SPRYCEL®
BRISTOL-M.S.
Inhibidor de quinasa.
Composición.
Cada comprimido recubierto contiene: x 20 mg: Dasatinib (como monohidrato) 20 mg. Lactosa monohidrato 27 mg. Celulosa microcristalina 27 mg. Croscarmelosa sódica 3,2 mg. Hidroxipropilcelulosa 2,4 mg. Estearato de magnesio 0,4 mg. Opadry blanco, YS-1-18177-A (hipromelosa, dióxido de titanio, polietilenglicol) 3,2 mg. Cada comprimido recubierto contiene: x 50 mg: Dasatinib (como monohidrato) 50 mg. Lactosa monohidrato 67,5 mg. Celulosa microcristalina 67,5 mg. Croscarmelosa sódica 8 mg. Hidroxipropilcelulosa 6 mg. Estearato de magnesio 1 mg. Opadry blanco, YS-1-18177-A (hipromelosa, dióxido de titanio, polietilenglicol) 7 mg. Cada comprimido recubierto contiene: x 70 mg: Dasatinib (como monohidrato) 70 mg. Lactosa monohidrato 94,5 mg. Celulosa microcristalina 94,5 mg. Croscarmelosa sódica 11,2 mg. Hidroxipropilcelulosa 8,4 mg. Estearato de magnesio 1,4 mg. Opadry blanco, YS-1-18177-A (hipromelosa, dióxido de titanio, polietilenglicol) 8,4 mg. Cada comprimido recubierto contiene: x 100 mg: Dasatinib (como monohidrato) 100 mg. Lactosa monohidrato 135 mg. Celulosa microcristalina 135 mg. Croscarmelosa sódica 16 mg. Hidroxipropilcelulosa 12 mg. Estearato de magnesio 2 mg. Opadry blanco, YS-1-18177-A (hipromelosa, dióxido de titanio, polietilenglicol) 12 mg.
Farmacología.
Descripción: SPRYCEL (dasatinib) es un inhibidor de quinasa. El nombre químico de dasatinib es monohidrato de N-(2-cloro-6-metilfenil)-2-[[6-[4-(2-hidroxietil)-1-piperazinil]-2-metil-4-pirimidinil]amino]-5-tiazolecarboxamida. La fórmula molecular es C22H26ClN7O2S•H2O, que corresponde a un peso de la fórmula de 506,02 (monohidrato). La base libre anhidra tiene un peso molecular de 488,01. Dasatinib tiene la siguiente estructura química: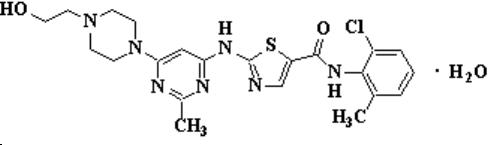
Dasatinib es un polvo blanco a casi blanco. El fármaco es insoluble en agua y apenas soluble en etanol y metanol. Farmacología: Mecanismo de Acción: Dasatinib, a concentraciones nanomolares, inhibe las siguientes quinasas: BCR-ABL, familia SRC (SRC, LCK, YES, FYN), c-KIT, EPHA2, y PDGFRb. Basado en estudios modelo, se predice que dasatinib se une a múltiples conformaciones de la quinasa ABL. In vitro, dasatinib resultó activo en las líneas de células leucémicas lo que representa variantes de la enfermedad sensible y resistente al mesilato de imatinib. Dasatinib inhibió el crecimiento de líneas de células de leucemia mieloide crónica, LMC, y leucemia linfoblástica aguda, LLA, con sobreexpresión de BCR-ABL. Bajo estas condiciones de los ensayos, dasatinib pudo superar la resistencia a imatinib originada a partir de las mutaciones de los dominios BCR-ABL quinasa, activación de vías alternativas de señalización con participación de las quinasas de la familia SRC (LYN, HCK), y sobreexpresión de genes de resistencia a fármacos múltiples. Farmacocinética: Absorción: Se observan concentraciones máximas en plasma (Cmáx) de dasatinib entre 0,5 y 6 horas (Tmáx) después de la administración oral. Dasatinib muestra aumentos proporcionales a la dosis en el área bajo la curva (Area Under Curve, AUC) y características de eliminación lineal en el rango de dosis de 15 a 240 mg/día. La vida media terminal promedio general de dasatinib es de 3 a 5 horas. Los datos de un ensayo de 54 sujetos sanos que recibieron una dosis única de 100 mg de dasatinib 30 minutos después del consumo de un alimento con alto contenido de grasa mostraron un aumento del 14% en la AUC promedio de dasatinib. Los efectos alimentarios observados no fueron clínicamente relevantes. Distribución: En los pacientes, dasatinib tiene un volumen de distribución aparente de 2505 L, lo que sugiere que el fármaco se distribuye extensamente en el espacio extravascular. La unión de dasatinib y su metabolito activo a las proteínas plasmáticas humanas in vitro fue de aproximadamente el 96% y 93%, respectivamente, independientemente de la concentración en el rango de 100 a 500 ng/mL. Metabolismo: Dasatinib se metaboliza extensamente en seres humanos, principalmente por la enzima 3A4 del citocromo P450. CYP3A4 fue la enzima primaria responsable de la formación del metabolito activo. Las enzimas uridina difosfato-glucuronosiltransferasa (UGT) y monooxigenasa 3 (FMO-3) que contiene flavina también participan de la formación de los metabolitos de dasatinib. La exposición del metabolito activo, que es equipotente a dasatinib, representa aproximadamente el 5% de la AUC de dasatinib. Esto indica que el metabolito activo de dasatinib probablemente no desempeñe un papel importante en la farmacología observada del fármaco. Dasatinib también presentó varios metabolitos oxidativos inactivos. Dasatinib es un inhibidor débil dependiente del tiempo de CYP3A4. A concentraciones clínicamente relevantes, dasatinib no inhibe CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 o 2E1. Dasatinib no es un inductor de enzimas CYP humanas. Eliminación: La eliminación se produce principalmente por la materia fecal. Después de una dosis oral única de dasatinib marcado con [14C], aproximadamente el 4% y 85% de la radiactividad administrada se recuperó en la orina y la materia fecal, respectivamente, dentro de los 10 días. El dasatinib sin alteraciones representó el 0,1% y el 19% de la dosis administrada en la orina y materia fecal, respectivamente, y el resto de la dosis consistió en metabolitos. Efectos de la Edad y el Género: Los análisis farmacocinéticos de los datos demográficos indican que no hay efectos clínicamente relevantes de edad y género en la farmacocinética de dasatinib. Disfunción Hepática: Las dosis de dasatinib de 50 mg y 20 mg se evaluaron en ocho pacientes con disfunción hepática moderada (clase B de Child-Pugh) y en siete pacientes con disfunción hepática grave (clase C de Child-Pugh). También se evaluaron individuos de control con función hepática normal (n=15) y recibieron una dosis de 70 mg de dasatinib. En comparación con los sujetos con función hepática normal, los pacientes con disfunción hepática moderada tuvieron disminuciones en los valores Cmáx y AUC normalizados según la dosis de 47% y 8%, respectivamente. Los pacientes con disfunción hepática grave tuvieron una disminución en los valores de Cmáx normalizados según la dosis de 43% y en los valores de AUC normalizados según la dosis de 28%, en comparación con los individuos de control normales. Estas diferencias en Cmáx y AUC no son clínicamente relevantes. El ajuste de la dosis no es necesario en pacientes con disfunción hepática. Estudios clínicos: LMC en Fase Crónica, recientemente diagnosticada: Se realizó un estudio abierto, multicéntrico, internacional, randomizado en pacientes adultos con LMC en fase crónica, recientemente diagnosticada. En total se randomizó a 519 pacientes para recibir SPRYCEL 100 mg una vez por día o imatinib 400 mg una vez por día. En este ensayo se incluyeron pacientes con antecedentes de patologías cardíacas, excepto aquellos que habían sufrido infarto de miocardio dentro de los últimos 6 meses, insuficiencia cardíaca congestiva dentro de los últimos 3 meses, arritmias significativas o prolongación del intervalo QTc. El criterio primario de valoración fue la tasa de respuesta citogenética completa confirmada (confirmed complete cytogenetic response, CCyR) a los 12 meses. La CCyR confirmada se definió como una CCyR observada en dos ocasiones consecutivas (con un intervalo de al menos 28 días). La mediana de la edad fue 46 años en el grupo de SPRYCEL y 49 años en los grupos de imatinib, con un 10% y un 11% de los pacientes con 65 años de edad o más, respectivamente. En ambos grupos había ligeramente más pacientes hombres que mujeres (59% frente a 41%). De todos los pacientes, 53% eran de raza caucásica y 39% eran asiáticos. En el nivel basal, la distribución de los puntajes de Hasford fue similar en los grupos de tratamiento con SPRYCEL e imatinib (riesgo bajo: 33% y 34%; riesgo intermedio: 48% y 47%; riesgo alto: 19% y 19%, respectivamente). Con un mínimo de 12 meses de seguimiento, el 85% de los pacientes randomizados a SPRYCEL y el 81% de los pacientes randomizados a imatinib aún seguían en el estudio. Con un mínimo de 24 meses de seguimiento, el 77% de los pacientes randomizados para recibir SPRYCEL y el 75% de los pacientes randomizados para recibir imatinib aún se encontraban en el estudio, y con un mínimo de 60 meses de seguimiento, el 61% y 62% de los pacientes, respectivamente, aún se encontraban bajo tratamiento al momento del cierre del estudio. Los resultados de la eficacia se resumen en la Tabla 9.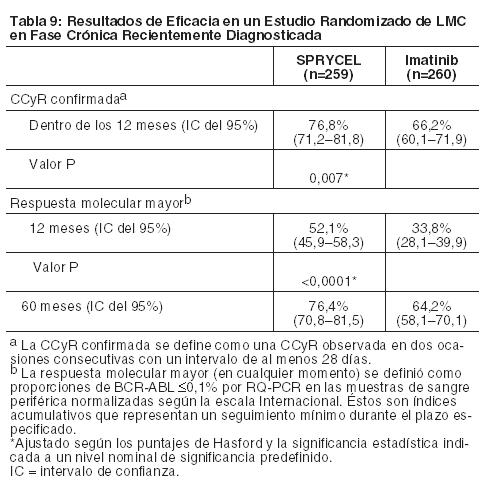
Las CCyR confirmadas dentro de los 24, 36 y 60 meses para las ramas de SPRYCEL versus imatinib fueron 80% versus 74%, 83% versus 77%, y 83% versus 79%, respectivamente. Las MMR a los 24 y 36 meses para las ramas de SPRYCEL versus imatinib fueron 65% versus 50% y 69% versus 56%, respectivamente. Luego de 60 meses de seguimiento, la mediana del tiempo hasta la CCyR confirmada fue de 3,1 meses en 215 pacientes que respondieron a SPRYCEL y de 5,8 meses en 204 pacientes que respondieron a imatinib. La mediana del tiempo hasta la MMR luego de 60 meses de seguimiento fue de 9,3 meses en 198 pacientes que respondieron a SPRYCEL y de 15,0 meses en 167 pacientes que respondieron a imatinib. Al cabo de 60 36 meses, en el grupo de dasatinib, 8 pacientes (3%) tuvieron progresión a la fase acelerada o una crisis blástica, mientras que en el grupo de imatinib, 15 pacientes (6%) tuvieron progresión a la fase acelerada o a una crisis blástica. Las tasas de sobrevida a los 60 meses estimadas para pacientes tratados con SPRYCEL e imatinib fueron 90,9% (IC: 86,6%-93,8%) y 89,6% (IC: 85,2%-92,8%), respectivamente. Sobre la base de los datos 5 años después de que el último paciente fuera enrolado en el ensayo, se sabía que el 83% y 77% de los pacientes estaban vivos en los grupos de tratamiento con dasatinib e imatinib, respectivamente, se sabía que el 10% habían muerto en ambos grupos de tratamiento, y el 7% y 13% tenían un estado de sobrevida desconocido en los grupos de tratamiento con dasatinib e imatinib, respectivamente. En el seguimiento de 60 meses, en la rama de SPRYCEL, el índice de MMR en cualquier momento en cada grupo de riesgo, determinado por el puntaje de Hasford era 90% (riesgo bajo), 71% (riesgo intermedio) y 67% (riesgo alto). En la rama de imatinib, el índice de MMR en cualquier momento en cada grupo de riesgo, determinado por el puntaje de Hasford, fue 69% (riesgo bajo), 65% (riesgo intermedio) y 54% (riesgo alto). Se llevó a cabo la secuenciación de BCR-ABL en muestras de sangre de pacientes del ensayo recientemente diagnosticados que discontinuaron la terapia con dasatinib o imatinib. Entre los pacientes tratados con dasatinib, las mutaciones detectadas fueron T315I, F317I/L y V299L. Sobre la base de los datos in vitro, dasatinib no parece ser activo contra la mutación T315I. LMC o LLA Ph+ Resistente o Intolerante a Imatinib: La eficacia y la seguridad de SPRYCEL se investigaron en pacientes adultos con LMC o LLA Ph+ con resistencia o intolerancia al imatinib: 1158 pacientes tenían LMC en fase crónica, 858 pacientes en fase acelerada, fase mieloblástica o fase linfoblástica y 130 pacientes tenían LLA Ph+. En un ensayo clínico en pacientes con LMC en fase crónica, la resistencia al imatinib se definió como la falta de logro de una respuesta hematológica completa [complete hematologic response, (CHR; (después de 3 meses)], respuesta citogenética mayor [major cytogenetic response, (MCyR; (después de 6 meses)], o respuesta citogenética completa [complete cytogenetic response, CCyR; (después de 12 meses)]; o pérdida de una respuesta molecular (con aumento concurrente de ≥10% en las metafases Ph+), citogenética o hematológica previas. La intolerancia al imatinib se definió como la incapacidad de tolerar 400 mg o más de imatinib por día o la discontinuación del imatinib por toxicidad. Los resultados que se describen a continuación se basan en un mínimo de 2 años de seguimiento después del comienzo del tratamiento con SPRYCEL en pacientes con una mediana de tiempo desde el diagnóstico inicial de aproximadamente 5 años. En todos los estudios, 48% de los pacientes eran mujeres, 81% eran blancos, 15% eran de raza negra o asiáticos, 25% tenían 65 años de edad o más, y 5% tenían 75 años de edad o más. La mayor parte de los pacientes tenían amplios antecedentes de la enfermedad y varios tratamientos previos, incluso con imatinib, quimioterapia citotóxica, interferón y trasplante de células madre. En total, el 80% de los pacientes tenían enfermedad resistente al imatinib y 20% de los pacientes mostraban intolerancia al imatinib. La dosis máxima de imatinib había sido de 400-600 mg/día en aproximadamente 60% de los pacientes y > 600 mg/día en 40% de los pacientes. La variable primaria de análisis de eficacia en la LMC en fase crónica fue la MCyR, definida como la eliminación (CCyR) o la considerable disminución (en al menos un 65%, respuesta citogenética parcial) de las células hematopoyéticas Ph+. El criterio primario de valoración de eficacia en la LMC en fase acelerada, mieloblástica, linfoblástica, y LLA Ph+ fue la respuesta hematológica mayor (major hematologic response, MaHR), definida como una respuesta hematológica completa (CHR) o la falta de evidencia de leucemia (no evidence of leukemia, NEL). LMC en Fase Crónica: Ensayo de optimización de dosis: Se realizó un ensayo abierto y randomizado en pacientes con LMC en fase crónica para evaluar la eficacia y seguridad de SPRYCEL administrado una vez por día en comparación con SPRYCEL administrado dos veces por día. Los pacientes con enfermedades cardíacas significativas, incluso infarto de miocardio en los 6 meses previos, insuficiencia cardíaca congestiva en los 3 meses previos, arritmias significativas o prolongación del intervalo QTc fueron excluidos del ensayo. El criterio primario de valoración de eficacia fue la MCyR en pacientes con LMC resistente al imatinib. Un total de 670 pacientes, de los cuales 497 tenían enfermedad resistente al imatinib, se randomizó para recibir SPRYCEL 100 mg una vez por día, 140 mg una vez por día, 50 mg dos veces por día, o 70 mg dos veces por día. La mediana de la duración del tratamiento fue de 22 meses. Se logró eficacia en todos los grupos de tratamiento con SPRYCEL con el régimen de tratamiento de una vez por día, lo que demostró una eficacia comparable (no inferioridad) con la del régimen diario de dos veces por día para el criterio primario de valoración de eficacia (diferencia en MCyR 1,9%; IC del 95% [-6,8%-10,6%]); sin embargo, el régimen de 100 mg una vez por día demostró mayor seguridad y tolerabilidad. Los resultados de eficacia se presentan en las Tablas 10, 11 y 12 para pacientes con LMC en fase crónica que recibieron la dosis inicial recomendada de 100 mg una vez por día.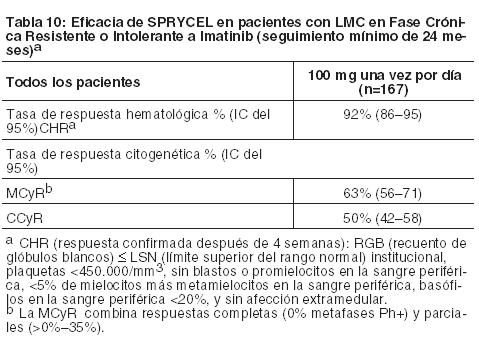
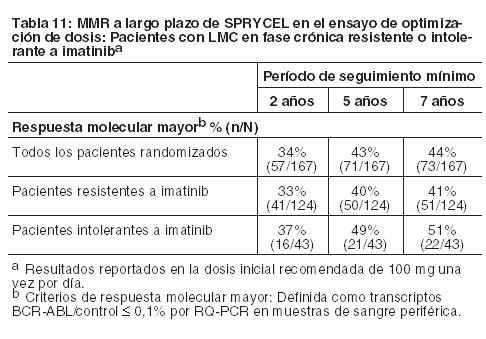
Sobre la base de los datos 7 años después de que el último paciente fuera enrolado en el estudio, se sabía que 44% estaban vivos, que 31% habían muerto y que 25% tenían un estado de sobrevida desconocido. A los 7 años, se produjo la transformación a fase acelerada o blástica en 9 pacientes durante el tratamiento en el grupo de 100 mg una vez por día. LMC en Fase Avanzada y LLA Ph+: Ensayo de optimización de dosis: Se realizó un ensayo abierto randomizado en pacientes con LMC en fase avanzada (LMC en fase acelerada, LMC en fase mieloblástica o LMC en fase linfoblástica) para evaluar la seguridad y la eficacia de SPRYCEL administrado una vez por día en comparación con SPRYCEL administrado dos veces por día. El criterio primario de valoración de eficacia fue la MaHR. Un total de 611 pacientes se randomizó ya sea para un grupo tratado con SPRYCEL 140 mg una vez por día o 70 mg dos veces por día. La mediana de la duración del tratamiento fue de aproximadamente 6 meses para ambos grupos de tratamiento. El régimen de una vez por día demostró una eficacia comparable (no inferioridad) al régimen de dos veces por día para el criterio primario de valoración. En la Tabla 12 se presentan los índices de respuesta para los pacientes del grupo de 140 mg una vez por día.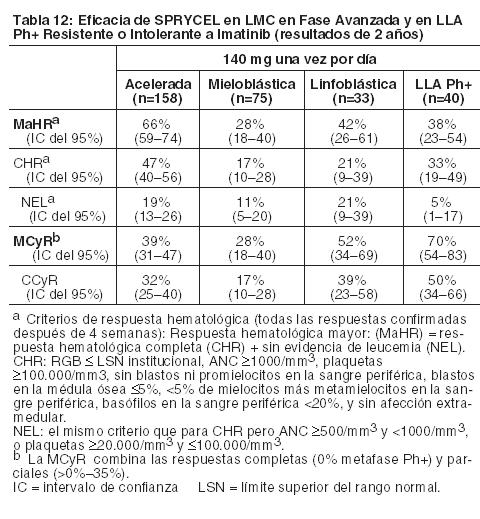
En el grupo de SPRYCEL 140 mg una vez por día, la mediana de tiempo hasta la MaHR fue de 1,9 meses (mín./máx.: 0,7-14,5) para los pacientes con LMC en fase acelerada, 1,9 meses (mín./máx.: 0,9-6,2) para los pacientes con LMC en fase mieloblástica y 1,8 meses (mín./máx.: 0,9-2,8) para los pacientes con LMC en fase linfoblástica. En pacientes con LMC en fase mieloblástica, la mediana de la duración de MaHR fue de 8,1 meses (mín./máx.: 2,7-21,1) y de 9,0 meses (mín./máx.: 1,8-23,1) para el grupo de 140 mg una vez por día y el de 70 mg dos veces por día, respectivamente. En pacientes con LMC en fase linfoblástica, la mediana de la duración de la MaHR fue de 4,7 meses (mín./máx.: 3,0-9,0) y 7,9 meses (mín./máx.: 1,6-22,1) para los grupos de 140 mg una vez por día y 70 mg dos veces por día, respectivamente. En pacientes con LLA Ph+ que fueron tratados con SPRYCEL 140 mg una vez por día, la mediana de la duración de la MaHR fue de 4,6 meses (mín./máx.: 1,4-10,2). Las medianas de sobrevida sin progresión para los pacientes con LLA Ph+ tratados con SPRYCEL 140 mg una vez por día y 70 mg dos veces por día fueron de 4,0 meses (mín./máx.: 0,4-11,1) y 3,1 meses (mín./máx.: 0,3-20,8), respectivamente.
Toxicología.
Carcinogénesis, Mutagénesis, Alteración de la Fertilidad: En un estudio de carcinogenicidad de 2 años de duración, se administraron dosis orales de dasatinib de 0,3, 1 y 3 mg/kg/día a ratas. La dosis mayor dio como resultado un nivel plasmático de exposición al fármaco (AUC) que es aproximadamente el 60% de la exposición humana con 100 mg una vez por día. Dasatinib indujo un aumento estadísticamente significativo en la incidencia combinada de carcinomas y papilomas de células escamosas en útero y cuello del útero en las hembras que recibieron la dosis alta, y adenoma de próstata en los machos que recibieron la dosis baja. Dasatinib fue clastogénico al ser probado in vitro en las células ováricas de hámster chino, con y sin activación metabólica. Dasatinib no fue mutagénico al ser probado en un ensayo celular bacterial in vitro (prueba Ames) y no fue genotóxico en un estudio de micronúcleo de rata in vivo. Dasatinib no afectó el apareamiento ni la fertilidad de ratas machos y hembras a una exposición plasmática al fármaco (AUC) similar a la exposición humana con 100 mg por día. En estudios de dosis repetidas, la administración de dasatinib causó una reducción del tamaño y secreción de las vesículas seminales, y próstata, vesículas seminales y testículos inmaduros. La administración de dasatinib provocó inflamación y mineralización uterina en monos, y quistes ováricos e hipertrofia ovárica en roedores.
Indicaciones.
SPRYCEL está indicado para el tratamiento de adultos con leucemia mieloide crónica (LMC) con cromosoma Philadelphia positivo (Ph+) en fase crónica de diagnóstico reciente. LMC Ph+ en fase crónica, acelerada, o mieloblástica o linfoblástica con resistencia o intolerancia a tratamiento previo, incluido imatinib. Leucemia linfoblástica aguda con cromosoma Philadelphia positivo (LLA Ph+) con resistencia o intolerancia a tratamiento previo.
Dosificación.
La dosis inicial recomendada de SPRYCEL para leucemia mieloide crónica (LMC) es de 100 mg administrado oralmente una vez al día. La dosis inicial recomendada de SPRYCEL para leucemia mieloide crónica (LMC) en fase acelerada, leucemia mieloide crónica (LMC) en fase mieloblástica o linfoblástica, o LLA Ph+ es de 140 mg administrado oralmente una vez al día. No se deben partir o triturar las tabletas/comprimidos recubiertos; deben tragarse enteros. SPRYCEL puede tomarse con o sin alimento, ya sea en la mañana o en la noche. En los estudios clínicos, se continuó el tratamiento con SPRYCEL hasta que se observó una progresión de la enfermedad o hasta que ya no era tolerado por el paciente. Se desconoce el efecto de suspender el tratamiento sobre el resultado de la enfermedad a largo plazo después de lograr una respuesta citogenética (incluida la respuesta citogenética completa [CCyR]) o una respuesta molecular mayor (MMR).
Modificación de la Dosis: Inductores concomitantes potentes de CYP3A4: El uso de inductores concomitantes potentes de CYP3A4 puede reducir las concentraciones plasmáticas de dasatinib y su uso debe evitarse (por e.j., dexametasona, fenitoína, carbamazepina, rifampicina, rifabutina y fenobarbital). La hierba de San Juan (St. John's Wort) puede disminuir las concentraciones plasmáticas de dasatinib de forma impredecible y su uso debe evitarse. De acuerdo con los estudios farmacocinéticos, en caso que se deba coadministrar un inductor potente de CYP3A4, se debe considerar un aumento de la dosis de SPRYCEL. Si se aumenta la dosis de SPRYCEL, debe monitorizarse cuidadosamente al paciente para detectar toxicidad [ver Interacciones Farmacológicas]. Inhibidores concomitantes potentes de CYP3A4: Los inhibidores de CYP3A4 (por e.j., ketoconazol, itraconazol, claritromicina, atazanavir, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina y voriconazol) pueden aumentar las concentraciones plasmáticas de dasatinib. El jugo de pomelo también puede aumentar las concentraciones plasmáticas de dasatinib y su uso debe evitarse. Se recomienda, si es posible, la selección de una medicación concomitante alternativa con un potencial nulo o mínimo de inhibición de enzimas. Si se debe administrar SPRYCEL con un inhibidor de CYP3A4 potente, se debe considerar una disminución de la dosis. Sobre la base de los estudios farmacocinéticos, debe considerarse una reducción de la dosis hasta 20 mg por día para pacientes que toman SPRYCEL 100 mg por día. Debe considerarse una reducción de la dosis hasta 40 mg por día para los pacientes que toman 140 mg de SPRYCEL por día. Se espera que estas reducciones de dosis de SPRYCEL ajusten el área bajo la curva (Area Under Curve, AUC) al intervalo observado sin inhibidores del CYP3A4. No obstante, no hay datos clínicos sobre estos ajustes de dosis obtenidos de pacientes en tratamiento con inhibidores potentes del CYP3A4. Si no se tolera SPRYCEL después de la disminución de la dosis, el uso de inhibidores potentes de CYP3A4 debe interrumpirse, o bien SPRYCEL debe suspenderse hasta que el tratamiento con el inhibidor haya terminado. Cuando se interrumpa el uso de los inhibidores potentes, debe permitirse un período de reposo farmacológico de aproximadamente 1 semana antes de aumentar la dosis de SPRYCEL. [ver Interacciones Farmacológicas]. Incremento Gradual de la Dosis: En los estudios clínicos de pacientes adultos con LMC y LLA Ph+, se permitió el incremento gradual de la dosis a 140 mg una vez al día (LMC en fase crónica) o 180 mg una vez al día (LMC en fase avanzada y LLA Ph+) en pacientes que no habían alcanzado una respuesta hematológica o citogenética a la dosis inicial recomendada. Ajuste de la Dosis para Reacciones Adversas. Mielosupresión: En los estudios clínicos, se controló la mielosupresión mediante la interrupción o reducción de la dosis o la descontinuación de la terapia del estudio. Se ha usado el factor de crecimiento hematopoyético en pacientes con mielosupresión resistente. Las normas para las modificaciones de la dosis están resumidas en la Tabla 1.
Reacciones Adversas no-hematológicas: Si se desarrolla una reacción adversa grave no hematológica con el uso de SPRYCEL, se debe suspender el tratamiento hasta que se haya resuelto o mejorado el evento. A partir de ese momento, se puede recomenzar el tratamiento de la manera apropiada con una dosis reducida que depende de la gravedad y la recurrencia del evento [ver Advertencias]. Formas de dosificación y concentraciones: Las tabletas/comprimidos de SPRYCEL (dasatinib) están disponibles como tabletas/comprimidos recubiertos de color blanco a casi blanco, biconvexos, en concentraciones de 20 mg, 50 mg, 70 mg, y 100 mg [ver Presentación].
Contraindicaciones.
SPRYCEL está contraindicado en pacientes con hipersensibilidad a dasatinib o a cualquier otro componente de SPRYCEL.
Reacciones adversas.
Las siguientes reacciones adversas se analizan con mayor detalle en otras secciones del prospecto: Mielosupresión [ver Dosificación y Advertencias]. Eventos relacionados con sangrado [ver Advertencias]. Retención de líquidos [ver Advertencias]. Eventos cardiovasculares [ver Advertencias]. Hipertensión arterial pulmonar [ver Advertencias]. Prolongación del intervalo QT [ver Advertencias]. Reacciones Dermatológicas Severas [ver Advertencias]. Síndrome de lisis tumoral [ver Advertencias]. Toxicidad embrio-fetal [ver Advertencias]. Dado que los estudios clínicos se llevan a cabo bajo condiciones muy variadas, los índices de reacciones adversas observadas en los estudios clínicos para un fármaco no pueden compararse directamente con los índices de estudios clínicos de otros fármacos y pueden no reflejar los índices observados en la práctica. Los datos que se describen a continuación reflejan la exposición a SPRYCEL en todas las dosis evaluadas en estudios clínicos que incluyeron 324 pacientes con LMC en fase crónica, recientemente diagnosticada, y en 2388 pacientes con LMC en fase crónica o avanzada o LLA Ph+, con resistencia o intolerancia a imatinib. La mediana de la duración del tratamiento en 2712 pacientes tratados con SPRYCEL fue de 19,2 meses (rango 0-93,2 meses). En un ensayo randomizado realizado en pacientes con LMC en fase crónica recientemente diagnosticada, la mediana de la duración del tratamiento fue de aproximadamente 60 meses. La mediana de la duración del tratamiento en 1618 pacientes con LMC en fase crónica fue de 29 meses (rango 0-92,9 meses). La mediana de la duración del tratamiento en 1094 pacientes con LMC en fase avanzada o LLA Ph+ fue de 6,2 meses (rango 0-93,2 meses). En la población general de 2712 pacientes tratados con SPRYCEL, el 88% de los pacientes experimentó reacciones adversas en algún momento, y el 19% experimentó reacciones adversas que condujeron a la discontinuación del tratamiento. En el estudio randomizado en pacientes con LMC en fase crónica recientemente diagnosticada, se discontinuó el uso del fármaco debido a reacciones adversas en el 16% de los pacientes tratados con SPRYCEL con un seguimiento mínimo de 60 meses. Luego de un seguimiento mínimo de 60 meses, el índice de discontinuación acumulativo fue del 39%. Entre los 1618 pacientes tratados con SPRYCEL con LMC en fase crónica, se reportaron eventos adversos relacionados con el fármaco que condujeron a la discontinuación en 329 pacientes (20,3%); entre los 1094 pacientes tratados con SPRYCEL con LMC en fase avanzada o LLA Ph+, se reportaron eventos adversos relacionados con el fármaco que condujeron a la discontinuación en 191 pacientes (17,5%). Las reacciones adversas reportadas en ≥10% de los pacientes, y otras reacciones adversas de interés, en un ensayo randomizado de pacientes con LMC en fase crónica recientemente diagnosticada con una mediana de seguimiento de aproximadamente 60 meses se presentan en la Tabla 2. Las reacciones adversas reportadas en ≥10% de los pacientes tratados con la dosis recomendada de 100 mg una vez por día (n=165), y otras reacciones adversas de interés, en un ensayo randomizado con optimización de dosis de pacientes con LMC en fase crónica resistentes o intolerantes a la terapia previa con imatinib con una mediana de seguimiento de aproximadamente 84 meses se presentan en la Tabla 4. Se reportaron eventos adversos serios (EAS) relacionados con el fármaco en el 16,7% de los pacientes tratados con SPRYCEL en el ensayo randomizado de pacientes con LMC en fase crónica recientemente diagnosticada. Las reacciones adversas serias reportadas en ≥5% de los pacientes incluyeron derrame pleural (5%). Se reportaron EAS relacionados con el fármaco en el 26,1% de los pacientes tratados con la dosis recomendada de 100 mg una vez por día en el ensayo randomizado con optimización de dosis de pacientes con LMC en fase crónica resistentes o intolerantes a la terapia previa con imatinib. Las reacciones adversas serias reportadas en ≥5% de los pacientes incluyeron derrame pleural (10%). Leucemia Mieloide Crónica (LMC): Las reacciones adversas (excepto las anormalidades de laboratorio) que se informaron en al menos un 10% de los pacientes se muestran en la Tabla 2 para pacientes con LMC en fase crónica recientemente diagnosticados y en las Tablas 4 y 6 para pacientes con LMC con resistencia o intolerancia a tratamiento previo con imatinib.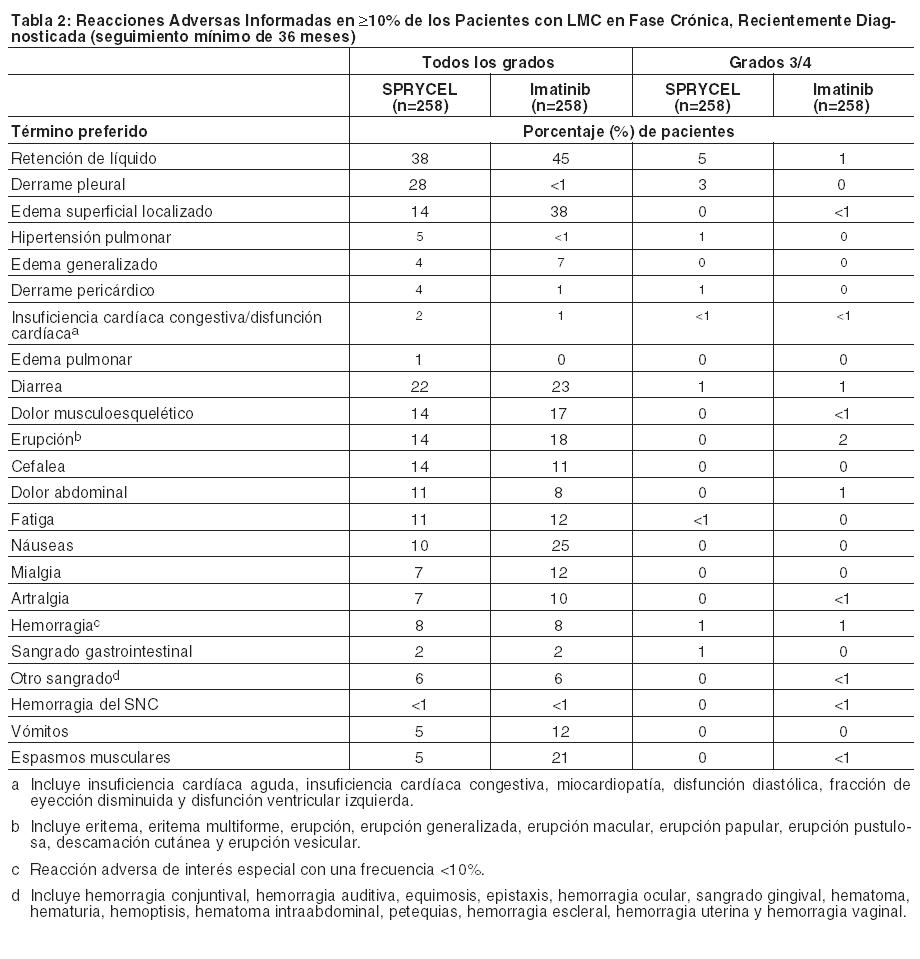
En la Tabla 3 se muestra una comparación de los índices acumulativos de reacciones adversas reportadas en ≥10% de los pacientes con un seguimiento mínimo de 1 y 5 años en un ensayo randomizado de pacientes con diagnóstico reciente de LMC en fase crónica tratados con SPRYCEL.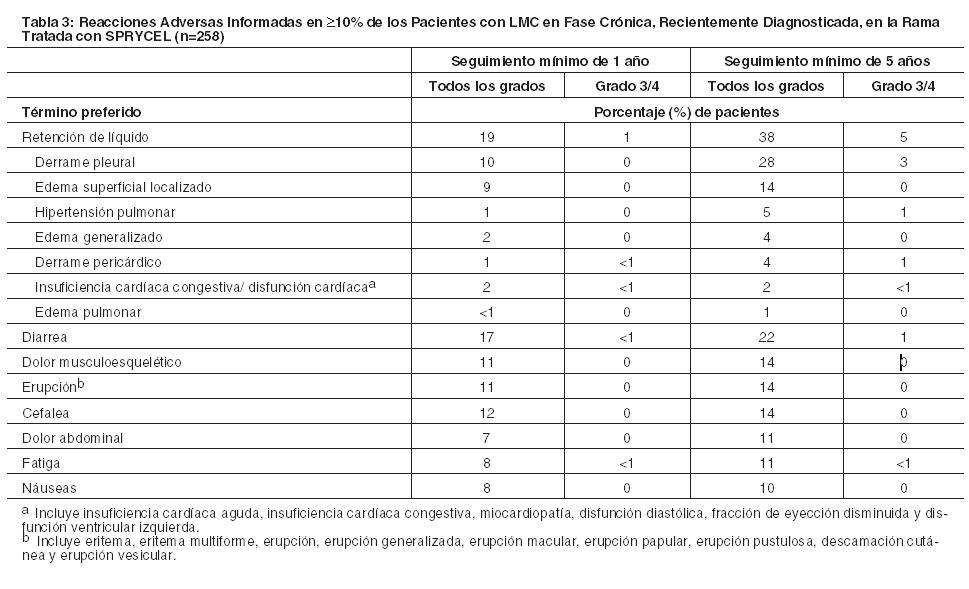
Tras 60 meses, hubo 26 muertes entre los pacientes tratados con dasatinib (10,1%) y 26 muertes entre pacientes tratados con imatinib (10,1%); 1 muerte de cada grupo fue evaluada por el investigador como relacionada con la terapia del estudio.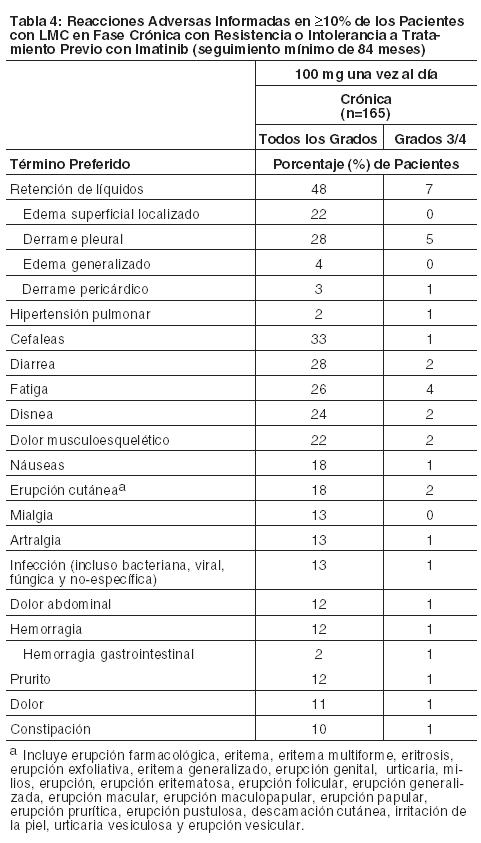
En la Tabla 5 se muestran los índices acumulativos de reacciones adversas seleccionadas que se reportaron en el tiempo en pacientes tratados con la dosis inicial recomendada de 100 mg una vez por día en un ensayo randomizado con optimización de dosis de pacientes resistentes o intolerantes a imatinib que presentaban LMC en fase crónica.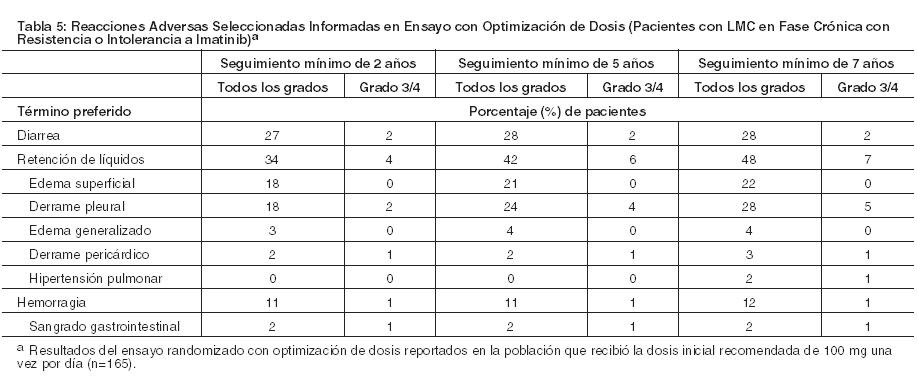
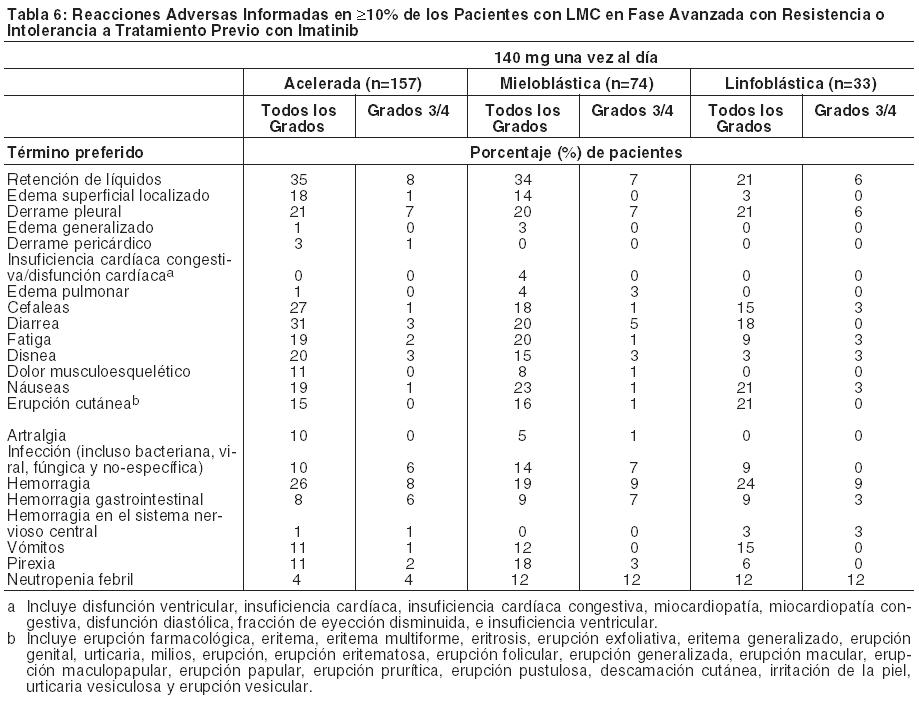
Anormalidades de Laboratorio: Las notificaciones de mielosupresión fueron comunes a todas las poblaciones de pacientes. La frecuencia de neutropenia, trombocitopenia y anemia de Grado 3 ó 4 fue mayor en los pacientes con LMC en fase avanzada que en los pacientes con LMC en fase crónica (Tablas 7 y 8). Se informó mielosupresión en pacientes con valores de laboratorio iniciales normales como así también en pacientes con anormalidades preexistentes de laboratorio. En los pacientes que experimentaron mielosupresión severa, la recuperación generalmente ocurrió después de la interrupción o reducción de la dosis; en un ensayo randomizado, la discontinuación permanente del tratamiento ocurrió en el 2% de los pacientes con LMC en fase crónica recientemente diagnosticada y en el 5% de los pacientes con resistencia o intolerancia a tratamiento previo con imatinib [ver Advertencias]. Se informaron elevaciones de transaminasas o bilirrubina de Grado 3 ó 4 e hipocalcemia, hipocalemia e hipofosfatemia de Grado 3 ó 4 en pacientes con cualquiera de las fases de LMC pero se informaron con una mayor frecuencia en pacientes con LMC en fase mieloblástica o linfoblástica. Las elevaciones en transaminasas o bilirrubina usualmente se manejaron con una reducción o interrupción de la dosis. Los pacientes con desarrollo de hipocalcemia de Grado 3 ó 4 durante el curso de la terapia con SPRYCEL a menudo se recuperaron con un suplemento de calcio por vía oral. Las anormalidades de laboratorio informadas en pacientes con LMC en fase crónica recientemente diagnosticada se muestran en la Tabla 7. No hubo discontinuaciones del tratamiento con SPRYCEL en esta población de pacientes debido a los parámetros bioquímicos de laboratorio.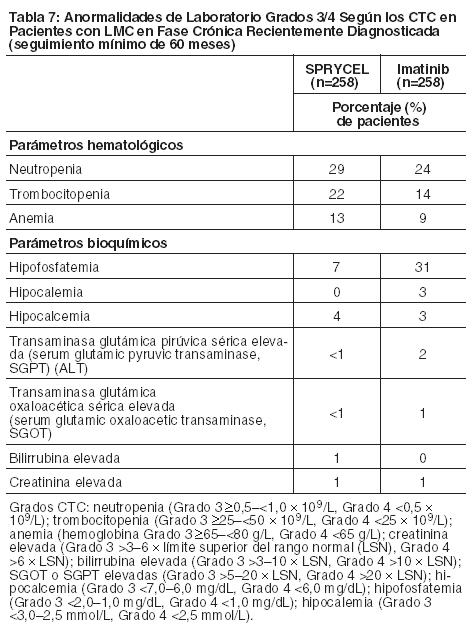
Las anormalidades de laboratorio informadas en pacientes con LMC con resistencia o intolerancia a imatinib que recibieron las dosis iniciales recomendadas de SPRYCEL se muestran clasificadas por fase de la enfermedad en la Tabla 8.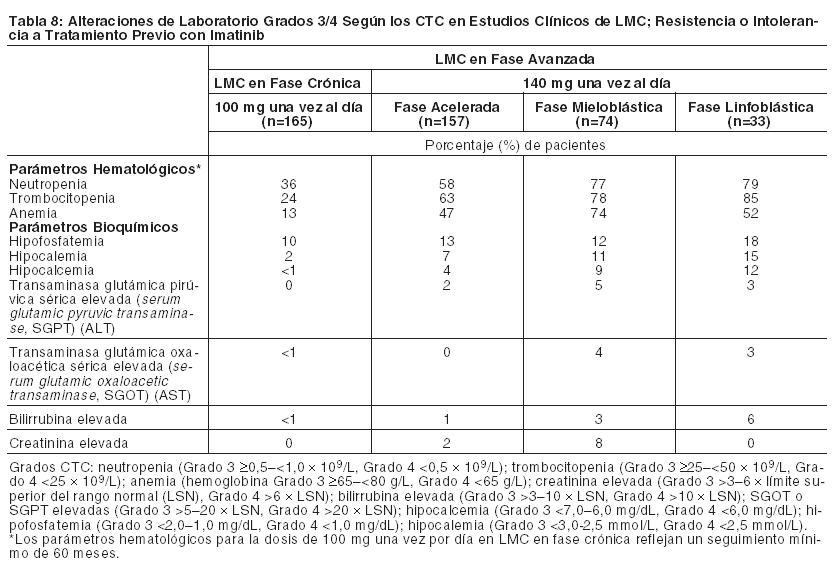
Entre los pacientes con LMC en fase crónica con resistencia o intolerancia al tratamiento previo con imatinib, los casos acumulativos de citopenia de Grado 3 ó 4 fueron similares a los 2 y 5 años, e incluyeron: neutropenia (36% vs. 36%), trombocitopenia (23% vs. 24%) y anemia (13% vs. 13%). Leucemia Linfoblástica Aguda de Cromosoma Philadelphia Positivo (LLA Ph+): Un total de 135 pacientes con LLA Ph+ fueron tratados con SPRYCEL en estudios clínicos. La mediana de la duración de tratamiento fue de 3 meses (intervalo: 0,03-31 meses). El perfil de seguridad de los pacientes con LLA Ph+ fue similar al de los pacientes con LMC en fase linfoblástica. Las reacciones adversas informadas con más frecuencia incluyeron eventos de retención de líquidos, tales como el derrame pleural (24%) y el edema superficial (19%), y trastornos gastrointestinales tales como diarrea (31%), náuseas (24%) y vómitos (16%). También se informaron con frecuencia hemorragia (19%), pirexia (17%), erupción cutánea (16%) y disnea (16%). Las reacciones adversas serias informadas en ≥5% de los pacientes incluyeron derrame pleural (11%), hemorragia gastrointestinal (7%), neutropenia febril (6%) e infección (5%). Datos Combinados Adicionales de los Ensayos Clínicos: Se informaron las siguientes reacciones adversas adicionales en pacientes de los estudios clínicos de SPRYCEL en LMC y LLA Ph+ con una frecuencia de ≥10%, 1%- < 10%, 0,1%- < 1%, o < 0,1%. Estos eventos están incluidos en base a la relevancia clínica. Trastornos gastrointestinales: 1%- < 10% - inflamación de mucosas (incluso mucositis/estomatitis), dispepsia, distensión abdominal, estreñimiento, gastritis, colitis (incluso colitis neutropénica), alteración de tejido blando oral; 0,1%- < 1% - ascitis, disfagia, fisura anal, úlcera gastrointestinal alta, esofagitis, pancreatitis, enfermedad de reflujo gastroesofágico; < 0,1% - gastroenteropatía con pérdida de proteínas, íleo, pancreatitis aguda, fístula anal. Trastornos generales y condiciones del sitio de administración: ≥ 10% - edema periférico, edema facial; 1%- < 10% - astenia, dolor en el pecho, escalofríos; 0,1%- < 1% - malestar general, otros edemas superficiales; < 0.1% - alteración de la marcha. Trastornos de la piel y del tejido subcutáneo: 1%- < 10% -alopecia, acné, piel seca, hiperhidrosis, urticaria, dermatitis (incluso eczema); 0,1%- < 1% - trastornos en la pigmentación, úlcera cutánea, condiciones ampollosas, reacción de fotosensibilidad, trastornos en las uñas, dermatosis neutrofílica, paniculitis, síndrome de eritrodisestesia palmar-plantar, trastornos del cabello; < 0,1% - vasculitis leucocitoclástica, fibrosis dérmica. Trastornos respiratorios, torácicos y mediastínicos: 1%- < 10% -infiltración pulmonar, neumonitis, tos; 0,1%- < 1% - asma, broncoespasmo, disfonía, hipertensión arterial pulmonar; < 0,1% - síndrome de dificultad respiratoria aguda, embolia pulmonar. Trastornos del sistema nervioso: 1%- < 10% - neuropatía (incluso neuropatía periférica), mareos, disgeusia, somnolencia; 0,1%- < 1% - amnesia, temblores, síncope, trastornos del equilibrio; < 0,1% - convulsiones, accidente cerebrovascular, ataque isquémico transitorio, neuritis óptica, parálisis del séptimo nervio, demencia, ataxia. Trastornos del sistema circulatorio y linfático: 0.1%- < 1% - linfadenopatía, linfopenia; < 0,1% - aplasia pura de eritrocitos. Trastornos musculoesqueléticos y del tejido conectivo: 1%- < 10% - debilidad muscular, rigidez musculoesquelética; 0,1%- < 1% - rabdomiólisis, tendinitis, inflamación muscular, osteonecrosis, artritis. Exploraciones complementarias: 1%- < 10% - aumento de peso, pérdida de peso; 0,1%- < 1% - aumento de la creatina fosfoquinasa en sangre, aumento de gamma-glutamiltransferasa. Infecciones e infestaciones: 1%- < 10% - neumonía (incluso bacteriana, viral, y fúngica), infección/inflamación del tracto respiratorio superior, infección por virus del herpes, infección por enterocolitis, sepsis (incluso con resultados fatales [0,2%]). Trastornos en el metabolismo y nutrición: 1%- < 10% -alteraciones en el apetito, hiperuricemia; 0,1%- < 1% - hipoalbuminemia, síndrome de lisis tumoral, deshidratación, hipercolesterolemia; < 0,1% - diabetes mellitus. Trastornos cardíacos: 1%- < 10% - arritmia (incluso taquicardia), palpitaciones; 0,1%- < 1% - angina de pecho, cardiomegalia, pericarditis, arritmia ventricular (incluso taquicardia ventricular), onda T anormal en el electrocardiograma, aumento de troponina; < 0,1% - cor pulmonale, miocarditis, síndrome coronario agudo, paro cardíaco, prolongación del intervalo PR en el electrocardiograma, enfermedad de arterias coronarias, pleuropericarditis. Trastornos oculares: 1%- < 10% - trastorno visual (incluso alteración visual, visión borrosa, y agudeza visual disminuida), ojo seco; 0,1%- < 1% - conjuntivitis, deterioro visual, fotofobia, aumento del lagrimeo. Trastornos vasculares: 1%- < 10% - enrojecimiento, hipertensión; < 0,1%- < 1% - hipotensión, tromboflebitis, trombosis; < 0,1% - livedo reticularis, trombosis venosa profunda, embolia. Trastornos psiquiátricos: 1%- < 10% - insomnio, depresión; 0,1%- < 1% - ansiedad, labilidad afectiva, estado de confusión, disminución de la libido. Afecciones durante el embarazo, el puerperio y el período perinatal: < 0,1% - aborto. Trastornos mamarios y del sistema reproductivo: 0,1%- < 1% - ginecomastia, trastornos menstruales. Lesión, intoxicación, y complicaciones de procedimiento: 1%- < 10% - contusión. Trastornos del oído y el laberinto: 1%- < 10% - acúfenos; 0,1%- < 1% - vértigo, pérdida de la audición. Trastornos hepatobiliares: 0,1%- < 1% - colestasis, colecistitis, hepatitis. Trastornos renales y urinarios: 0,1%- < 1% - aumento de la frecuencia urinaria, insuficiencia renal, proteinuria; < 0.1% - deterioro renal. Trastornos del sistema inmunológico: < 0,1%- < 1% - hipersensibilidad (incluso eritema nodoso). Desórdenes Endocrinos: 0.1%- < 1% - hipotiroidismo; < 0.1% - hipertiroidismo, tiroiditis. Experiencia Posterior a la Comercialización: Se han identificado las siguientes reacciones adversas adicionales durante el uso de SPRYCEL posterior a la aprobación. Debido a que estas reacciones se informan en forma voluntaria en una población de tamaño incierto, no siempre es posible estimar confiablemente su frecuencia ni establecer una relación causal con la exposición al fármaco. Infecciones: reactivación del virus de la hepatitis B. Trastornos cardíacos: fibrilación auricular/aleteo auricular. Trastornos respiratorios, torácicos y mediastínicos: neumopatía intersticial. Trastornos de la piel y el tejido subcutáneo: síndrome de Stevens-Johnson.
Advertencias.
Mielosupresión: El tratamiento con SPRYCEL está asociado a trombocitopenia, neutropenia y anemia graves (Criterios de Toxicidad Común (CTC) del NIC Grado 3 ó 4), que se producen más temprano y con mayor frecuencia en pacientes con LMC en fase avanzada o LLA Ph+ que en pacientes con LMC en fase crónica. En pacientes con LMC en fase crónica, los recuentos completos de sangre deben realizarse cada 2 semanas durante 12 semanas, y a partir de entonces cada 3 meses, o según esté clínicamente indicado. En pacientes con LMC en fase avanzada o LLA Ph+, los recuentos completos de sangre deben realizarse semanalmente durante los primeros dos meses y mensualmente de ahí en adelante, o según