TALZENNA
PFIZER
Inhibidor de las enzimas poli ADP-ribopolimerasa (PARP, por sus siglas en inglés).
Composición.
Cada cápsula de TALZENNA 0,25 mg contiene: Talazoparib(como Talazoparib Tosilato 0,363 mg) 0,25 mg. Excipientes: Celulosa microcristalina silicificada (tipo 50) 33,855 mg, Celulosa microcristalina silicificada (tipo 90) 50,782 mg, Hidroxipropilmetilcelulosa 37,238 mg, Dióxido de Titanio 0,748 mg, Óxido de hierro amarillo 0,015 mg y TekPrint Tinta Negra SW-9008 c.s. Cada cápsula de TALZENNA 1 mg contiene: Talazoparib(como Talazoparib Tosilato 1,453 mg) 1 mg. Excipientes: Celulosa microcristalina silicificada (tipo 50) 33,419 mg, Celulosa microcristalina silicificada (tipo 90) 50,128 mg, Hidroxipropilmetilcelulosa 37,211 mg, Dióxido de Titanio 0,657 mg, Óxido de hierro rojo 0,067 mg, Óxido de hierro amarillo 0,064 mg y TekPrint Tinta Negra SW-9008 c.s.
Farmacología.
Descripción: El talazoparib es un inhibidor de la enzima poliadenosina 5'-difosforibosa polimerasa (PARP) de los mamíferos. El nombre químico del tosilato de talazoparib es (8S,9R)-5-Fluoro-8-(4-fluorofenil)-9-(1- metiyl-1H-1,2,4-triazol-5-il)-2,7,8,9-tetrahidro-3H-pirido[4,3,2-de]ftalazina-3-ona 4- metilbencenosulfonato (1:1). La fórmula química del tosilato de talazoparib es C26H22F2N6O4S, y su peso molecular relativo es 552,56 Daltons. La estructura química del talazoparib se muestra a continuación: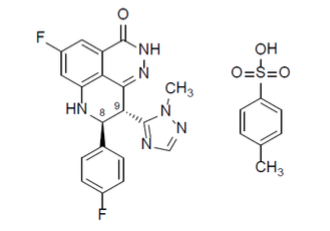
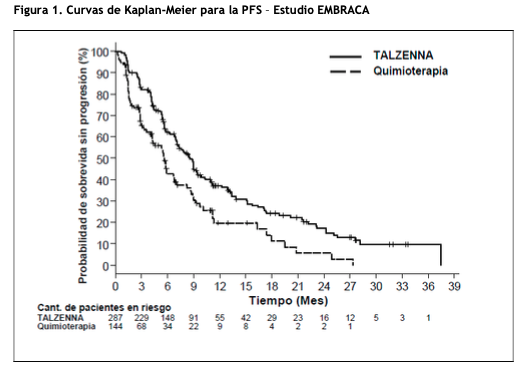
Mecanismo de Acción: El Talazoparibes un inhibidor de las enzimas poli ADP-ribosa polimerasa (PARP, por sus siglas en inglés), incluidas PARP1 y PARP2, que desempeñan un papel en la reparación del ADN. Los estudios in vitro con líneas celulares de cáncer que albergaban defectos en los genes de reparación del ADN, incluyendo BRCA 1 y 2, han demostrado que la citotoxicidad inducida por Talazoparibpuede implicar la inhibición de la actividad enzimática del PARP y la formación incrementada de complejos de ADN-PARP que resultan dañinas para el ADN, reducción de la proliferación celular y apoptosis. Se observó actividad antitumoral de Talazopariben modelos de tumores de cáncer de mama de xenoinjerto derivados de pacientes humanos que expresaban BRCA 1 y 2 mutados o de tipo salvaje. Propiedades farmacodinámicas: Electrofisiología Cardíaca: El efecto de Talazopariben la repolarización cardíaca se evaluó en 37 pacientes con tumores sólidos avanzados. El Talazoparibno tuvo una prolongación QTc grande (es decir, > 20 ms) a la dosis recomendada. Propiedades farmacocinéticas: Después de la administración oral de la dosis recomendada de 1 mg de TALZENNA una vez al día en pacientes, la media geométrica [% del coeficiente de variación (CV%)] del ABC y la concentración plasmática máxima observada (Cmax) de Talazopariben el estado estacionario fue de 208 (37%) ng.hr/mL y 16,4 (32%) ng/mL, respectivamente. La farmacocinética de Talazoparibes lineal desde 0,025 mg a 2 mg (2 veces la dosis recomendada). La mediana de la tasa de acumulación de Talazoparibdespués de la administración oral repetida de 1 mg una vez al día estuvo en el rango de 2,3 a 5,2. Las concentraciones plasmáticas de Talazoparibalcanzaron el estado estacionario entre las 2 a 3 semanas. Absorción: Después de la administración oral de Talazoparib, el tiempo medio hasta la Cmáx (Tmax) fue generalmente entre 1 y 2 horas después de la administración. Efecto de los alimentos: Después de una dosis oral única de 0,5 mg de TALZENNA con alimentos altos en grasa y en calorías (aproximadamente 800 a 1000 calorías con 150, 250 y 500 a 600 calorías de proteínas, carbohidratos y grasas, respectivamente), la Cmax media de Talazoparibse redujo en un 46%, la Tmax media se retrasó de 1 a 4 horas y el ABCinf no se vio afectada. Distribución: El volumen medio aparente de distribución de Talazoparibes de 420 L. In vitro, la unión a proteínas de Talazoparibes del 74% y es independiente de la concentración de Talazoparib. Eliminación: La vida media plasmática (± desviación estándar) de Talazoparibes de 90 (± 58) horas, y el clearance oral aparente medio (variabilidad entre los sujetos) es de 6,45 L/h (31,1%) en pacientes con cáncer. Metabolismo: Talazoparibsufre un metabolismo hepático mínimo. Las vías metabólicas identificadas de Talazopariben humanos incluyen monooxidación, deshidrogenación, conjugación con cisteína de mono-desfluoro-Talazopariby conjugación con glucurónido. Excreción: La excreción de Talazopariba través de la orina fue la principal vía de eliminación. Aproximadamente el 68,7% (54,6% sin cambios) de la dosis radiactiva [C14] total administrada de Talazoparibse recuperó en la orina, y el 19,7% (13,6% sin cambios) se recuperó en las heces. Poblaciones especiales: Edad, sexo y peso corporal: La edad (18 a 88 años), el sexo, la raza (361 caucásicos, 41 asiáticos, 16 de raza negra, 9 de otras y 63 no informados) y el peso corporal (36 a 162 kg) no tuvieron un efecto clínicamente relevante en la farmacocinética del Talazoparib. Población pediátrica: La farmacocinética de Talazoparibno se ha evaluado en pacientes ≤18 años. Insuficiencia hepática: La insuficiencia hepática leve (bilirrubina total ≤1,0 × ULN y AST > ULN, o bilirrubina total > 1,0 a 1,5 × ULN y cualquier AST) no tuvo efecto en la farmacocinética del Talazoparib. La farmacocinética del Talazoparibno se ha estudiado en pacientes con insuficiencia hepática moderada (bilirrubina total > 1,5 a 3,0 × y cualquier AST) o insuficiencia hepática grave (bilirrubina total > 3,0 × ULN y cualquier AST). Insuficiencia renal: El CL/F del Talazoparibse redujo en un 14,4% en pacientes con insuficiencia renal leve (CLcr 60 89 ml/min) y en un 37,1% en pacientes con insuficiencia renal moderada (CLcr 30 - 59 ml/min), en comparación con pacientes con función renal normal (CLcr ≥ 90 mL/min). La farmacocinética del Talazoparibno se ha estudiado en pacientes con insuficiencia renal grave (CLcr < 30 ml/min) o en pacientes que requieren hemodiálisis. Estudios de interacciones medicamentosas: Efecto de otras drogas sobre Talazoparib: Efecto de los inhibidores de la P-gp: la coadministración de inhibidores de la P-gp, incluidos amiodarona, carvedilol, claritromicina, itraconazol y verapamilo en estudios clínicos, aumentó la exposición a Talazopariben un 45% (ver Posología y forma de administración e Interacciones medicamentosas). La coadministración con inhibidores de la P-gp, que incluyen azitromicina, atorvastatina, diltiazem, felodipina, fluvoxamina y quercetina en estudios clínicos, aumentó la exposición a Talazopariben un 8% (ver Posología y forma de administración e Interacciones medicamentosas). Efecto de los inductores de P-gp: no se ha estudiado el efecto de los inductores de P-gp en la farmacocinética de Talazoparib. Efecto de los inhibidores de la BCRP: No se ha estudiado el efecto de los inhibidores de BCRP en la farmacocinética de Talazoparib. La coadministración con inhibidores de BCRP puede aumentar la exposición al Talazoparib(ver Interacciones Medicamentosas). Efecto de los agentes reductores del ácido en talazoparib: la coadministración de agentes reductores del ácido, incluidos los inhibidores de la bomba de protones (PPI), los antagonistas del receptor de histamina 2 (H2RA) u otros agentes reductores del ácido, no tienen efecto sobre la absorción de Talazoparib. Estudios in vitro. El Talazoparibes un sustrato de los transportadores de P-gp y BCRP. El Talazoparibno es un sustrato del polipéptido transportador de aniones orgánico [OATP] 1B1, OATP1B3, transportador catiónico orgánico [OCT] 1, OCT2, transportador de aniones orgánico [OAT] 1, OAT3, bomba de exportación de sales biliares [BSEP], multidroga y extrusión de toxina [MATE] 1, y MATE2-K. El Talazoparibno es un inhibidor del citocromo (CYP) 1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 o CYP3A4/5, o inductor de CYP1A2, CYP2B6 o CYP3A4. El Talazoparibno es un inhibidor de los transportadores que incluyen P gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, BSEP, MATE1 y MATE2-K. El Talazoparibno es un inhibidor de las isoformas de uridina-difosfato glucuronosiltransferasa (UGT) (1A1, 1A4, 1A6, 1A9, 2B7 y 2B15). Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad: No se han realizado estudios de carcinogenicidad con Talazoparib. El Talazoparibfue clastogénico en un ensayo de aberración cromosómica in vitro en linfocitos de sangre periférica humana y en un ensayo in vivo de micronúcleos de médula ósea en ratas. Esta clastogenicidad es consistente con la inestabilidad genómica resultante de la farmacología primaria de Talazoparib, lo que indica el potencial de genotoxicidad en humanos. Talazoparibno fue mutagénico en una prueba de mutación reversa bacteriana (Ames). No se han realizado estudios de fertilidad en animales con Talazoparib. En estudios de toxicidad a dosis repetidas de hasta 3 meses de duración, los hallazgos relacionados con el Talazopariben el testículo y el epidídimo a dosis ≥0,04 mg/kg/día en ratas y ≥0,01 mg/kg/día en perros incluyeron disminución del peso de los órganos, restos celulares luminales, esperma reducido, y degeneración/atrofia. Estas dosis en ratas y perros resultaron en aproximadamente 1,0 veces y 0,2 veces, respectivamente, la exposición (ABC) en humanos a la dosis recomendada. Se observó atresia folicular del ovario en ratas con dosis ≥1 mg/kg/día de Talazoparib, aproximadamente 9,5 veces el ABC en humanos a la dosis recomendada. Estudios Clínicos: Estudio EMBRACA (NCT01945775): Cáncer de mama, localmente avanzado o metastásico, con mutaciones deletéreas germinales de BRCA posibles o confirmadas (gBRCAm) negativos para HER2: EMBRACA (NCT01945775) fue un estudio abierto en el que se aleatorizó a pacientes (N=431) con cáncer de mama localmente avanzado o metastásico con gBRCAm negativos para HER2 en una proporción 2:1 para recibir TALZENNA 1 mg o la quimioterapia de elección del profesional médico (capecitabina, eribulina, gemcitabina o vinorelbina) hasta la progresión de la enfermedad o una toxicidad inaceptable. La aleatorización se estratificó según la quimioterapia previa para la enfermedad metastásica (0 frente a 1, 2 o 3), según el estado triple negativo de la enfermedad (cáncer de mama triple negativo [TNBC] frente a cáncer no TNBC) y los antecedentes de metástasis del sistema nervioso central (CNS; sí frente a no). Los pacientes recibieron no más de 3 regímenes quimioterapéuticos citotóxicos previos para la enfermedad metastásica o avanzada localmente. Se requirió a los pacientes tratamiento previo con una antraciclina o un taxano (excepto en caso de contraindicación) a modo de tratamiento neoadyuvante, adyuvante o para las metastásis. Se permitió el tratamiento de primera línea para la enfermedad avanzada o metastásica sin quimioterapia adyuvante previa si el investigador determinaba que 1 de las 4 opciones de quimioterapia del grupo de control sería una opción de tratamiento adecuada para el paciente. A los pacientes que recibieron tratamiento anterior con un compuesto de platino para la enfermedad avanzada se les requirió ausencia de signos de progresión de la enfermedad durante el tratamiento con el compuesto de platino. No se permitió ningún tratamiento anterior con inhibidores de la PARP. De los 431 pacientes aleatorizados en el estudio EMBRACA, en 408 (95%) se confirmó una gBRCAm deletérea posible o confirmada a nivel central usando un ensayo del estudio clínico; de estos, 354 (82%) se confirmaron con BRACAnalysis CDx®. El estado de la mutación de BRCA (positivo para el gen de susceptibilidad al cáncer de mama 1 [BRCA1] o el gen de susceptibilidad al cáncer de mama 2 [BRCA2]) fue similar en ambos grupos de tratamiento. La mediana de edad de los pacientes tratados con TALZENNA fue de 45 años (intervalo de 27 a 84) y entre los pacientes tratados con quimioterapia, de 50 años (intervalo de 24 a 88). Entre todos los pacientes aleatorizados, 1% frente a 2% eran de sexo masculino, 67% frente a 75% eran blancos; 11% frente a 11% eran asiáticos y 4% frente a 1% eran negros o afroamericanos en los grupos de TALZENNA y de quimioterapia, respectivamente. Prácticamente todos los pacientes (98%) de los dos grupos presentaban un estado funcional según el Grupo Cooperativo de Oncología del Este (Eastern Cooperative Oncology Group, ECOG) de 0 o 1. Aproximadamente el 56% presentaba enfermedad positiva para receptores de estrógenos o positiva para receptores de progesterona; el 44% de los pacientes presentaba la forma triple negativa de la enfermedad y las proporciones en ambos grupos de tratamiento estuvieron equilibradas. El quince (15%) de los pacientes en el grupo de TALZENNA y el 14% de los pacientes en el grupo de quimioterapia tenían antecedentes de metástasis del CNS. El noventa y uno (91%) de los pacientes del grupo de TALZENNA había recibido tratamiento previo con taxanos, y el 85% había recibido tratamiento previo con antraciclina para cualquier cuadro. El dieciséis (16%) de los pacientes del grupo de TALZENNA y el 21% de los pacientes del grupo de quimioterapia habían recibido tratamiento anterior con un compuesto de platino para cualquier cuadro. La mediana de la cantidad de regímenes citotóxicos anteriores en los pacientes con cáncer de mama avanzado fue uno; el 38% no recibió regímenes citotóxicos anteriores para la enfermedad avanzada o metastásica, el 37% recibió uno, el 20% recibió dos y el 5% recibió tres o más regímenes citotóxicos anteriores. La medida del criterio de eficacia principal fue la supervivencia sin progresión (progression-free survival, PFS) según los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST), versión 1.1, determinada por revisión central independiente a ciego (blinded independent central review, BICR). Se demostró una mejora significativa, desde el punto de vista estadístico, en la PFS con TALZENNA en comparación con la quimioterapia. Un análisis de sensibilidad de la PFS determinada por el investigador fue coherente con los resultados de PFS determinada por el BICR. Se observaron resultados coherentes en cuanto a PFS entre los subgrupos de pacientes definidos por los factores de estratificación del estudio (línea de tratamiento, estado de TNBC y antecedentes de metástasis en el CNS). Los datos de sobrevida general (overall survival, OS) no eran definitivos al momento del análisis de PFS (38% de los pacientes habían fallecido). Los datos de eficacia del estudio EMBRACA se resumen en la Tabla 1 y las curvas de PFS de Kaplan-Meier se muestran en la Figura 1.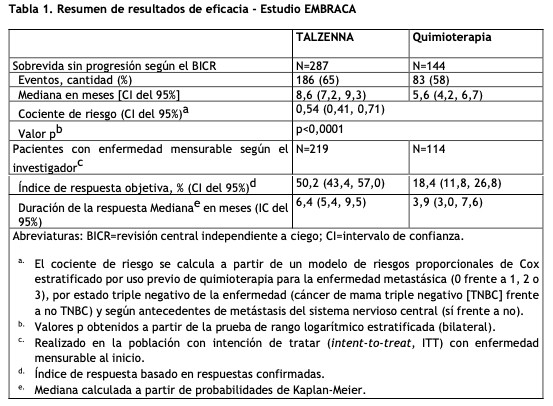
Indicaciones.
TALZENNA está indicado para el tratamiento de pacientes con cáncer de mama, metastásico o localmente avanzado, que presenten mutación deletérea germinal posible o confirmada del gen de susceptibilidad para el cáncer de mama (BRCA) (gBRCAm) y negatividad para el receptor 2 del factor de crecimiento epidérmico humano (HER2).
Dosificación.
La selección de pacientes para el tratamiento con Talzenna está basada en la presencia de mutaciones germinales de BRCA determinadas por un método de diagnóstico validado. Dosis recomendada y esquema: La dosis recomendada de TALZENNA es de 1 mg por vía oral una vez al día, con o sin alimentos. La cápsula de 0,25 mg está disponible para la reducción de la dosis. Los pacientes deben recibir tratamiento hasta que se produzca una progresión de la enfermedad o se produzca una toxicidad inaceptable.Las cápsulas duras deben tragarse enteras y no deben abrirse o disolverse. Si el paciente vomita o pierde una dosis, no debe tomarse una dosis adicional. La siguiente dosis prescripta debe tomarse a la hora habitual. Modificación de la dosis debido a reacciones adversas: Para controlar las reacciones adversas, considere la interrupción del tratamiento con o sin reducción de la dosis según la gravedad y el cuadro clínico. Las reducciones de dosis recomendadas se indican en la Tabla 2 y la Tabla 3. El tratamiento con TALZENNA debe interrumpirse si se requieren más de tres reducciones de dosis.
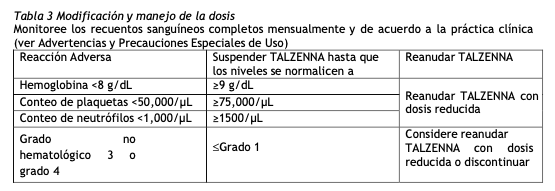
Modificación de la dosis en pacientes con insuficiencia renal: Para los pacientes con insuficiencia renal moderada (CLcr 30 - 59 ml/min), la dosis recomendada de TALZENNA es de 0,75 mg una vez al día (ver Uso en Poblaciones Específicas y Características Farmacológicas). Modificación de la dosis para administración con inhibidores de la glicoproteína P (P-gp): Reduzca la dosis de TALZENNA a 0,75 mg una vez al día cuando se administra conjuntamente con ciertos inhibidores de la P-gp. Para obtener información adicional sobre la interacción de los inhibidores de la P-gp, (ver Interacciones con medicamentos). Cuando se suspende el inhibidor de la P-gp, aumente la dosis de TALZENNA (después de 3 a 5 vidas medias del inhibidor de la P-gp) a la dosis utilizada antes del inicio del inhibidor de la P-gp (ver Interacciones con otros medicamentos).
Contraindicaciones.
Hipersensibilidad al principio activo o a algunos de los excipientes.
Reacciones adversas.
Las siguientes reacciones adversas clínicamente significativas se describen en otras secciones del prospecto: Síndrome mielodisplásico/leucemia mieloide aguda (ver Advertencia y Precauciones de Especiales de Uso). Mielosupresión (ver Advertencias y Precauciones Especiales de Uso). Experiencia en estudios clínicos: Debido a que los estudios clínicos se realizan en condiciones muy diversas, las tasas de reacciones adversas observadas en los estudios clínicos de un medicamento no pueden compararse directamente con las tasas en los estudios clínicos de otro fármaco y es posible que no reflejen las tasas observadas en la práctica. Tratamiento del cáncerdemama local avanzado o metastásicogBRCAm HER2 negativo: Embraca: La seguridad de TALZENNA como monoterapia se evaluó en pacientes con cáncer de mama localmente avanzado o metastásico c o n e l gen BRCA mutado, HER2 negativo que habían recibido previamente no más de 3 líneas de quimioterapia para el tratamiento de enfermedad local avanzada/metastásica. EMBRACA fue un estudio aleatorizado, abierto, multicéntrico, en el que 412 pacientes recibieron TALZENNA 1 mg una vez al día (n = 286) o un agente de quimioterapia (capecitabina, eribulina, gemcitabina o vinorelbina) a elección del proveedor de atención médica (n=126) hasta progresión de la enfermedad o toxicidad inaceptable. La duración media del tratamiento del estudio fue de 6,1 meses en pacientes que recibieron TALZENNA y de 3,9 meses en pacientes que recibieron quimioterapia. Las interrupciones de las dosis debidas a una reacción adversa de cualquier grado ocurrieron en el 65% de los pacientes que recibieron TALZENNA y en el 50% de los que recibieron quimioterapia; las reducciones de dosis debidas a cualquier causa se produjeron en el 53% de los pacientes con TALZENNA y en el 40% de los pacientes con quimioterapia. La interrupción permanente debida a reacciones adversas se produjo en el 5% de los pacientes con TALZENNA y en el 6% de los pacientes con quimioterapia. Las tablas 4 y 5 resumen las reacciones adversas y anormalidades de laboratorio más comunes, respectivamente, en pacientes tratados con TALZENNA o quimioterapia en el estudio EMBRACA.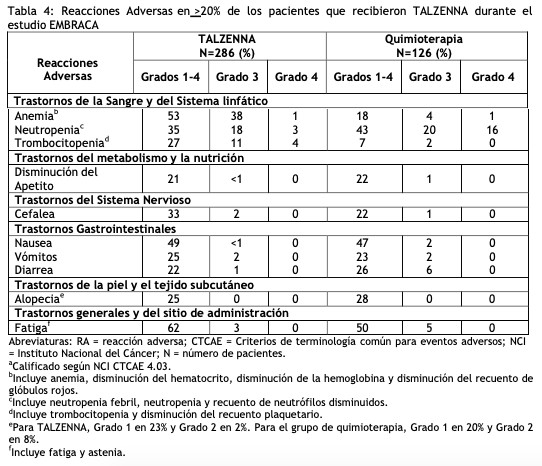
Se identificaron las siguientes reacciones adversas en < 20% de los 286 pacientes que recibieron TALZENNA y, por lo tanto, no se incluyeron en la Tabla 4: dolor abdominal (19%), mareo (17%), leucopenia (17%), disgeusia (10%), dispepsia (10%), estomatitis (8%) y linfopenia (7%).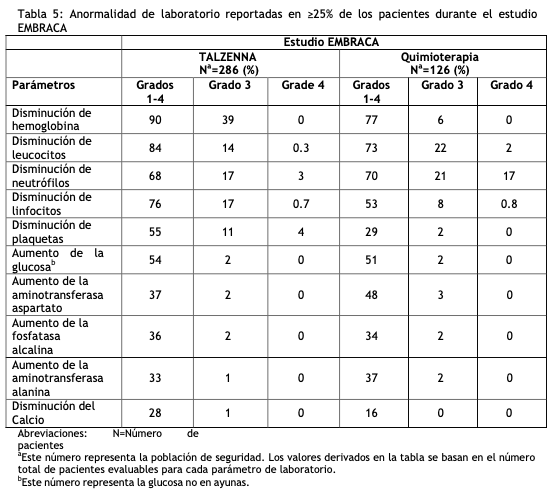
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia al siguiente link: http://sistemas.anamt.gov.ar/aplicaciones_net/fvg_eventos_adversos_nuevo/index.html
Advertencias.
Síndrome mielodisplásico/leucemia mieloide aguda: El síndrome mielodisplásico/leucemia mieloide aguda (MDS/AML por sus siglas en inglés) se ha reportado en pacientes que recibieron TALZENNA. En general, se ha informado MDS/AML en 2 de 584 pacientes (0,3%) con tumores sólidos tratados con TALZENNA en estudios clínicos. La duración del tratamiento con TALZENNA en estos dos pacientes antes de desarrollar MDS/AML fue de 4 meses y 24 meses, respectivamente. Ambos pacientes habían recibido quimioterapia previa con agentes de platino y u otros agentes que dañan el ADN, incluida la radioterapia. No inicie el tratamiento con TALZENNA hasta que los pacientes se hayan recuperado adecuadamente de la toxicidad hematológica causada por la quimioterapia previa. Monitoree los recuentos sanguíneos completos para detectar citopenia al inicio y mensualmente a partir de entonces. Para toxicidades hematológicas prolongadas, interrumpa TALZENNA y monitoree los recuentos sanguíneos semanalmente hasta la recuperación. Si los niveles no se han recuperado después de 4 semanas, remita al paciente a un hematólogo para que realice más investigaciones, incluidos análisis de médula ósea y una muestra de sangre para citogenética. Si se confirma MDS/AML, suspenda TALZENNA. Mielosupresión: Se notificó mielosupresión consistente en anemia, leucopenia neutropenia y/o trombocitopenia en pacientes tratados con TALZENNA (ver Reacciones Adversas). Se informaron anemia, neutropenia y trombocitopenia de Grado ≥3 en 39%, 21% y 15%, respectivamente, de los pacientes que recibieron TALZENNA. La descontinuación debida a anemia, neutropenia y trombocitopenia ocurrió, respectivamente, en el 0,7%, el 0,3% y el 0,3% de los pacientes. Monitoree el hemograma completo para detectar citopenia al inicio y mensualmente a partir de entonces. No inicie TALZENNA hasta que los pacientes se hayan recuperado adecuadamente de la toxicidad hematológica causada por la terapia previa. Si esto ocurre, se recomiendan modificaciones de la dosis (interrupción de la dosis con o sin reducción de la dosis) (ver Modificaciones de la Dosis). Toxicidad embrio-fetal: Basado en su mecanismo de acción y los hallazgos de los datos en animales, TALZENNA puede causar daño fetal cuando se administra a una mujer embarazada. En un estudio de reproducción en animales, la administración de Talazopariba ratas preñadas durante el período de organogénesis causó malformaciones fetales y variaciones estructurales esqueléticas, y muerte embrio-fetal a exposiciones que fueron 0,24 veces el área bajo la curva de concentración-tiempo (ABC) en pacientes que recibieron la dosis recomendada en humanos de 1 mg al día. Se debe informar a las mujeres embarazadas y a las mujeres con potencial reproductivo del riesgo potencial para el feto. Aconseje a las mujeres con potencial reproductivo para que usen anticonceptivos eficaces durante el tratamiento y durante al menos 7 meses después de la última dosis de TALZENNA (ver Uso en Poblaciones Específicas). En base a los hallazgos de toxicidad genética y estudios de reproducción en animales, informe a los pacientes masculinos con parejas femeninas con potencial reproductivo o que están embarazadas para que usen un método anticonceptivo eficaz durante el tratamiento y durante al menos 4 meses después de la última dosis de TALZENNA (ver Fertilidad, embarazo y Lactancia).
Interacciones.
Efecto de los medicamentos que afectan a TALZENNA: Efecto de los inhibidores de la P-gp: La coadministración con inhibidores de la P-gp puede aumentar la exposición a Talazoparib. En los estudios clínicos, la administración concomitante de inhibidores de la P-gp, incluyendo amiodarona, carvedilol, claritromicina, itraconazol y verapamilo, produjo un aumento aproximado de 45% en la exposición a Talazopariby un aumento en la tasa de reducción de la dosis de TALZENNA. Si no se puede evitar la administración conjunta de TALZENNA con estos inhibidores de la P-gp, reduzca la dosis de TALZENNA (ver Posología y forma de administración). Cuando se suspende el inhibidor de P-gp, aumente la dosis de TALZENNA (después de 3 a 5 semividas del inhibidor) a la dosis utilizada antes del inicio del inhibidor de P-gp (ver Posología y forma de administración y Características Farmacológicas). Cuando se administre conjuntamente TALZENNA con inhibidores de la P-gp no incluidos en la lista anterior, vigile a los pacientes para detectar posibles reacciones adversas (ver Posología y forma de administración y Características Farmacológicas). Efecto de los inhibidores del BCRP: La coadministración con inhibidores del BCRP puede aumentar la exposición a Talazoparib. Si no se puede evitar la administración conjunta, vigile a los pacientes para detectar posibles reacciones adversas cuando realice la coadministración (ver Características Farmacológicas). Uso en poblaciones especificas: Fertilidad, embarazo y lactancia Embarazo: Resumen de Riesgos: En base a los hallazgos de estudios en animales y su mecanismo de acción (ver Características Farmacológicas), TALZENNA puede causar daños embrio-fetales cuando se administra a una mujer embarazada. No hay datos disponibles sobre el uso de TALZENNA en mujeres embarazadas para informar un riesgo asociado con el fármaco. En un estudio de reproducción en animales, la administración de Talazopariba ratas preñadas durante el período de organogénesis causó malformaciones fetales y variaciones estructurales esqueléticas y muerte embrionaria fetal a exposiciones maternas que fue 0,24 veces mayor que el ABC en pacientes que recibieron la dosis recomendada de 1 mg al día (ver Datos). Se debe informar a las mujeres embarazadas y a las mujeres con potencial reproductivo del riesgo potencial para el feto. Datos: Datos en animales: En un estudio sobre toxicidad del desarrollo embrio-fetal, las ratas preñadas recibieron dosis orales de 0,015; 0,05 y 0,15 mg/kg/día de Talazoparibdurante el período de organogénesis. El Talazoparibcausó muerte fetal embrionaria en dosis ≥0,015 mg kg/día (aproximadamente 0,24 veces el ABC en pacientes a la dosis recomendada). Una dosis de 0,015 mg/kg/día causó una disminución de los pesos corporales fetales y un aumento en la incidencia de malformaciones fetales (abultamiento ocular deprimido, ojo pequeño, esternebra dividida y arco vertebral cervical fusionado) y variaciones estructurales que incluyen deformaciones u osificación incompleta de la esternebra, cráneo, costilla y vértebra. Lactancia: Resumen de Riesgos: No hay datos sobre la presencia de Talazopariben la leche humana, los efectos de la droga en la producción de leche, o los efectos de la droga en el niño amamantado. Debido a la posibilidad de reacciones adversas graves en un niño amamantado con Talazoparib, se aconseja a las mujeres lactantes que no amamanten durante el tratamiento con TALZENNA y durante al menos 1 mes después de la dosis final. Potencial reproductivo en hombres y mujeres: Pruebas de embarazo: Se recomienda a las mujeres con potencial reproductivo, realizar una prueba de embarazo antes de comenzar con el tratamiento de TALZENNA. Anticoncepción: Mujeres: TALZENNA puede ocasionar daños al feto, cuando se administra a una mujer embarazada (ver Fertilidad, embarazo y lactancia). Se debe informar a las mujeres con potencial reproductivo, para que utilicen un método anticonceptivo eficaz durante el tratamiento con TALZENNA y durante al menos 7 meses después de la última dosis. Hombres: En base a los estudios de genotoxicidad y reproducción animal, se aconseja a los pacientes masculinos con parejas femeninas con potencial reproductivo y las parejas embarazadas que utilicen métodos anticonceptivos eficaces durante el tratamiento con TALZENNA y durante al menos 4 meses después de la última dosis. Infertilidad: Hombres: En base a los estudios animales, TALZENNA puede ocasionar infertilidad en hombres con potencial reproductivo (ver Carcinogénesis, mutagénesis, deterioro de la fertilidad). Uso en pediatría: La seguridad y eficacia de TALZENNA no se ha establecido en pacientes pediátricos. Uso en Geriatría: En los estudios clínicos de TALZENNA que incluyeron a 494 pacientes con tumores sólidos avanzados que recibieron TALZENNA 1 mg al día como monoterapia, 85 (17%) pacientes tenían ≥65 años de edad, y esto incluía 19 (4%) pacientes que tenían ≥75 años de edad. Hubo 5 pacientes ≥85 años. No se observaron diferencias generales en la seguridad o la eficacia de TALZENNA entre estos pacientes y pacientes más jóvenes, pero no se puede descartar una mayor sensibilidad de algunas personas mayores. Insuficiencia renal: Se debe reducir la dosis recomendada de TALZENNA en pacientes con insuficiencia renal moderada (CLcr 30-59 ml/min) (ver Posología y forma de administración y Características Farmacológicas). No se requiere ajuste de la dosis en pacientes con insuficiencia renal leve (CLcr 60-89 ml/min). TALZENNA no se ha estudiado en pacientes con insuficiencia renal grave (CLcr < 30 ml/min) o en pacientes que requieren hemodiálisis (ver Características Farmacológicas). Insuficiencia hepática: TALZENNA no se ha estudiado en pacientes con insuficiencia hepática moderada (bilirrubina total > 1,5 a 3,0 veces el límite superior normal [LSN] y cualquier aspartato aminotransferasa [AST]) o insuficiencia hepática grave (bilirrubina total > 3,0 × ULN y cualquier AST). No se requiere un ajuste de la dosis en pacientes con insuficiencia hepática leve (bilirrubina total ≤1 × ULN y AST > LSN, o bilirrubina total > 1,0 a 1,5 × LSN y cualquier AST) (ver Características Farmacológicas).
Conservación.
Conservar a una temperatura entre 20°C y 25 °C.
Sobredosificación.
No hay un tratamiento específico en caso de sobredosis de TALZENNA y no se han establecido los síntomas de sobredosis. En caso de sobredosis, suspenda el tratamiento con TALZENNA, considere lavado gástrico, siga las medidas de apoyo generales y trate los síntomas. Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse a los Centros de Toxicología: Hospital de Pediatría Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital A. Posadas: (011) 4658-7777 / 4654-6648.
Presentación.
TALZENNA 0,25 mg se presenta en envases que contienen 30 cápsulas. TALZENNA 1 mg se presenta en envases que contienen 30 cápsulas.
Revisión.
Mayo 2019. LPD: 16/Oct/2018.