SOMAVERT
PFIZER
Análogo de la hormona de crec. humana.
Composición.
Cada frasco ampolla contiene: Pegvisomant 10 mg y 15 mg. Excipientes: Glicina, Manitol, Fosfato de sodio dibásico anhidro, Fosfato de sodio monobásico monohidrato. Cada frasco ampolla de disolvente contiene: Agua estéril para inyectables 8 ml.
Farmacología.
Descripción: SOMAVERT contiene pegvisomant inyectable, un análogo de la hormona de crecimiento humana GH que ha sido modificada estructuralmente para actuar como un antagonista del receptor de la GH. Pegvisomant es una proteína originada mediante ADN recombinante que contiene 191 residuos de aminoácidos a los cuales varios polímeros de polietilenglicol (PEG) están covalentemente unidos (predominantemente 4 a 6 PEG/molécula proteica). El peso molecular de la proteína pegvisomant es 21.998 Daltons. El peso molecular de la porción de PEG de pegvisomant es de aproximadamente 5000 Daltons. Los pesos moleculares predominantes de pegvisomant son, de este modo, aproximadamente 42.000, 47.000 y 52.000 Daltons. El esquema muestra la secuencia de aminoácidos de la proteína pegvisomant. (Los polímeros PEG se muestran adjuntos a los 5 sitios de unión más probables). Pegvisomant es sintetizado por una cepa específica de la bacteria Escherichia coli que ha sido modificada genéticamente por la incorporación de un plásmido que transporta un gen para ser un antagonista del receptor de la GH. La potencia biológica es determinada usando un bioensayo de proliferación celular.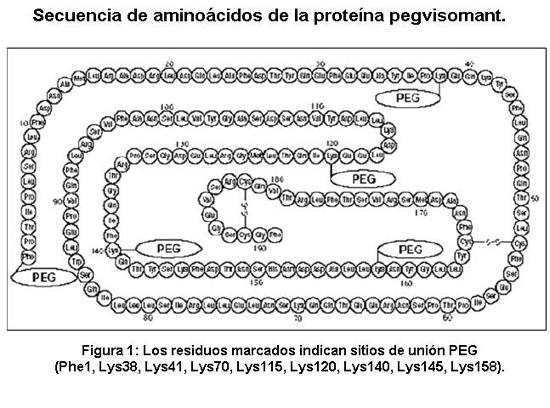
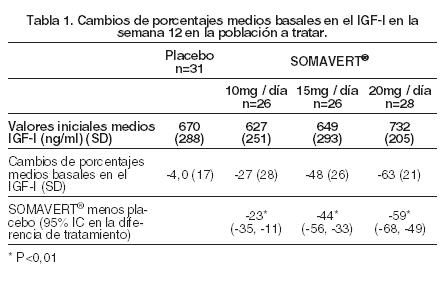
Farmacología: Mecanismo de Acción: Pegvisomant se une selectivamente a los receptores de la hormona de crecimiento (GH) en las superficies celulares, donde bloquea la unión de la hormona de crecimiento endógena, y de este modo afecta la transducción de señales de la GH. La inhibición de la acción de la GH da como resultado concentraciones séricas menores del factor de crecimiento análogo a la insulina I (IGF-I), así como también otras proteínas séricas inducidas por GH, incluyendo la proteína de unión IGF-3 (IGFBP-3), y la subunidad ácido lábil (ALS). Farmacocinética: Absorción: Luego de la administración subcutánea, las concentraciones de pegvisomant séricas pico, generalmente se alcanzan de 33 a 77 horas después de la administración. El alcance medio de absorción de una dosis subcutánea de 20 mg fue del 57%, respecto de una dosis intravenosa de 10 mg. Distribución: El volumen aparente medio de distribución de pegvisomant es 7L (coeficiente del 12% de variación), indicando que pegvisomant no se distribuye extensamente en los tejidos. Luego de una administración subcutánea única, la exposición (Cmáx, AUC) a pegvisomant aumenta en forma no proporcional con dosis cada vez mayores. Las concentraciones de pegvisomant séricas medias ± SEM luego de 12 semanas de tratamiento con dosis diarias de 10, 15 y 20 mg fueron 6600 ± 1330; 16000 ± 2200; y 27000 ± 3100 ng/mL, respectivamente. Metabolismo y Eliminación: La molécula de pegvisomant contiene polímeros de polietilenglicol covalentemente unidos para reducir el índice del clearance. El clearance de pegvisomant, luego de dosis múltiples, es menor que los observados luego de una dosis única. El clearance sistémico corporal total medio de pegvisomant luego de dosis múltiples es estimado en un rango de 36 a 28 mL/h en dosis subcutáneas que oscilan de 10 a 20 mg/día, respectivamente. El clearance de pegvisomant resultó aumentar con el peso corporal. Pegvisomant es eliminado del suero con una vida media promedio de aproximadamente 6 días luego de dosis múltiples o únicas. Menos del 1% de la droga administrada se recupera en la orina durante 96 horas. La vía de eliminación de pegvisomant no ha sido estudiada en humanos. Interacciones con otros Medicamentos: En ensayos clínicos, los pacientes en tratamiento con opiáceos, a menudo necesitaron concentraciones séricas mayores de pegvisomant para lograr la supresión del IGF-I apropiada, en comparación con los pacientes que no recibieron opiáceos. El mecanismo de esta interacción se desconoce. (Ver Precauciones, Interacciones del Medicamento). Poblaciones Especiales: Renal: No se han realizados estudios farmacocinéticos en pacientes con insuficiencia renal. Hepática: No se han realizados estudios farmacocinéticos en pacientes con insuficiencia hepática. Ancianos: No se han realizados estudios farmacocinéticos en pacientes ancianos. Pediátrico: No se han realizados estudios farmacocinéticos en pacientes pediátricos. Sexo: No se encontró ningún efecto del sexo sobre la farmacocinética de pegvisomant en un análisis farmacocinético de población. Raza: No se ha estudiado el efecto de la raza sobre la farmacocinética de pegvisomant. Estudios clínicos: Un total de ciento doce pacientes (63 hombres y 49 mujeres) con acromegalia participaron en un estudio multicéntrico, aleatorizado y de doble ciego de 12 semanas de duración, que comparó placebo con SOMAVERT. La edad media ±SD fue de 48±14 años y la duración media de la acromegalia fue de 8±8 años. A noventa y tres se les había realizado una cirugía pituitaria anterior, de los cuales 57 también recibieron tratamiento convencional con radioterpia. Seis pacientes recibieron radioterapia sin cirugía, nueve habían recibido solamente tratamiento farmacológico y cuatro no habían recibido ningún tratamiento previo. Al inicio del estudio, la media del tiempo ±SD desde la última cirugía o tratamiento con radioterapia de los sujetos, respectivamente, fue de 6,8 ± 0,93 años (n= 63) y 5,6 ± 0,57 años (n= 93). Los sujetos calificaron para la participación en el estudio si su IGF-I sérica, extraída después del período de reposo farmacológico, fue ≥1,3 veces el límite superior del rango normal ajustado para la edad. Fueron asignados aleatoriamente en la visita inicial a uno de los cuatro grupos de tratamiento: placebo (n= 32), 10 mg/día (n= 26), 15 mg/día (n= 26) o 20 mg/día (n= 28) de SOMAVERT subcutáneo. El principal criterio de evaluación de la eficacia fue el cambio porcentual en las concentraciones de IGF-I desde el inicio y hasta la semana 12. Los tres grupos que recibieron SOMAVERT demostraron reducciones estadísticamente significativas (p < 0,01) en los niveles séricos de IGF-I comparados con el grupo placebo (Tabla 1).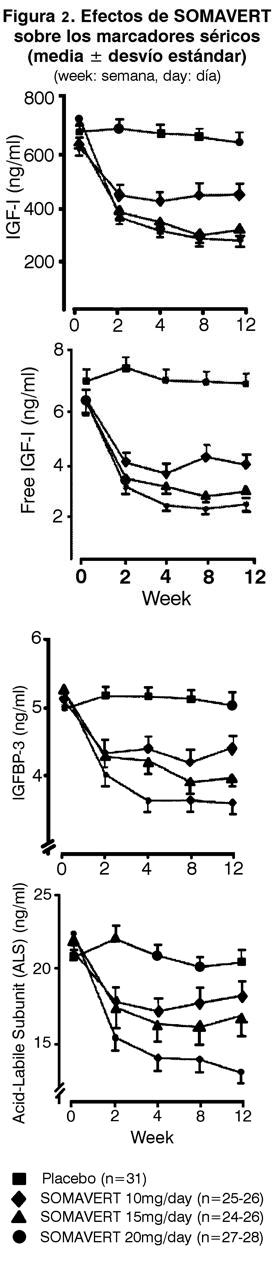
También hubo reducciones en los niveles séricos de IGF-I libre, IGFBP-3 y ALS en comparación con el placebo en todas las visitas posteriores al inicio (Figura 2). Luego de 12 semanas de tratamiento, los niveles del IGF-I séricos fueron normales en el 10%, 39%, 75% y 82% de los sujetos tratados con placebo, 10, 15 ó 20 mg/día de SOMAVERT, respectivamente. (Figura 3).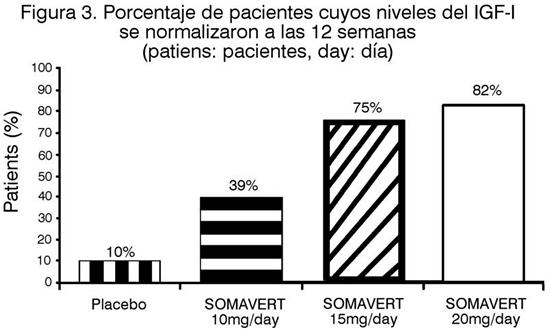
La Tabla 2 muestra el efecto del tratamiento con SOMAVERT sobre el tamaño del anillo (tamaños estándares de joyería convertidos en puntajes numéricos de 1 a 63), y sobre los puntajes totales e individuales en signos y síntomas de acromegalia. Cada puntaje individual (en edema leve del tejido, artralgia, dolor de cabeza, perspiración y fatiga) estuvo basado en una escala de estimación ordinal de nueve puntos (0= ausente y 8= severo e incapacitado), y el puntaje total derivó de una suma de puntajes individuales. Los puntajes basales medios fueron los siguientes: tamaño del anillo = 47,1; signos y síntomas totales = 15,2; edema leve del tejido = 2,5; artralgia = 3,2; dolor de cabeza = 2,4; perspiración = 3,3 y fatiga = 3,7.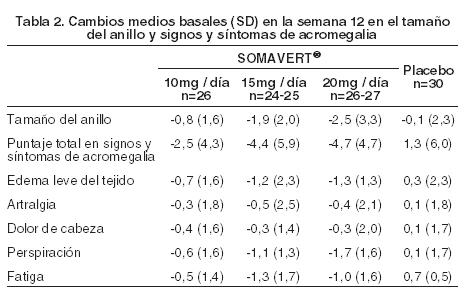
El tamaño del anillo en la semana 12 fue menor (mejor) en los grupos tratados con 15 ó 20 mg de SOMAVERT, en comparación con placebo. El puntaje total medio en signos y síntomas en la semana 12 fue menor (mejor) en cada uno de los grupos tratados con SOMAVERT, en comparación con el grupo tratado con placebo. Las concentraciones séricas de la hormona de crecimiento (GH), como las medidas por los ensayos de investigación utilizando anticuerpos que no provocan reacciones en forma cruzada con pegvisomant aumentan dentro de las dos semanas de comenzado el tratamiento con SOMAVERT. La respuesta de la GH más importante fue observada en pacientes tratados con SOMAVERT con dosis mayores a 20 mg/día. Este efecto es, presumiblemente, el resultado de la menor inhibición de la secreción de la GH a medida que los niveles del IGF-I descienden. Como se muestra en la Figura 4, cuando a los pacientes con acromegalia se les dio una dosis de ataque de SOMAVERT seguida por una dosis diaria fija, este aumento en la GH fue inversamente proporcional a la disminución en el IGF-I y generalmente se estabilizó hacia la semana 2. Las concentraciones de la GH séricas también permanecieron estables en pacientes tratados con SOMAVERT durante el promedio de 43 semanas (rango de 0-82 semanas).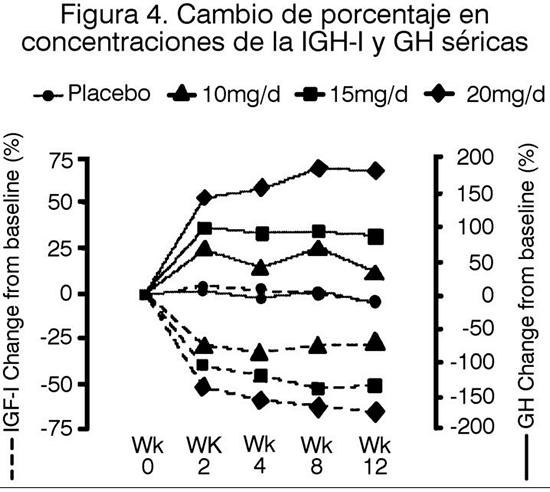
En la extensión abierta del estudio clínico, participaron 109 sujetos (incluso 6 pacientes nuevos) con una exposición media al tratamiento de 42,6 semanas (rango de 1 día a 82 semanas). 93 sujetos (85,3%) tuvo/sufrió un evento adverso, 16 (14,7%) un SAE (evento adverso serio) y 4 interrumpieron (3,7%) el tratamiento debido a un EA (cefaleas, resultados elevados en la prueba de función hepática, cáncer de páncreas y aumento de peso). Un total de 100 (92,6%) de los 108 sujetos con datos de IGF-I disponibles tenían una concentración IGF-I normal durante cualquier visita del estudio.
Indicaciones.
SOMAVERT se encuentra indicado para el tratamiento de la acromegalia en pacientes que no hayan respondido en forma adecuada al tratamiento con cirugía o radiación o para quienes estos tratamientos no sean apropiados. El objetivo del tratamiento es normalizar los niveles séricos del factor de crecimiento análogo de la insulina (IGF-I).
Dosificación.
Una dosis de ataque de 40 mg de SOMAVERT debe ser administrada en forma subcutánea bajo supervisión médica. Luego, al paciente se le debe informar que debe comenzar con inyecciones subcutáneas diarias de 10 mg de SOMAVERT. Las concentraciones del IGF-I séricas deben ser medidas cada cuatro o seis semanas, en las cuales la dosificación de SOMAVERT debe ser ajustada en incrementos de 5 mg/día si los niveles del IGF-I todavía son elevados (o disminuciones de 5 mg, si los niveles del IGF-I han disminuido por debajo del rango normal). Aunque los objetivos del tratamiento son lograr (y luego mantener) las concentraciones del IGF-I séricas dentro del rango normal de ajuste por edad y aliviar los signos y síntomas de acromegalia, la titulación de la dosis debe ser en base a los niveles del IGF-I. Se desconoce si los pacientes que permanecen sintomáticos mientras se logran niveles de IGF-I normalizados, se beneficiarían con una dosis mayor de SOMAVERT. El rango de dosificación se encuentra entre 10 y 30 mg por vía subcutánea una vez al día y la dosis diaria máxima es de 30 mg aplicados por vía subcutánea una vez al día. Antes de comenzar el tratamiento con SOMAVERT, se les deberá realizar a los pacientes una evaluación de los niveles iniciales de las pruebas hepáticas [alanina aminotransferasa en suero (ALT), aspartato aminotransferasa (AST), bilirrubina total en suero (BT) y fosfatasa alcalina (Fal)]. Para recibir recomendaciones en cuanto al inicio del tratamiento con SOMAVERT con base en las pruebas hepáticas iniciales y las recomendaciones para controlar las pruebas hepáticas mientras se encuentra bajo tratamiento con SOMAVERT, consulte la Tabla 3 en Advertencia y precauciones. Preparación de la dosis SOMAVERT se presenta como un polvo liofilizado. Cada frasco-ampolla de SOMAVERT debe ser reconstituido con 1 mL de diluyente proporcionado en el envase (Agua Estéril para Inyectables). Las instrucciones con respecto a la reconstitución y administración se encuentran incluidas en el envase de SOMAVERT y se deben seguir cuidadosamente. Para preparar la solución, retirar 1 mL de Agua Estéril para Inyectables e inyectarlo dentro del frasco-ampolla de SOMAVERT, dirigiendo el chorro del líquido sobre las paredes de vidrio del frasco-ampolla. Sostener el frasco-ampolla entre las palmas de ambas manos y lentamente hacerlo girar para disolver el polvo. No sacudir el frasco-ampolla, dado que esto podría causar la desnaturalización de pegvisomant. Desechar el frasco-ampolla del diluyente que contenga agua para inyectables sobrante. Luego de la reconstitución, cada frasco-ampolla de SOMAVERT contiene 10, 15 ó 20 mg de la proteína de pegvisomant en 1 mL de solución. Los medicamentos parenterales deben ser inspeccionados visualmente para detección de partículas y decoloración, previa a la administración. La solución debe estar clara después de la reconstitución. Si la solución está turbia, no inyectarla. Se debe administrar una sola dosis de cada frasco-ampolla. SOMAVERT debe ser administrado dentro de las seis horas después de la reconstitución. Pegvisomant puede ser dado en el muslo, glúteo, parte superior del brazo, o abdomen; el sitio de inyección subcutánea debe rotarse diariamente para ayudar a prevenir la lipohipertrofia.
Contraindicaciones.
SOMAVERT se encuentra contraindicado en pacientes con antecedentes de hipersensibilidad a cualquiera de sus componentes. El tapón del frasco-ampolla de SOMAVERT contiene látex.
Reacciones adversas.
Cambios de Laboratorios: Elevaciones de concentraciones séricas de alanina aminotransferasa (ALT) y aspartato aminotransferasa (AST) mayores a 10 veces el límite superior de lo normal (LSN) fueron informadas en dos pacientes (0,8%) expuestos a SOMAVERT durante estudios clínicos previos a la comercialización. A un paciente se le volvió a dar SOMAVERT, y la recurrencia de los niveles de transaminasas elevados indicaron una relación causal probable entre la administración del medicamento y la elevación de las enzimas del hígado. Una biopsia del hígado llevada a cabo en el segundo paciente fue consistente con hepatitis crónica de etiología desconocida. En ambos pacientes, las elevaciones de transaminasas se normalizaron luego de la interrupción del medicamento. Las elevaciones en los niveles de la ALT y AST no estuvieron asociadas con los niveles mayores de bilirrubina total sérica (BT) y fosfatasa alcalina (Fal), con la excepción de dos pacientes con mínimos aumentos asociados en niveles de Fal (a saber, menos de 3 veces el LSN). Las elevaciones de transaminasas no parecieron estar relacionadas con la dosis administrada de SOMAVERT, generalmente ocurrieron dentro de las 4 a 12 semanas del inicio del tratamiento y no estuvieron asociadas con ninguno de los factores predictivos genéticos, de fenotipo o bioquímicos identificables. General: Ocho pacientes con acromegalia (5,3%) fueron retirados de los estudios clínicos previos a la comercialización debido a eventos adversos, incluyendo dos pacientes con marcadas elevaciones de transaminasas (ver Alteraciones en el hepatograma), un paciente con lipohipertrofia en los sitios de inyección, y un paciente con aumento de peso substancial. La mayoría de los eventos adversos informados fueron de intensidad leve a moderada y de duración limitada. La mayoría de los eventos adversos no parecieron ser dependientes de la dosis. La Tabla 5 muestra la incidencia de los eventos adversos emergentes del tratamiento que fueron informados en por lo menos dos pacientes tratados con SOMAVERT y con frecuencias mayores que con placebo durante estudios controlados con placebo de 12 semanas.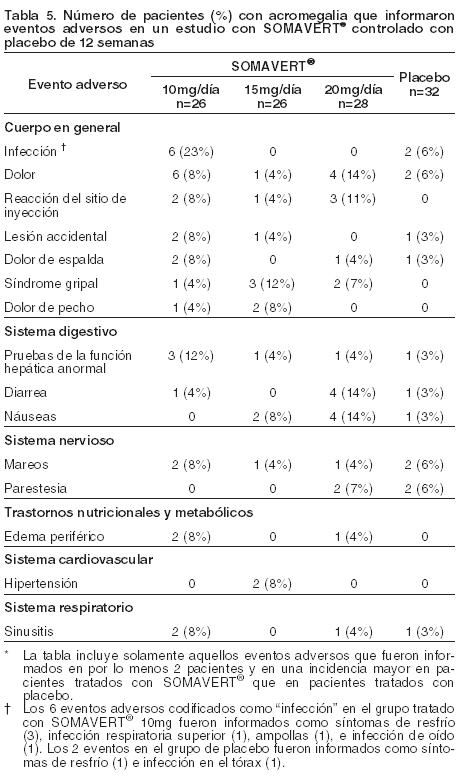
Inmunogenicidad: En estudios clínicos previos a la comercialización, aproximadamente el 17% de los pacientes desarrolló anticuerpos anti-GH no neutralizantes de bajo título. Aunque la presencia de estos anticuerpos no pareció influenciar la eficacia de SOMAVERT, se desconoce la importancia clínica de estos anticuerpos durante tiempo prolongado. No se encuentra disponible comercialmente ninguna determinación de anticuerpos anti-pegvisomant para pacientes que reciben SOMAVERT. Experiencia post-comercialización: Registro de pacientes con acromegalia tratados con SOMAVERT ACROSTUDY es un registro de observación internacional que obtiene los datos de seguridad a largo plazo en pacientes con acromegalia tratados con SOMAVERT, según se utiliza en la práctica clínica. La dosis de tratamiento y el cronograma se determinaron a criterio de cada médico tratante. Si bien era obligatorio el control de la seguridad según el cronograma estipulado, no se realizaron todas las evaluaciones en todos los puntos temporales para cada paciente. Como consecuencia de esto, las tasas de comparación de los eventos adversos con relación al ensayo clínico original no resultan apropiadas. En un informe provisorio, hubo 1288 pacientes inscritos (duración media del tratamiento de 3,7 años). Al inicio del tratamiento con SOMAVERT 648 pacientes se encontraban bajo monoterapia con SOMAVERT por acromegalia. De los 454 pacientes que tuvieron valores normales de AST y ALT al comienzo, 4 pacientes tuvieron resultados elevados > 3 veces el LSN, dos de los cuales tuvieron pruebas elevadas > 5 veces el LSN. Lipohipertrofia fue reportada en 6 pacientes (0,5%). Se compararon imágenes por resonancia magnética con imágenes previas, y un cambio en el volumen del tumor fue reportado como localmente significativo solo si el diámetro aumentaba más de 3 mm para los microadenomas o si el volumen aumentaba más del 20% para los macroadenomas. Todos los cambios en las imágenes por resonancia magnética considerados significativos en la lectura local fueron reanalizados centralmente. De los 747 pacientes que tuvieron una imagen reportada al nivel basal y al menos una durante el estudio de seguimiento, 51 pacientes (7%) tuvieron un incremento en la imagen local. De estos, 16 pacientes (2%) tuvieron confirmación de este incremento, 6 pacientes tuvieron un decrecimiento y 12 pacientes no tuvieron cambios; hubo un paciente con resultados insuficientes y 16 pacientes no tuvieron lectura central.
Advertencias.
General: Metabolismo de la Glucosa: El efecto de la GH se opone al efecto de la insulina sobre el metabolismo de los carbohidratos disminuyendo la sensibilidad a la insulina; de este modo, la tolerancia a la glucosa puede aumentar en algunos pacientes tratados con SOMAVERT. Aunque ninguno de los pacientes con acromegalia y diabetes mellitus que fueron tratados con SOMAVERT durante los estudios clínicos tuvieron hipoglucemia clínicamente relevante, estos pacientes deberán ser cuidadosamente monitoreados y las dosis de medicamentos antidiabéticos reducidas cuando sea necesario. Alteraciones en el hepatograma: Los niveles séricos basales de Fal, BT, AST y ALT deben obtenerse previo al inicio del tratamiento con SOMAVERT. La Tabla 3 enumera las recomendaciones con respecto al inicio del tratamiento con SOMAVERT en base a los resultados del laboratorio hepático. Se han observado elevaciones asintomáticas y transitorias de las transaminasas de hasta 15 veces el LSN en < 2% de los pacientes en la experiencia posterior a la comercialización entre dos ensayos abiertos (con un total de 147 pacientes). Estos informes no estuvieron asociados con un aumento en la bilirrubina y no tuvieron consecuencias clínicas para estos pacientes. Los aumentos de las transaminasas se normalizaron con el tiempo, con frecuencia después de suspender el tratamiento (SOMAVERT se debía utilizar conforme a la información de la Tabla 4 en cuanto a las anormalidades hepáticas).
Si un paciente desarrolla aumentos en el hepatograma, o cualquier otro signo o síntoma de disfunción hepática, mientras recibe SOMAVERT, se recomienda el siguiente control del paciente (Tabla 4).
Lipohipertrofia: Ha habido casos de lipohipertrofia en pacientes tratados con SOMAVERT. En un estudio doble ciego y controlado con placebo de 12 semanas de duración, se informó un caso (1,35) de lipohipertrofia en el lugar de la inyección en un sujeto que recibía 10 mg/día. El sujeto se recuperó mientras duraba el tratamiento. Entre dos estudios abiertos (con un total de 147 pacientes), hubo dos sujetos, ambos recibían 10 mg/día, que desarrollaron lipohipertrofia. Un caso se recuperó durante el tratamiento y el otro culminó con el abandono del tratamiento. Se deben alternar los lugares de la inyección a diario para ayudar a prevenir la lipohipertrofia (aplicar en un lugar diferente al de la última inyección). Información para pacientes: Los pacientes y todas las personas que puedan administrar SOMAVERT deben ser cuidadosamente informados por el profesional de la salud sobre cómo reconstituir correctamente e inyectar el producto (ver instrucciones adjuntas). A los pacientes se les debe informar acerca de la necesidad del monitoreo periódico del hepatograma. Se les debe decir que interrumpan inmediatamente el tratamiento y que se contacten con su médico si comienzan con ictericia. Además, a los pacientes se los debe concientizar sobre los niveles de IGF-I periódicos que se van a necesitar obtener para permitirle al médico ajustar correctamente la dosis de SOMAVERT. Pruebas de Laboratorio: Pruebas Hepáticas: Las recomendaciones para el monitoreo del hepatograma se encuentran establecidas anteriormente (ver Alteraciones en el hepatograma). Niveles del IGF-I: El tratamiento de SOMAVERT debe ser evaluado con el monitoreo de las concentraciones séricas de IGF-I cuatro a seis semanas luego de que el tratamiento se inicie o que cualquier ajuste de dosis sea realizado y por lo menos cada seis meses después de que los niveles del IGH-I se hayan normalizado. Los objetivos del tratamiento deben ser los de mantener la concentración sérica de IGH-I del paciente dentro del rango normal de ajuste por edad y controlar los signos y síntomas de acromegalia. Niveles de GH: Pegvisomant afecta las mediciones de las concentraciones séricas de GH realizadas con las determinaciones de GH disponibles comercialmente (ver Interacciones con Pruebas de Laboratorio). Además, aun cuando sean determinados con exactitud, los niveles de la GH usualmente aumentan durante el tratamiento con SOMAVERT. Por lo tanto, el tratamiento con SOMAVERT no debe ser ajustado en base a las concentraciones séricas de GH. Interacciones Medicamentosas: Los pacientes con acromegalia y diabetes mellitus que están siendo tratados con insulina y/o agentes hipoglucemiantes orales pueden requerir reducciones de dosis de estos agentes terapéuticos después del inicio del tratamiento con SOMAVERT. En estudios clínicos, los pacientes tratados con opiáceos, a menudo, necesitaron concentraciones séricas de pegvisomant mayores, para lograr la supresión del IGF-I apropiado, comparados con pacientes que no recibieron opiáceos. El mecanismo de esta interacción se desconoce. Interacciones con Pruebas de Laboratorio: Pegvisomant posee una estructura significativamente similar a la GH que provoca una reacción cruzada en las determinaciones de GH disponibles comercialmente. Dado que las concentraciones séricas de pegvisomant en dosis efectivas terapéuticamente son generalmente 100 a 1000 veces mayores que los niveles séricos endógenos de GH vistos en pacientes con acromegalia, las determinaciones de GH disponibles comercialmente exagerarán los niveles verdaderos de GH. Por lo tanto, el tratamiento con SOMAVERT no debe ser monitoreado o ajustado en base a las concentraciones séricas de GH informadas por estos ensayos. En cambio, el monitoreo y los ajustes de dosis deben ser en base a los niveles séricos de IGF-I. Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad: Pegvisomant se administró por vía subcutánea a ratas, diariamente durante 2 años, en dosis de 2, 8 y 20 mg/kg (alrededor de 2, 10 y 25 veces la dosis única de 20 mg en humanos basada en el AUC). El tratamiento a largo plazo con dosis de pegvisomant de 8 y 20 mg/kg causó un aumento de histiocitoma fibroso maligno en los sitios de inyección en ratones macho. No se observaron tumores en los sitios de inyección en ratas hembras en las mismas dosis. La mayor incidencia de tumores en los sitios de inyección fue muy probablemente causada por la irritación y la alta sensibilidad de la rata a repetidas inyecciones subcutáneas. Pegvisomant no causó alteraciones genéticas en los ensayos in vitro estándar (mutación bacteriana, aberraciones cromosómicas en linfocitos humanos). Pegvisomant resultó no tener efectos sobre la fertilidad o función reproductiva de conejas con dosis subcutáneas de hasta 10 mg/kg/día (10 veces la dosis recomendada en humanos en base al área de superficie corporal). Embarazo: Categoría B de embarazo. El desarrollo embrionario temprano y los estudios teratológicos fueron realizados en conejas preñadas administrando dosis subcutáneas de pegvisomant de 1, 3 y 10 mg/kg/día. No hubo evidencias de efectos teratogénicos asociados con el tratamiento de pegvisomant durante la organogénesis. Con la dosis de 10 mg/kg/día (10 veces la dosis terapéutica humana máxima en base al área de superficie corporal), un aumento leve, reproducible, en la pérdida post-implantación fue observado en ambos estudios. No se cuenta con estudios adecuados, bien controlados, en mujeres embarazadas. Debido a que los estudios de reproducción en animales no siempre son predictores de las respuestas en humanos, SOMAVERT debe ser utilizado durante el embarazo sólo si claramente se lo necesita. Madres en período de lactancia: Se desconoce si pegvisomant es excretado en la leche humana. Dado que muchas drogas son excretadas en la leche, se deberá tener cuidado cuando SOMAVERT sea administrado a una mujer en período de lactancia. Uso pediátrico: La seguridad y efectividad de SOMAVERT en pacientes pediátricos no han sido establecidas. Uso geriátrico: Los estudios clínicos de SOMAVERT no incluyeron números suficientes de individuos de 65 años de edad y mayores para determinar si ellos responden de manera diferente a los individuos más jóvenes. En general, la selección de dosis en un paciente mayor debe ser cuidadosa, usualmente comenzando con la menor dosis del rango de dosificación, reflejando la mayor frecuencia de disminución de la función cardiaca, renal o hepática y de enfermedad o tratamiento médico concomitante.
Dependencia.
Los datos disponibles no demuestran potencial abuso del medicamento ni dependencia psíquica de SOMAVERT. Pegvisomant radiomarcado no cruza la barrera hematoencefálica en ratas.
Conservación.
Previo a la reconstitución, SOMAVERT debe ser almacenado en un refrigerador a temperaturas de 2 a 8°C. Proteger del congelamiento. Luego de la reconstitución, SOMAVERT debe ser administrado dentro de las seis horas. Se debe administrar una sola dosis de cada frasco-ampolla. La solución reconstituida se puede mantener a temperatura ambiente (no mayor de 30°C) dentro del frasco ampolla o de la jeringa, pero se la debe inyectar dentro de las 6 (seis) horas. Descartar la solución si no se la ha utilizado dentro de las 6 (seis) horas.
Sobredosificación.
En un incidente de sobredosis aguda con SOMAVERT informado durante los estudios clínicos previos a la comercialización, un paciente se autoadministró 80 mg/día durante siete días. El paciente experimentó un leve aumento de la fatiga, no tuvo otras complicaciones y no mostró anormalidades de laboratorio clínicamente significativas. En casos de sobredosis, la administración de SOMAVERT debe suspenderse y no se debe continuar hasta que los niveles de IGF-I vuelvan dentro o por encima del rango normal. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital A. Posadas: (011) 4658-7777 / 4654-6648.
Presentación.
SOMAVERT se encuentra disponible en empaques 30 frascos ampolla de vidrio de dosis únicas, con las siguientes concentraciones: Frasco-ampolla de 10 mg de Pegvisomant (como proteína). Frasco-ampolla de 15 mg de Pegvisomant (como proteína). Cada empaque de SOMAVERT también incluye 30 frascos ampolla de disolvente (respectivamente) de dosis única que contiene Agua Estéril para Inyectables. El tapón del frasco ampolla de SOMAVERT contiene látex.
Revisión.
Diciembre 2014. LPD: 10/Diciembre/2013.