de la infección con el virus de la coriomeningitis linfocítica.
Indicaciones.
Carcinoma urotelial: Tecentriq (atezolizumab) está indicado para el tratamiento de pacientes con carcinoma urotelial localmente avanzado o metastásico que: han experimentado progresión de la enfermedad durante o después de una quimioterapia basada en platino; han experimentado progresión de la enfermedad dentro de los 12 meses de haber recibido quimioterapia adyuvante o neoadyuvante con platino. Cáncer de pulmón de células no pequeñas metastásico: Tecentriq está indicado para el tratamiento de pacientes con cáncer de pulmón de células no pequeñas (CPCNP) metastásico que han experimentado progresión de la enfermedad durante o después de la quimioterapia basada en platino. Los pacientes con aberraciones genómicas en EGFR o ALK deben haber experimentado progresión a una terapia previa aprobada para estas aberraciones.
Dosificación.
Tecentriq debe administrarse bajo la supervisión de un profesional de la salud entrenado. El reemplazo por cualquier otro agente biológico requiere el consentimiento del médico prescriptor. Posología: Dosis recomendada: La dosis recomendada de Tecentriq es 1.200 mg administrados como infusión intravenosa durante 60 minutos cada 3 semanas hasta la progresión de la enfermedad o toxicidad inaceptable. Si el paciente tolera la primera infusión, las infusiones posteriores pueden administrarse durante 30 minutos. No administrar Tecentriq como pulso o bolo intravenoso. Retraso u omisión de dosis: Si se olvida administrar una dosis de Tecentriq, deberá administrarse lo antes posible, sin esperar hasta la próxima dosis planificada. Se debe ajustar el cronograma de administración para dejar un período de 3 semanas entre las dosis. Ajustes de dosis: No se recomienda reducir las dosis de Tecentriq. Interrumpir Tecentriq en cualquiera de los siguientes casos: Neumonitis Grado 2. Aspartato aminotransferasa (AST) o alanina aminotransferasa (ALT) mayor que 3 y hasta 5 veces el límite superior normal (ULN) o bilirrubina total mayor que 1,5 y hasta 3 veces el ULN. Diarrea o colitis Grados 2 o 3. Casos sintomáticos de hipofisitis, insuficiencia suprarrenal, hipotiroidismo, hipertiroidismo o hiperglucemia Grados 3 o 4. Toxicidad ocular inflamatoria Grado 2. Pancreatitis Grados 2 o 3, o aumentos Grados 3 o 4 en los niveles de amilasa o lipasa (mayores que 2.0 veces el ULN). Infección Grados 3 o 4. Reacciones relacionadas con la infusión Grado 2. Erupción cutánea Grado 3. Tecentriq puede reiniciarse en pacientes cuyas reacciones adversas se normalizan a Grado 0-1. Interrumpir Tecentriq en forma permanente en caso de cualquiera de las siguientes: Neumonitis Grados 3 o 4 AST o ALT mayores que 5 veces el ULN o bilirrubina total mayor que 3 veces el ULN. Diarrea o colitis Grado 4. Hipofisitis Grado 4. Síndrome miasténico/miastenia grave, Guillain-Barré o meningoencefalitis (todos los Grados). Toxicidad ocular inflamatoria Grados 3 o 4. Pancreatitis recurrente de cualquier grado o Grado 4. Reacciones relacionadas con la infusión Grados 3 o 4. Erupción cutánea Grado 4. Indique a los pacientes que contacten de inmediato a su médico en caso de signos o síntomas de erupción cutánea. Formas de administración: Administrar la primera infusión durante 60 minutos a través de una vía intravenosa con o sin un filtro en vía estéril, no pirogénico, de baja unión a proteínas (tamaño de poro: 0,2-0,22 micrones). Si el paciente tolera la primera infusión, el resto de las infusiones pueden ser administradas durante 30 minutos. No coadministrar otros medicamentos a través de la misma vía.
Contraindicaciones.
Tecentriq está contraindicado en pacientes con hipersensibilidad conocida a atezolizumab o a cualquiera de los excipientes.
Reacciones adversas.
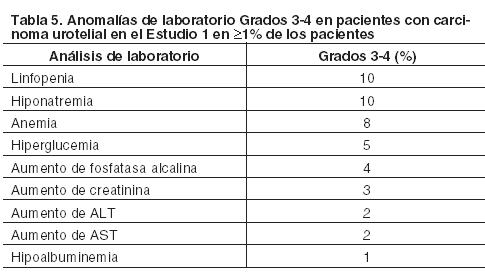
En otras secciones del prospecto se describen en detalle las siguientes reacciones adversas: Neumonitis inmunorrelacionada. Hepatitis inmunorrelacionada. Colitis inmmunorrelacionada. Endocrinopatías inmunorrelacionadas. Otras reacciones adversas inmunorrelacionadas. Infecciones. Reacciones relacionadas con la infusión. Experiencia en estudios clínicos: Debido a que los estudios clínicos se realizan bajo condiciones ampliamente variables, las tasas de reacciones adversas de un fármaco determinado no pueden compararse directamente con las tasas de estudios clínicos de otro medicamento, y podrían no reflejar las cifras observadas en la práctica. Carcinoma urotelial: Los datos presentados en la Tabla 4 muestran la exposición de Tecentriq en la cohorte 2 del Estudio 1. En esta cohorte se incorporó a 310 pacientes en un estudio de una sola rama con carcinoma urotelial localmente avanzado o metastásico, que experimentaron progresión de la enfermedad durante o después de al menos un régimen de quimioterapia basada en platino o progresión de la enfermedad dentro de los 12 meses de tratamiento con un régimen de quimioterapia neoadyuvante o adyuvante basado en platino. Los pacientes recibieron 1.200 mg de Tecentriq por vía intravenosa cada 3 semanas hasta toxicidad inaceptable o progresión radiográfica o clínica. La mediana de la duración de la exposición fue de 12,3 semanas (rango: 0,1 a 46 semanas). Las reacciones adversas más frecuentes (≥20%) fueron, fatiga (52%), falta de apetito (26%), náuseas (25%), infección urinaria (22%), pirexia (21%) y constipación (21%). Las reacciones adversas Grados 3-4 más frecuentes (≥ 2%) fueron infecciones urinarias, anemia, fatiga, deshidratación, obstrucción intestinal, obstrucción urinaria, hematuria, disnea, injuria renal aguda, dolor abdominal, tromboembolismo venoso, sepsis y neumonía. Tres (0,9%) pacientes que fueron tratados con Tecentriq experimentaron sepsis, neumonitis u obstrucción intestinal, que condujeron a la muerte. Tecentriq fue discontinuado por reacciones adversas en el 3,2% (10/310) de los pacientes. La sepsis requirió discontinuar el tratamiento en el 0,6% (2/310) de los pacientes. Las reacciones adversas que obligaron a interrumpir Tecentriq se manifestaron en el 27%; las más frecuentes ( > 1%) fueron: aumento de las enzimas hepáticas, infección urinaria, diarrea, fatiga, estado de confusión, obstrucción urinaria, pirexia, disnea, tromboembolismo venoso y neumonitis. El 45% padeció reacciones adversas serias de las cuales las más frecuentes ( > 2%) fueron infección urinaria, hematuria, injuria renal aguda, obstrucción intestinal, pirexia, tromboembolismo venoso, obstrucción urinaria, neumonía, disnea, dolor abdominal, sepsis y estado de confusión. La Tabla 4 resume las reacciones adversas que ocurrieron en ≥ 10% de los pacientes, mientras que la Tabla 5 presenta las anomalías de laboratorio Grados 3-4 seleccionadas que aparecieron en ≥ 1% de los pacientes tratados con Tecentriq en la cohorte 2 del Estudio 1.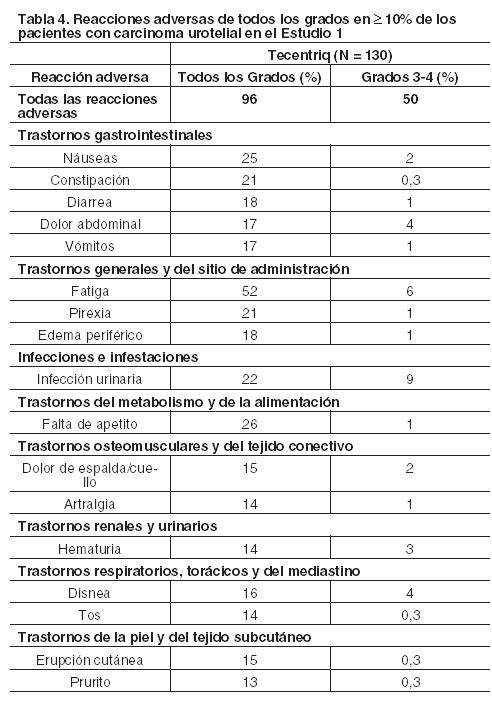
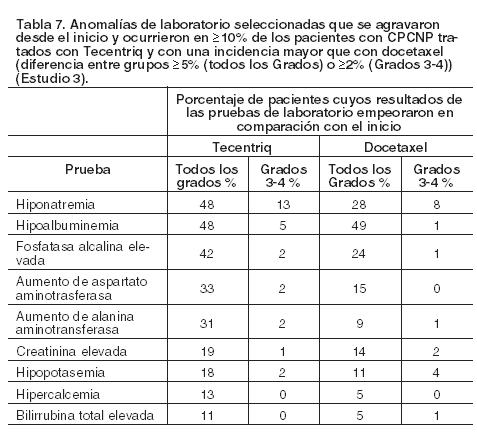
CPCNP: La seguridad de Tecentriq fue evaluada en el Estudio 3, multicéntrico, internacional, aleatorizado, abierto, en pacientes con CPCNP metastásico, que progresaron durante o después de un régimen basado en platino, independientemente de la expresión de PD-L1. Los pacientes recibieron 1.200 mg de Tecentriq (n=142) por vía intravenosa cada 3 semanas hasta toxicidad inaceptable o progresión radiográfica o clínica, o docetaxel (n==135) administrado por vía intravenosa a 75 mg/m2 cada 3 semanas hasta toxicidad inaceptable o progresión de la enfermedad. La mediana de la duración de la exposición fue de 3,7 meses (rango: 0-19 meses) en los pacientes tratados con Tecentriq y de 2,1 meses (rango: 0-17 meses) en los tratados con docetaxel. Las reacciones adversas más frecuentes (≥20%) en pacientes tratados con Tecentriq fueron: fatiga (46%), falta de apetito (35%), disnea (32%), tos (30%), náuseas (22%), dolor osteomuscular (22%) y constipación (20%). Las reacciones adversas Grados 3-4 más frecuentes (≥2%) fueron: disnea, neumonía, hipoxia, hiponatremia, fatiga, anemia, dolor osteomuscular, aumento de AST y de ALT, disfagia y artralgia. Nueve (6,3%) pacientes que fueron tratados con Tecentriq experimentaron embolia pulmonar (2), neumonía (2), neumotórax, úlcera hemorrágica, caquexia secundaria a disfagia, infarto de miocardio o perforación intestinal mortal. Tecentriq fue discontinuado por reacciones adversas en el 4% (6/142) de los pacientes. En el 24% las reacciones adversas obligaron a interrumpir el tratamiento con Tecentriq; las más frecuentes ( > 1%) fueron neumonía, pruebas anormales de la función hepática, infección de las vías respiratorias altas, neumonitis, injuria renal aguda, hipoxia, hipotiroidismo, disnea, anemia y fatiga. El 37% padeció reacciones adversas serias. Las reacciones adversas serias más frecuentes ( > 2%) fueron neumonía, disnea, derrame pleural, pirexia y tromboembolismo venoso. La Tabla 6 enumera las reacciones adversas que ocurrieron en al menos el 10% de los pacientes tratados con Tecentriq y con una incidencia mayor en comparación con docetaxel. La Tabla 7 detalla las anomalías de laboratorio seleccionadas que se agravaron desde el inicio y que se manifestaron en ≥10% de las pacientes tratados con Tecentriq, y con una incidencia mayor que con docetaxel.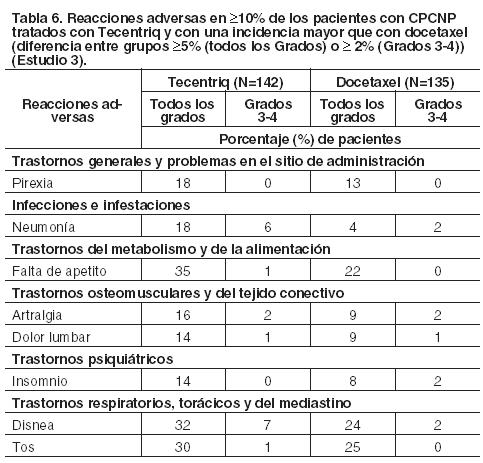
Otras reacciones adversas inmunorrelacionadas: Se han reportado otras reacciones adversas inmunorrelacionadas, incluida meningoencefalitis, síndrome miasténico/miastenia gravis, Guillain-Barré, toxicidad ocular inflamatoria y pancreatitis, incluidos aumentos en los niveles séricos de amilasa y lipasa, en ≤ 1,0% de los pacientes tratados con Tecentriq. Indique a los pacientes que se contacten de inmediato con su médico en caso de signos o síntomas de toxicidad ocular inflamatoria. Meningitis/encefalitis: Monitorear a los pacientes para identificar signos y síntomas clínicos de meningitis o encefalitis. Discontinuar Tecentriq en forma permanente en caso de meningitis o encefalitis de cualquier grado. Tratar con esteroides IV (l-2 mg/kg/día de metilprednisolona o equivalente) y convertir a corticosteroides orales (prednisona 60 mg/día o equivalente) una vez que el paciente ha mejorado. Cuando los síntomas mejoren a ≤ Grado 1, retirar los corticosteroides gradualmente durante ≥ 1 mes. Indique a los pacientes que se contacten con su médico de inmediato en caso de signos o síntomas de meningitis. Neuropatía motora y sensorial: Monitorear a los pacientes para identificar síntomas de neuropatía motora y sensorial. Discontinuar Tecentriq en forma permanente en caso de síndrome miasténico/miastenia grave o síndrome de Guillain-Barré de cualquier grado. Proceder a la intervención médica según sea necesario. Considerar iniciar corticosteroides sistémicos a una dosis de 1-2 mg/kg/día de prednisona. Indique a los pacientes que se contacten con su médico de inmediato en caso de síndrome miasténico/miastenia gravis o síndrome de Guillain-Barré. Pancreatitis: El 0,1% (2/1978) de los pacientes en los estudios clínicos experimentaron pancreatitis sintomática sin una etiología alternativa. Monitorear a los pacientes para identificar signos y síntomas de pancreatitis aguda. Suspender Tecentriq en caso de niveles séricos de amilasa o lipasa ≥ Grado 3 ( > 2,0 ULN) o pancreatitis Grados 2 o 3. Tratar con metilprednisolona 1-2 mg/kg IV o equivalente por día. Una vez que mejoren los síntomas, continuar con 1-2 mg/kg de prednisona oral o equivalente por día. Reiniciar el tratamiento con Tecentriq cuando los niveles séricos de amilasa y lipasa hayan mejorado a ≤ Grado l dentro de las 12 semanas o los síntomas de pancreatitis se hayan resuelto, y la dosis de corticosteroides se haya reducido a ≤ 10 mg de prednisona oral o equivalente por día. Discontinuar Tecentriq en forma permanente en caso de pancreatitis Grado 4 o recurrente de cualquier Grado. Indique a los pacientes que se contacten de inmediato con su médico en caso de signos y síntomas de pancreatitis. Infecciones: Se han comunicado casos de infecciones severas, entre ellas, sepsis, encefalitis herpética e infección micobacteriana que condujo a hemorragia retroperitoneal, en pacientes tratados con Tecentriq. Monitorear a los pacientes para identificar signos y síntomas de infección y tratar con antibióticos en caso de infecciones bacterianas sospechadas o confirmadas. Suspender Tecentriq en caso de infección ≥ Grado 3. En los estudios clínicos, el 38,4% (759/1978) de los pacientes presentaron infecciones. Indique a los pacientes que se contacten de inmediato con su médico en caso de signos o síntomas de infección. Carcinoma urotelial: De 523 pacientes con carcinoma urotelial que recibieron Tecentriq, 197 (37,7%) presentaron infecciones. En 60 (11,5%) la infección era Grados 3 o 4, mientras que 3 fallecieron por infecciones. La causa más frecuente de infecciones Grado 3 o mayor fueron las infecciones urinarias, en 37 (7,1%) pacientes. CPCNP: En el Estudio 3, aleatorizado en pacientes con CPCNP, las infecciones fueron más frecuentes en pacientes tratados con Tecentriq (43%), en comparación con aquellos que recibieron docetaxel (34%). El 9,2% de los pacientes tratados con Tecentriq experimentaron infecciones Grados 3 o 4, en comparación con el 2,2% de los tratados con docetaxel. Dos (1,4%) pacientes tratados con Tecentriq y tres (2,2%) de los que recibieron docetaxel fallecieron por infecciones. La causa más frecuente de infecciones Grado 3 o mayor fue neumonía que ocurrió en el 7,7% de los pacientes tratados con Tecentriq. Comunicación de reportes de reacciones adversas: Es importante comunicar las presuntas reacciones adversas después de la autorización del medicamento. Esto permite la monitorización continua de la relación riesgo/beneficio. Se solicita a los profesionales de la salud informar de cualquier sospecha de eventos adversos asociados con el uso de Tecentriq® al Área de Farmacovigilancia de Roche al siguiente teléfono 0800-77-ROCHE (76243). En forma alternativa, esta información puede ser reportada ante ANMAT. Ante cualquier inconveniente con el producto, el paciente puede llenar la ficha que está en la Página Web de la ANMAT: http://www.anmat.gov.ar/farmacovigilancia/Notificar.asp o llamar a ANMAT responde al 0800-333-1234.
Precauciones.
Para mejorar la trazabilidad de los medicamentos biológicos, el nombre comercial y el número de lote del producto administrado deben estar claramente registrados (o mencionados) en la historia clínica del paciente. Neumonitis inmunorrelacionada: En los pacientes tratados con Tecentriq se reportaron casos de neumonitis o enfermedad pulmonar intersticial inmunomediadas, definida como aquella que requiere el uso de corticosteroides y no presenta una etiología alternativa clara. Los pacientes deben ser monitoreados con imágenes radiográficas para identificar signos y síntomas de neumonitis. Se deben administrar corticosteroides a una dosis de 1-2 mg/kg/día de equivalentes de prednisona para la neumonitis Grado 2 o mayor, seguido del retiro gradual de corticosteroides. Interrumpir Tecentriq hasta la resolución de la neumonitis Grado 2. Discontinuar permanentemente Tecentriq en caso de neumonitis Grados 3 o 4. En los estudios clínicos, el 2,6% (51/1978) de los pacientes desarrollaron neumonitis. En 2 pacientes la neumonitis fue fatal. Indique a los pacientes que se comuniquen de inmediato con su médico en caso de aparición de tos, dolor de pecho o dificultad para respirar o si estos empeoran. Carcinoma urotelial: De 523 pacientes con carcinoma urotelial que recibieron Tecentriq, 6 (1,1%) experimentaron neumonitis. De estos, uno presentó neumonitis mortal, uno desarrolló neumonitis Grado 3, tres Grado 2 y uno Grado 1. Tecentriq fue suspendido en todos los casos y 5 pacientes recibieron tratamiento con corticosteroides. La neumonitis se resolvió en tres pacientes. La mediana del tiempo hasta el inicio fue de 2,6 meses (rango: 15 días a 4,2 meses). La mediana de la duración fue de 15 días (rango: 6 días a 3,1+ meses). CPCNP: De 1027 pacientes con CPCNP que recibieron Tecentriq, 38 (3,7%) desarrollaron neumonitis. De estos pacientes, uno presentó neumonitis mortal, 2 pacientes neumonitis Grado 4, 13 Grado 3, 11 pacientes Grado 2 y 11 Grado 1. Tecentriq fue suspendido en 24 pacientes y 21 fueron tratados con corticosteroides. La neumonitis se resolvió en 26 de los 38 pacientes. La mediana del tiempo hasta el inicio fue de 3,3 meses (rango: 3 días a 18,7 meses). La mediana de la duración fue de 1,4 meses (rango: 0 días a 12,6+ meses). Hepatitis inmunorrelacionada: La hepatitis inmunomediada, definida como hepatitis que requiere del uso de corticosteroides y no tiene una etiología alternativa clara ocurrió en pacientes tratados con Tecentriq. Los pacientes que recibieron Tecentriq manifestaron anomalías en las pruebas hepáticas. Se debe monitorear a los pacientes para identificar signos y síntomas de hepatitis. Monitorear los niveles de AST, ALT y bilirrubina antes y durante el tratamiento con Tecentriq. Administrar corticosteroides a una dosis de 1-2 mg/kg/día de equivalentes de prednisona en caso de aumentos Grado 2 o mayor de transaminasa, con o sin incrementos concurrentes en la bilirrubina total, seguido del retiro gradual de los corticosteroides. Suspender Tecentriq en caso de Grado 2 y discontinuar Tecentriq en forma permanente en caso de hepatitis inmunomediada Grados 3 o 4. En los estudios clínicos (n=1978), se notificaron aumentos Grados 3 o 4 en los valores de ALT (2,5%), AST (2,3%) y bilirrubina total (1.6%). Indique a sus pacientes que se contacten de inmediato con su médico en caso de ictericia, náuseas o vómitos severos, dolor en el hemiabdomen derecho, letargia o propensión a hematomas o sangrado. Carcinoma urotelial: En pacientes con carcinoma urotelial (n=523) se informaron casos de aumentos Grados 3 o 4 en ALT (2,5%), AST (2,5%) y bilirrubina total (2,1%). El 1,3% de los pacientes experimentaron hepatitis inmunomediada. De estos casos, un paciente falleció por hepatitis, cinco contrajeron hepatitis Grado 3 y otro hepatitis Grado 2. La mediana del tiempo hasta el inicio fue de 1,1 meses (rango: 0,4 a 7,7 meses). Tecentriq fue interrumpido temporalmente en 4 pacientes; ninguno experimentó recurrencia de la hepatitis luego de reiniciar Tecentriq. CPCNP: En pacientes con CPCNP, se reportaron aumentos Grados 3 o 4 en ALT (1,4%), AST (1,3%) y bilirrubina total (0,6%). El 0,9% (9/1027) presentó hepatitis inmunomediada. De estos 9 pacientes, 1 desarrolló hepatitis inmunomediada Grado 4, 4 Grado 3, 3 pacientes Grado 2 y uno Grado 1. La mediana del tiempo hasta el inicio fue de 28 días (rango: 15 días a 4,2 meses). Tecentriq fue temporalmente interrumpido en 7 pacientes; ninguno desarrolló recurrencia de hepatitis luego de reiniciar Tecentriq. Colitis inmunorrelacionada: Los pacientes tratados con Tecentriq desarrollaron colitis o diarrea inmunomediada, definida como aquella que requiere del uso de corticosteroides y no tiene una etiología alternativa clara. Se debe monitorear a los pacientes para identificar signos y síntomas de diarrea o colitis. Interrumpir el tratamiento con Tecentriq en caso de diarrea o colitis Grado 2. Si los síntomas persisten por más de 5 días o recurren, administrar 1-2 mg/kg de prednisona o equivalente por día. Interrumpir el tratamiento con Tecentriq en caso de diarrea o colitis Grado 3. Tratar con metilprednisolona IV 1-2 mg/kg por día y convertir a corticosteroides orales una vez que el paciente haya mejorado. En caso de diarrea o colitis Grados 2 y 3, cuando los síntomas mejoren a Grado 0 o Grado 1, retirar gradualmente los esteroides durante ≥ 1 mes. Reiniciar el tratamiento con Tecentriq si el evento mejora a Grado 0 o 1 dentro de las 12 semanas y si se ha reducido la dosis de corticosteroides al equivalente de ≤ 10 mg de prednisona oral por día. Discontinuar Tecentriq en forma permanente en caso de diarrea o colitis Grado 4. En los estudios clínicos, el 19,7% (398/1978) de todos los pacientes experimentaron colitis o diarrea. Indique a los pacientes que se contacten de inmediato con su médico en caso de diarrea o dolor abdominal intenso. Carcinoma urotelial: De 523 pacientes con carcinoma urotelial tratados con Tecentriq, 98 (18,7%) desarrollaron colitis o diarrea. Diez (1,9%) tuvieron diarrea Grados 3 o 4. Cuatro (0,8%) presentaron colitis o diarrea inmunomediada con una mediana del tiempo hasta el inicio de 1,7 meses (rango: 1,1 a 3,1 meses). La colitis inmunomediada mejoró con la administración de corticosteroides en 3 de estos pacientes, mientras que el otro paciente falleció sin que se resolviera la colitis en el contexto de insuficiencia renal asociada con diarrea. CPCNP: De 1027 pacientes con CPCNP que recibieron Tecentriq, 198 (19,3%) desarrollaron colitis o diarrea. Doce (1,2%) experimentaron colitis o diarrea Grado 3. Cinco (0,5%) presentaron colitis o diarrea inmunomediada, con una mediana del tiempo hasta el inicio de 21 días (rango: 12 días a 3,4 meses). De estos pacientes, uno tenía colitis o diarrea inmunomediada Grado 3, dos Grado 2 y dos Grado 1. La colitis o diarrea inmunomediadas mejoraron con la administración de corticosteroides en cuatro de estos pacientes, mientras que el quinto paciente falleció por progresión de la enfermedad antes de la resolución de la colitis. Endocrinopatías inmunorrelacionadas: Se han comunicado casos de trastornos inmunorrelacionados de la tiroides, insuficiencia suprarrenal y diabetes mellitus tipo 1, incluso cetoacidosis diabética, en pacientes tratados con Tecentriq. Monitorear a los pacientes en caso de signos y síntomas clínicos de endocrinopatías. Indique a sus pacientes que se contacten de inmediato con su médico para identificar signos o síntomas de hipofisitis, hipertiroidismo, hipotiroidismo, insuficiencia suprarrenal o diabetes mellitus tipo 1, incluida la cetoacidosis diabética. Hipofisitis: Se reportaron casos de hipofisitis en el 0,2% (1/523) de los pacientes con cáncer urotelial tratados con Tecentriq. Monitorear a los pacientes para identificar signos y síntomas de hipofisitis. Administrar terapia de reemplazo hormonal y corticosteroides según indicación clínica. Suspender Tecentriq en caso de Grados 2 o 3 y discontinuar en forma permanente en hipofisitis Grado 4. Trastornos de la tiroides: La función tiroidea fue evaluada en forma rutinaria sólo al inicio y al final del estudio. La función tiroidea debe monitorearse antes y periódicamente durante el tratamiento con Tecentriq. Los pacientes asintomáticos con resultados anormales en las pruebas de la función tiroidea pueden recibir Tecentriq. En el caso del hipotiroidismo sintomático, suspender Tecentriq e iniciar la terapia de reemplazo hormonal según sea necesario. Manejar los casos de hipotiroidismo aislado con terapia de reemplazo y sin corticosteroides. En el caso de hipertiroidismo sintomático suspender Tecentriq e iniciar un antitiroideo según sea necesario. Reiniciar el tratamiento con Tecentriq cuando los síntomas de hipotiroidismo o hipertiroidismo estén controlados y la función tiroidea esté mejorando. En los estudios clínicos, se reportaron casos de hipotiroidismo e hipertiroidismo en el 3,9% (77/1978) y el 1,0% (20/1978) de los pacientes, respectivamente. Carcinoma urotelial: En 523 pacientes con carcinoma urotelial tratados con Tecentriq, se informaron casos de hipotiroidismo en el 2,5% (13/523) de los pacientes. Un paciente presentó hipotiroidismo Grado 3 y 12 Grados 1-2. La mediana del tiempo hasta la primera aparición fue 5,4 meses (rango: 21 días a 11,3 meses). La concentración de hormona estimulante de la tiroides (TSH, por sus siglas en inglés) era elevada y estaba por encima del valor inicial en el 16% (21/131) de los pacientes con una medición en el seguimiento. El 0,6% (3/523) de los pacientes con carcinoma urotelial desarrollaron hipertiroidismo. De los tres con carcinoma urotelial, uno presentó Grado 2 y dos pacientes Grado 1. La mediana del tiempo hasta el inicio fue de 3,2 meses (rango: 1,4 a 5,8 meses). La TSH se redujo y estaba por debajo del valor inicial en el 3,8% (5/131) de los pacientes con una medición en el seguimiento. CPCNP: En 1027 pacientes con CPCNP que recibieron Tecentriq, el 4,2% (43/1027) desarrolló hipotiroidismo. Tres presentaron hipotiroidismo Grado 3 y 40 Grados 1-2. La mediana del tiempo hasta el inicio fue 4,8 meses (rango: 15 días a 31 meses). La TSH estaba elevada y por encima del valor inicial en el 17% (54/315) de los pacientes con una medición en el seguimiento. El 1,1% (11/1027) con CPCNP desarrolló hipertiroidismo, en 8 de Grado 2 y en 3 de Grado 1. La mediana del tiempo hasta el inicio fue de 4,9 meses (rango: 21 días a 31 meses). La TSH se redujo y estaba debajo de