SOGROYA
NOVO NORDISK
Sustitución de la hormona de crecimiento (GH).
Composición.
Sogroya® 5 mg/1,5 ml: Un ml de solución contiene 3,3 mg de somapacitán*. Cada lapicera prellenada contiene 5 mg de somapacitán en 1,5 ml de solución. Sogroya® 10 mg/1,5 ml: Un ml de solución contiene 6,7 mg de somapacitán*. Cada lapicera prellenada contiene 10 mg de somapacitán en 1,5 ml de solución. Sogroya® 15 mg/1,5 ml: Un ml de solución contiene 10 mg de somapacitán*. Cada lapicera prellenada contiene 15 mg de somapacitán en 1,5 ml de solución. *Producido mediante tecnología de ADN recombinante en Escherichia coli seguido de la unión de un fragmento de unión a albúmina. Excipientes: histidina, manitol, poloxamer 188, fenol, ácido clorhídrico/hidróxido de sodio (para ajuste del pH), agua para inyectables. Forma farmacéutica: Solución inyectable. Líquido transparente a ligeramente opalescente, incoloro a ligeramente amarillento y libre de partículas visibles.
Farmacología.
Propiedades farmacodinámicas: Mecanismo de acción: Somapacitán es un derivado de la hormona de crecimiento humana recombinante de acción prolongada. Consiste en 191 aminoácidos similares a la hormona de crecimiento humana endógena con una única sustitución en la cadena principal de aminoácidos (L101C) al cual se ha unido una fracción de unión a la albúmina. La fracción de unión a la albúmina (cadena lateral) consiste en una fracción de ácido graso y un espaciador hidrofílico unido a la posición 101 de la proteína. El mecanismo de acción de somapacitán tiene lugar directamente a través del receptor de GH y/o indirectamente a través del IGF-I producido en los tejidos de todo el cuerpo, pero predominantemente por el hígado. Cuando se trata la deficiencia de hormona de crecimiento con somapacitán, se logra una normalización de la composición corporal (es decir, disminución de la masa grasa corporal, aumento de la masa magra corporal) y de la acción metabólica. Somapacitán estimula el crecimiento esquelético en pacientes pediátricos con GHD como resultado de los efectos en las placas de crecimiento (epífisis) de los huesos. Efectos farmacodinámicos: IGF-I IGF-I es un biomarcador universalmente aceptado para evaluar la eficacia en GHD. Tras la administración de somapacitán, se induce una respuesta de IGF-I dependiente de la dosis. Tras 1-2 dosis semanales se alcanza un patrón de estado estacionario en la respuesta de IGF-I. Los niveles de IGF-I fluctúan durante la semana. La respuesta de IGF-I es máxima después de 2 a 4 días. En comparación con el tratamiento diario con GH, el perfil de IGF-I de somapacitán difiere; ver Figura 1. En pacientes pediátricos con GHD, somapacitán produce una respuesta de IGF-I lineal a la dosis, con un cambio de 0,02 mg/kg en promedio, resultando en un cambio en la puntuación de desviación estándar (SDS) de IGF-I de 0,32.
Datos de eficacia clínica y seguridad: GHD en pediatría: REAL 4 (fase 3): La eficacia y seguridad de somapacitán una vez a la semana se evaluó en un estudio clínico de fase 3 (REAL 4) de 52 semanas, aleatorizado, multicéntrico, abierto, con control activo, por grupos paralelos en 200 pacientes pediátricos con GHD sin tratamiento previo. Los pacientes fueron aleatorizados a 0,16 mg/kg/semana de somapacitán una vez a la semana (N=132) o a 0,034 mg/kg/día de somatotropina una vez al día (N=68). Al inicio del estudio, los 200 pacientes tenían una media de edad de 6,4 años (rango: 2,5 a 11 años). El 74,5% de los pacientes eran varones. La velocidad de crecimiento anualizada a la semana 52 fue similar para somapacitán y somatotropina (Tabla 1).
De acuerdo con esto, los cambios a la semana 52 en comparación con el inicio con respecto a la SDS de altura y la SDS de IGF-I también fueron similares para somapacitán y somatotropina (Tabla 2).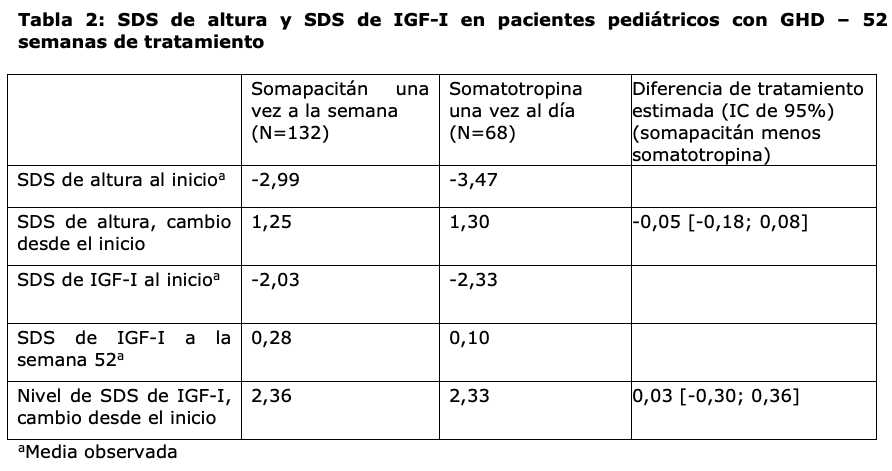
La gran mayoría de los pacientes pediátricos (96,9%) en el estudio alcanzó un nivel promedio de SDS de IGF-I dentro del rango normal (-2 a +2) después de 52 semanas de tratamiento con somapacitán una vez a la semana (Tabla 3). Un bajo número de pacientes tuvo una SDS de IGF-I promedio por encima de +2 (2,3%) y ningún paciente tuvo una SDS de IGF-I promedio por encima de +3.
REAL 3 (fase 2): Un total de 59 pacientes pediátricos con deficiencia de GH sin tratamiento previo de GH completaron un estudio de grupos paralelos de 4 brazos con un período principal de 26 semanas y una extensión de 26 semanas, con somapacitán una vez a la semana a niveles de dosis de 0,04, 0,08 y 0,16 mg/kg/semana y un brazo de control activo con 0,034 mg/kg/día de somatotropina una vez al día. Los pacientes continuaron en una extensión de seguridad de 104 semanas abierta, de brazos paralelos, con somapacitán 0,16 mg/kg/semana y somatotropina 0,034 mg/kg/día. Luego, todos los pacientes fueron transferidos a 0,16 mg/kg/semana de somapacitán una vez a la semana en una extensión de seguridad a largo plazo de 208 semanas. El tratamiento con somapacitán una vez a la semana condujo a beneficios del tratamiento continuos hasta al menos la semana 208. La SDS de la altura fue -1,06 (cambio desde el inicio: 2,85) en 38 pacientes. Los resultados de altura obtenidos a la semana 208 en pacientes que cambiaron de 0,034 mg/kg/día de somatotropina diaria a 0,16 mg/kg/semana de somapacitán semanal en la semana 156 indicaron que los beneficios del tratamiento con GH diaria se mantienen luego de cambiar a somapacitán una vez a la semana. El promedio de SDS de IGF-I se mantuvo dentro del rango normal para todos los grupos. GHD en adultos: En un estudio con control activo (abierto) y controlado con placebo (doble ciego) de 34 semanas de duración, se aleatorizaron 301 pacientes adultos con GHD sin tratamiento previo (2:1:2) y 300 fueron expuestos a somapacitán una vez a la semana o a placebo o a somatotropina diaria durante un período de 34 semanas de tratamiento (fase principal del estudio). La población de pacientes tenía una edad media de 45,1 años (rango de 23-77 años; 41 pacientes tenían 65 años o más), el 51,7% eran mujeres, y el 69,7% tenía GHD iniciada en la adultez. Un total de 272 pacientes adultos con GHD que completaron la fase principal de 34 semanas continuaron con una fase de extensión abierta de 53 semanas. Los pacientes con placebo cambiaron a somapacitán y los pacientes con somatotropina volvieron a ser aleatorizados (1:1) a somapacitán o a somatotropina. A continuación se presentan los efectos clínicos observados para los criterios de valoración principales en la fase principal del tratamiento (Tabla 4) y en la fase de extensión del tratamiento (Tabla 5).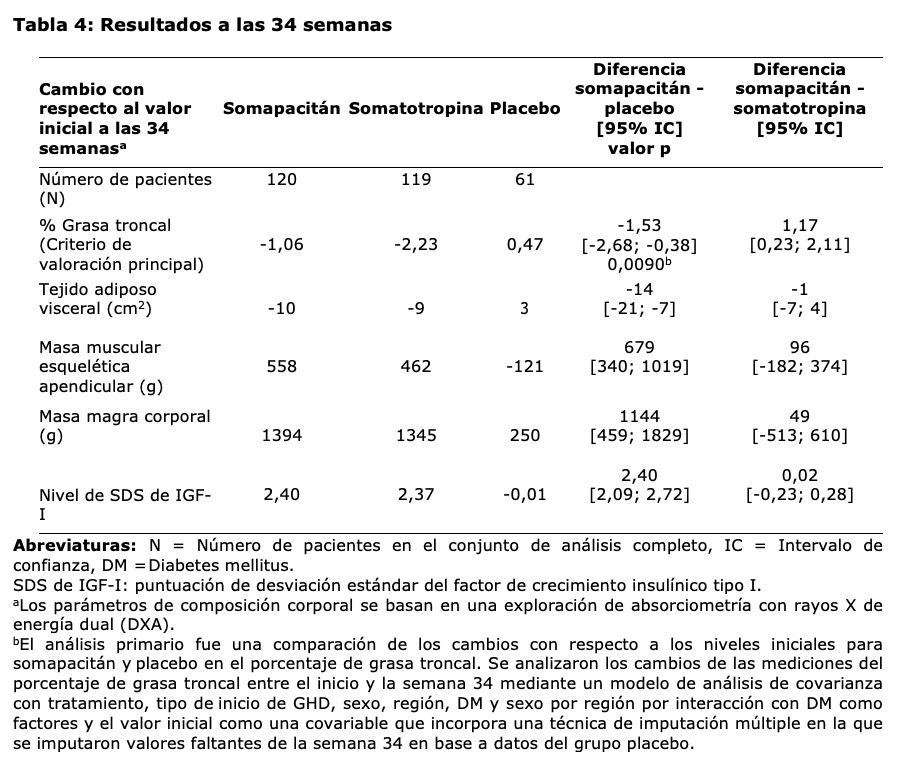
El análisis de subgrupos post-hoc de los cambios desde el valor inicial en el porcentaje de grasa troncal (%) en comparación con el placebo en la semana 34 mostró una diferencia de tratamiento estimada (somapacitán-placebo) de -2,49% [-4,19; -0,79] en hombres, -0,80% [-2,99; 1,39] en mujeres sin tratamiento con estrógenos orales, -1,44% [-3,97; 1.09] en mujeres en tratamiento con estrógenos orales.
Niveles de SDS de IGF-I observados y simulados en el estudio clínico En la fase principal del estudio clínico, en general se alcanzaron valores de SDS de IGF-I de 0 y superiores en el 53% de los pacientes adultos con GHD tratados con somapacitán en el estudio después de un período de ajuste de la dosis de 8 semanas. Sin embargo, esta proporción fue menor en subgrupos particulares como las mujeres en tratamiento con estrógenos orales (32%) y los pacientes con inicio en la niñez (39%) (Tabla 6). Los análisis de simulación post-hoc indicaron que se espera que las proporciones de pacientes adultos con GHD que alcancen niveles de SDS de IGF-I por encima de 0 sean mayores si se permitiera un ajuste de la dosis de somapacitán después de las 8 semanas. En este análisis de simulación, se asumió que el ajuste de la dosis de somapacitán fue bien tolerado en todos los pacientes hasta que se alcanzó el rango objetivo de SDS de IGF-I o una dosis de somapacitán de 8 mg por semana.
Dosis de mantenimiento La dosis de mantenimiento varía de persona a persona y entre hombres y mujeres. La dosis media de mantenimiento de somapacitán observada en los estudios clínicos de fase 3 fue 2,4 mg/semana. GHD en pediatría y en adultos: Seguridad clínica: El perfil de seguridad de somapacitán fue similar al perfil de seguridad bien conocido de somatotropina. No se identificaron nuevos problemas de seguridad; ver sección Reacciones adversas. Inmunogenicidad: Se detectaron con poca frecuencia anticuerpos antimedicamento (ADA, por sus siglas en inglés) en pacientes pediátricos (16/132). Ninguno de esos anticuerpos fue neutralizante. No se observó evidencia de que los ADA tuvieran impacto en la farmacocinética, eficacia o seguridad. No se detectaron anticuerpos antimedicamento en pacientes adultos. Propiedades farmacocinéticas: Somapacitán tiene propiedades farmacocinéticas compatibles con una administración de una vez a la semana. La unión reversible a la albúmina endógena retrasa la eliminación de somapacitán y, por lo tanto, prolonga la vida media in vivo y la duración de la acción. Se ha investigado la farmacocinética de somapacitán tras la administración subcutánea a niveles de dosis de 0,02 a 0,16 mg/kg/semana en población pediátrica (2,5 a 14 años), a niveles de dosis de 0,01 a 0,32 mg/kg en adultos sanos y en dosis de hasta 0,12 mg/kg en pacientes adultos con GHD. En general, somapacitán muestra una farmacocinética no lineal en el rango de dosis investigado. Sin embargo, en el rango de dosis clínicamente relevante de somapacitán en adultos con GHD, la farmacocinética de somapacitán es aproximadamente lineal. En GHD pediátrica, una dosis de 0,16 mg/kg/semana de somapacitán corresponde a una concentración promedio de 80,2 ng/ml, y en GHD en adultos, las dosis de somapacitán en el rango clínicamente relevante corresponden a concentraciones promedio de 0,1-36,2 ng/ml. Absorción: En pacientes adultos y pediátricos con GHD, la mediana de la tmáx fue de 4 a 25,5 horas en dosis desde 0,02 mg/kg/semana hasta 0,16 mg/kg/semana. El estado estacionario se alcanzó tras 1-2 administraciones semanales. No se ha investigado la biodisponibilidad absoluta de somapacitán en humanos. Distribución: Somapacitán se encuentra ampliamente unido ( > 99%) a proteínas plasmáticas y se espera que se distribuya como la albúmina. Según los análisis farmacocinéticos poblacionales, el volumen de distribución estimado (V/F) fue de 1,7 litros en pacientes pediátricos con GHD y de 14,6 litros en pacientes adultos con GHD. Eliminación: Tras una dosis única y dosis repetidas de 0,16 mg/kg/semana, la vida media terminal fue de aproximadamente 34 horas en pacientes pediátricos con GHD. La vida media terminal se estimó con las medias geométricas que fueron de aproximadamente 2 a 3 días en estado estacionario en pacientes pediátricos y adultos con GHD (dosis: 0,02 a 0,12 mg/kg). Somapacitán estará presente en la circulación durante aproximadamente 2 semanas tras la última dosis. Se ha observado poca o ninguna acumulación (índice de acumulación medio: 1-2) de somapacitán tras dosis múltiples. Biotransformación: Somapacitán se metaboliza en gran medida por degradación proteolítica y escisión de la secuencia que une el péptido y la fracción de unión a albúmina. Somapacitán se metabolizó en gran medida antes de la excreción y no se encontró somapacitán intacto en la orina, que fue la principal vía de excreción (81%) ni en las heces, donde se encontró el 13% del material relacionado con somapacitán, lo que indica una biotransformación completa antes de la excreción. Poblaciones especiales: Pacientes pediátricos con GHD: En base al análisis farmacocinético poblacional, el sexo, la raza y el peso corporal no tienen un efecto clínicamente significativo en la farmacocinética tras la dosificación en base al peso. Pacientes adultos con GHD: Edad: Los pacientes mayores de 60 años tienen una exposición superior (29%) que los pacientes más jóvenes con la misma dosis de somapacitán. En la sección Posología y modo de administración se describe una dosis de inicio menor para pacientes mayores de 60 años. Sexo: Las mujeres y en particular las mujeres en tratamiento con estrógenos orales tienen una exposición más baja (53% en mujeres en tratamiento con estrógenos orales y 30% en mujeres sin tratamiento con estrógenos orales) que los hombres con la misma dosis de somapacitán. En la sección Posología y modo de administración se describe una dosis de inicio más alta en mujeres en tratamiento con estrógenos orales. Raza: No hubo diferencia en la exposición a somapacitán y la respuesta de IGF-I entre los pacientes japoneses y caucásicos. A pesar de haber una exposición mayor en la población asiática no japonesa en comparación con la caucásica con la misma dosis de somapacitán, las poblaciones caucásica, japonesa y asiática no japonesa necesitaron las mismas dosis para alcanzar niveles similares de IGF-I. Por lo tanto, no hay una recomendación de ajuste de la dosis según la raza. Etnia: No se investigaron los grupos étnicos (hispano o latino 4,5% (15 pacientes recibieron somapacitán)) debido al reducido tamaño de la muestra en el programa de desarrollo. Peso corporal A pesar de existir una mayor exposición en los pacientes con un peso corporal inferior en comparación con los pacientes con un peso corporal superior con la misma dosis de somapacitán, los pacientes necesitaron las mismas dosis para alcanzar niveles similares de IGF-I en el rango de peso corporal de 35 kg a 150 kg. Por lo tanto, no hay una recomendación de ajuste de la dosis según el peso corporal. Insuficiencia renal: Una dosis de somapacitán de 0,08 mg/kg en estado estacionario dio lugar a exposiciones mayores en pacientes con insuficiencia renal, más pronunciada en pacientes con insuficiencia renal grave y en pacientes que requieren hemodiálisis, donde los índices de AUC0-168h con respecto a la función renal normal fueron 1,75 y 1,63, respectivamente. En general, la exposición a somapacitán tendió a aumentar con la disminución de la TFG. Se observaron niveles más altos de AUC0-168h de IGF-I en pacientes con insuficiencia renal moderada y grave y en pacientes que requerían hemodiálisis, con índices con respecto a una función renal normal de 1,35, 1,40 y 1,24 respectivamente. Debido al aumento moderado observado en IGF-I combinado con las bajas dosis de inicio recomendadas y el ajuste individual de la dosis de somapacitán, no se recomienda hacer ningún ajuste de la dosis en los pacientes con insuficiencia renal. Insuficiencia hepática: Una dosis de somapacitán de 0,08 mg/kg en estado estacionario dio lugar a exposiciones mayores en pacientes con insuficiencia hepática moderada con índices con respecto a una función hepática normal de 4,69 para AUC0-168h y 3,52 para Cmáx. Se observaron niveles más bajos de IGF-I estimulados por somapacitán en los pacientes con insuficiencia hepática leve y moderada en comparación con los pacientes con una función hepática normal (el índice con respecto a la función normal fue 0,85 para leve y 0,75 para moderada). Debido a la disminución moderada observada en IGF-I combinada con el ajuste de la dosis individual de somapacitán, no se recomienda hacer ningún ajuste de la dosis en los pacientes con insuficiencia hepática. Datos preclínicos de seguridad: Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad o desarrollo prenatal y posnatal. No se han realizado estudios de carcinogenicidad con somapacitán. No se han observado efectos adversos en la fertilidad de ratas macho y hembra a dosis resultando en una exposición al menos 13 y 15 veces superior a la exposición clínica máxima esperada a 8 mg/semana para machos y hembras, respectivamente. No obstante, se ha observado un ciclo estral irregular en las hembras en todas las dosis de tratamiento. No se identificaron evidencias de daño fetal al administrar somapacitán subcutáneo en ratas y conejas preñadas durante la organogénesis con dosis que conducían a exposiciones muy por encima de la exposición esperada a la dosis clínica máxima de 8 mg/semana (al menos 18 veces). En dosis altas que conducían a exposiciones de al menos 130 veces por encima de la exposición clínica máxima esperada a 8 mg/semana, se hallaron huesos largos acortados/doblados/engrosados en crías de ratas hembra que recibieron somapacitán. Se sabe que estos hallazgos en ratas se resuelven tras el nacimiento y deben valorarse como malformaciones menores, no como anomalías permanentes. Se redujo el crecimiento fetal tras la administración de somapacitán subcutáneo a conejas preñadas en exposiciones al menos 9 veces por encima de la exposición esperada con la dosis clínica máxima de 8 mg/semana. En el caso de las ratas lactantes, se secretó material relacionado con somapacitán en la leche pero en un nivel inferior al observado en plasma (hasta un 50% del nivel en plasma).
Indicaciones.
Sogroya® está indicado como sustitución de la hormona de crecimiento (GH) endógena en niños a partir de los 3 años de edad y adolescentes con falla en el crecimiento debido a deficiencia de hormona de crecimiento (GHD en pediatría) y en adultos con deficiencia de hormona de crecimiento (GHD en adultos).
Dosificación.
El tratamiento con somapacitán se debe iniciar y monitorizar por médicos debidamente calificados y con la experiencia adecuada en el diagnóstico y el tratamiento de pacientes con deficiencia de hormona de crecimiento (por ejemplo, endocrinólogos). Posología:
GHD en pediatría: Ajuste de la dosis La dosis de somapacitán puede individualizarse y ajustarse en base a la velocidad de crecimiento, reacciones adversas, peso corporal y concentraciones séricas del factor de crecimiento insulínico tipo I (IGF-I). Los niveles promedio de la puntuación de desviación estándar (SDS) de IGF-I (tomados 4 días después de la dosificación) pueden guiar el ajuste de la dosis. Los ajustes de la dosis deben apuntar a alcanzar niveles promedio de SDS de IGF-I en el rango normal, es decir, entre -2 y +2 (preferiblemente cerca de SDS 0). Si el IGF-I (SDS) es > 2, se debe reevaluar después de la siguiente administración de somapacitán. Si el valor permanece > 2, se recomienda reducir la dosis en 0,04 mg/kg/semana. En algunos pacientes, puede requerirse más de una reducción de la dosis. En pacientes que tuvieron una reducción de dosis pero no están creciendo bien, la dosis puede aumentarse gradualmente según la tolerancia hasta una dosis máxima de 0,16 mg/kg/semana. Los aumentos de dosis no deberían exceder los 0,02 mg/kg por semana. Evaluación del tratamiento: Se debe considerar la evaluación de la eficacia y seguridad a intervalos de aproximadamente 6 a 12 meses, y puede evaluarse mediante parámetros auxológicos, bioquímica (niveles de IGF-I, hormonas, glucosa y lípidos) y estado puberal. Deben considerarse evaluaciones más frecuentes durante la pubertad. Debe interrumpirse el tratamiento en pacientes que hayan alcanzado la altura final o cerca de la altura final, es decir, una velocidad de crecimiento anualizada < 2 cm/año y una edad ósea > 14 años en mujeres o > 16 años en varones, que corresponde al cierre de las placas de crecimiento epifisarias; ver sección Contraindicaciones. Una vez que las epífisis están fusionadas, los pacientes deben ser reevaluados clínicamente para determinar la necesidad del tratamiento con hormona de crecimiento. Si la deficiencia de hormona de crecimiento persiste después de completado el crecimiento, se debería continuar el tratamiento con hormona de crecimiento para alcanzar el desarrollo somático adulto completo, incluyendo la masa magra corporal y el depósito mineral óseo (para información sobre la dosificación ver la dosis recomendada para adultos (Tabla 7)). GHD en adultos: Ajuste de la dosis: La dosis de somapacitán se debe ajustar individualmente para cada paciente. Se recomienda aumentar la dosis de manera gradual en intervalos de 2-4 semanas en incrementos de 0,5 mg a 1,5 mg según la respuesta clínica y las reacciones adversas experimentadas por los pacientes, hasta una dosis de 8 mg de somapacitán por semana. Se pueden utilizar los niveles séricos del factor de crecimiento insulínico tipo I (IGF-I) (extraído 3-4 días tras la administración) como guía para ajustar la dosis. El objetivo es que la puntuación de la desviación estándar (SDS) del IGF-I se encuentre en el rango normal superior sin exceder 2 SDS. Los niveles de SDS de IGF-I en el rango objetivo normalmente se alcanzan dentro de las 8 semanas de ajuste de la dosis. Se puede necesitar un ajuste de la dosis más prolongado en algunos pacientes adultos con GHD (ver a continuación y Propiedades farmacodinámicas). Evaluación del tratamiento: Usando el SDS de IGF-I como biomarcador para el ajuste de dosis, el objetivo es alcanzar niveles de SDS de IGF-I en el rango superior de referencia ajustado a la edad (rango superior de referencia de SDS de IGF-I: 0 a +2) en 12 meses de ajuste. Si no se alcanza dicho rango en este período o el paciente no obtiene la respuesta clínica deseada, se deben considerar otras opciones de tratamiento. Se debe considerar una evaluación de eficacia y seguridad durante el tratamiento de mantenimiento con somapacitán en intervalos de 6 a 12 meses aproximadamente, y se puede evaluar analizando la bioquímica (niveles de IGF-I, glucosa y lípidos), composición corporal e índice de masa corporal. GHD en pediatría y adultos: Cambio desde otros productos de hormona de crecimiento: Se recomienda que los pacientes que cambian desde una hormona de crecimiento semanal a somapacitán continúen la administración en su día habitual de dosificación semanal. Los pacientes que cambian desde una hormona de crecimiento humana diaria a somapacitán una vez por semana deberían elegir el día de su preferencia para la dosis semanal, e inyectarse la dosis final del tratamiento diario el día anterior (o al menos 8 horas antes) a inyectarse la primera dosis de somapacitán semanal. Los pacientes deben seguir las instrucciones de dosis presentadas en la Tabla 7. Tratamiento con estrógenos orales: Las mujeres en tratamiento con estrógenos orales pueden tener niveles reducidos de IGF-I y pueden requerir un ajuste de la dosis de hormona de crecimiento para alcanzar el objetivo del tratamiento (ver sección Advertencias y precauciones especiales de uso). En GHD pediátrica, no se han estudiado y no se recomiendan dosis superiores a 0,16 mg/kg/semana. Dosis omitida: Se debe advertir a los pacientes que omitan una dosis que se inyecten somapacitán semanal tan pronto como sea posible, dentro de los 3 días posteriores a la dosis omitida, y que después reanuden su esquema de dosificación habitual una vez a la semana. En caso de que hayan transcurrido más de 3 días, se debe saltear la dosis y administrar la siguiente dosis de forma habitual en el día programado. Si se han omitido dos o más dosis, se debe reanudar la dosificación de forma habitual en el día programado. Cambio del día de administración: Se puede cambiar el día de inyección semanal siempre que el tiempo entre dos dosis sea de al menos 4 días. Una vez seleccionado el nuevo día de administración, se debe continuar con el esquema de dosificación de una vez a la semana. Flexibilidad en el momento de administración: En ocasiones en que no es posible la administración en el día programado, somapacitán semanal puede administrarse hasta 2 días antes o 3 días después del día de la semana programado, siempre que el tiempo entre dos dosis sea de al menos 4 días (96 horas). La administración semanal de la próxima dosis puede reanudarse en el día programado habitual. Poblaciones especiales: Pacientes de edad avanzada (60 años o más): Generalmente se necesitan dosis menores de somapacitán en los pacientes de edad avanzada. Para más información, ver sección Propiedades farmacocinéticas. Población pediátrica: Hay datos limitados sobre los efectos clínicos de somapacitán en pacientes pediátricos con GHD menores de 3 años de edad. Los datos actualmente disponibles se describen en las secciones Propiedades farmacodinámicas y Propiedades farmacocinéticas, pero no puede recomendarse una posología. Sexo: Los hombres muestran un incremento en la sensibilidad a IGF-I con el tiempo. Esto implica que existe un riesgo de que los hombres sean sobretratados. Las mujeres, especialmente aquellas en tratamiento con estrógenos orales, pueden necesitar dosis más altas y un período de ajuste de dosis más prolongado que los hombres; ver secciones Propiedades farmacodinámicas y Propiedades farmacocinéticas. En mujeres en tratamiento con estrógenos orales, se debe considerar cambiar la vía de administración del estrógeno (por ejemplo, transdérmica, vaginal); ver sección Advertencias y precauciones especiales de uso. Insuficiencia renal: No se necesita ajustar la dosis de inicio en pacientes con insuficiencia renal. Puede que los pacientes con insuficiencia renal necesiten dosis inferiores de somapacitán, pero dado que la dosis de somapacitán se ajusta de forma individual en función de las necesidades de cada paciente, no es necesario un ajuste adicional de la dosis; ver sección Propiedades farmacocinéticas. Insuficiencia hepática: No se necesita ajustar la dosis de inicio en pacientes con insuficiencia hepática. Puede que los pacientes con insuficiencia hepática moderada necesiten dosis superiores de somapacitán, pero dado que la dosis de somapacitán se ajusta de forma individual en función de las necesidades de cada paciente, no es necesario un ajuste adicional de la dosis. No hay información disponible sobre el uso de somapacitán en pacientes con insuficiencia hepática grave. Se debe extremar la precaución al tratar a estos pacientes con somapacitán; ver sección Propiedades farmacocinéticas. Modo de administración: Somapacitán se debe administrar una vez a la semana a cualquier hora del día. Somapacitán debe inyectarse por vía subcutánea en el abdomen, los muslos, los glúteos o la parte superior de los brazos sin ajustar la dosis. Se debe rotar el sitio de inyección cada semana para prevenir lipoatrofia local. La lapicera de Sogroya® 5 mg/1,5 ml (3,3 mg/ml) administra dosis de 0,025 mg (0,0075 ml) a 2 mg (0,6 ml) en incrementos de 0,025 mg. La lapicera de Sogroya® 10 mg/1,5 ml (6,7 mg/ml) administra dosis de 0,05 mg (0,0075 ml) a 4 mg (0,6 ml) en incrementos de 0,05 mg. La lapicera de Sogroya® 15 mg/1,5 ml (10 mg/ml) administra dosis de 0,10 mg (0,01 ml) a 8 mg (0,8 ml) en incrementos de 0,10 mg. Para instrucciones acerca del medicamento antes de la administración, ver sección Precauciones especiales de descarte y manipulación.
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes listados en la sección Composición. No se debe utilizar somapacitán cuando exista evidencia de actividad de un tumor. Los tumores intracraneales deben estar inactivos y se debe completar el tratamiento antitumoral antes de iniciar el tratamiento con somapacitán. Se debe interrumpir el tratamiento si hay evidencia de crecimiento tumoral; ver sección Advertencias y precauciones especiales de uso. No se debe utilizar somapacitán para la promoción del crecimiento longitudinal en niños con epífisis cerradas; ver sección Posología y modo de administración. Los pacientes con enfermedad crítica aguda que presenten complicaciones tras una cirugía a corazón abierto, cirugía abdominal, traumatismo accidental múltiple, insuficiencia respiratoria aguda o condiciones similares, no deben ser tratados con somapacitán (en cuanto a los pacientes sometidos a tratamiento de sustitución, ver sección Advertencias y precauciones especiales de uso).
Reacciones adversas.
Resumen del perfil de seguridad: Las reacciones adversas al medicamento notificadas con mayor frecuencia son (en orden decreciente, [GHD pediátrica, GHD en adultos]: cefalea (12%, 12%), dolor en extremidades (9%, N/A), hipotiroidismo (5%, 2%), reacciones en el sitio de inyección (5%, 1%), edema periférico (3%, 4%), artralgia (2%, 7%), hiperglucemia (2%, 1%), fatiga (2%, 6%) e insuficiencia adrenocortical (1,5%, 3%). Tabla de reacciones adversas: Las reacciones adversas listadas en la Tabla 8 se basan en los datos de seguridad de un estudio pivotal de fase 3 en curso (52 semanas) en pacientes pediátricos con GHD (edad al inicio: 2,5 a 11 años) y las reacciones adversas del tratamiento con somapacitán. Las frecuencias de las reacciones adversas fueron calculadas en base a las frecuencias en el estudio pivotal de fase 3. Las reacciones adversas que se mencionan en la Tabla 9 se basan en los datos de seguridad recopilados de tres estudios de fase 3 completados en pacientes adultos con GHD (edad al inicio: 19 a 77 años). Las reacciones adversas están enumeradas según la clasificación de órganos del sistema MedDRA y las categorías de frecuencia se definen del siguiente modo: muy frecuentes (≥1/10); frecuentes (≥1/100 a < 1/10); poco frecuentes (≥1/1.000 a < 1/100); raras (≥1/10.000 a < 1/1.000); muy raras ( < 1/10.000).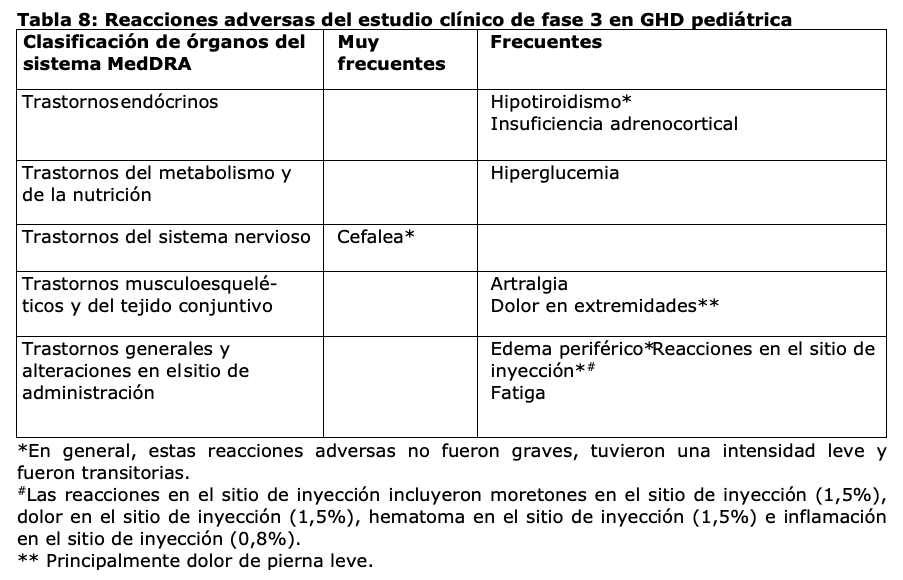
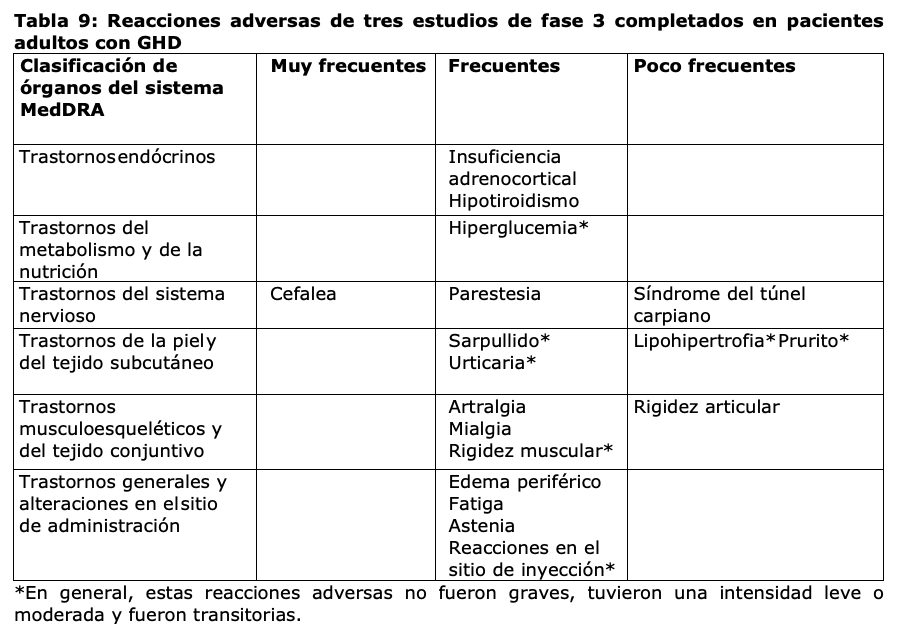
Descripción de reacciones adversas seleccionadas: Edema periférico: El edema periférico se observó frecuentemente (3% en GHD pediátrica y 4% en GHD en adultos). Los pacientes con deficiencia de hormona de crecimiento se caracterizan por tener un déficit de volumen extracelular. Este déficit se corrige cuando se inicia el tratamiento con hormona de crecimiento. Puede aparecer retención de líquidos con edema periférico. Los síntomas son normalmente transitorios, dependientes de la dosis y pueden requerir una reducción transitoria de la dosis. Insuficiencia adrenocortical Se observó insuficiencia adrenocortical frecuentemente (1,5% en GHD pediátrica y 3% en GHD en adultos); ver sección Advertencias y precauciones especiales de uso. Población pediátrica: La seguridad de somapacitán fue establecida en niños y adolescentes a partir de los 3 años de edad con falla en el crecimiento debido a GHD. El perfil de seguridad de somapacitán en pacientes con GHD menores de 3 años de edad no está establecido.
Advertencias.
Insuficiencia adrenocortical: La introducción del tratamiento con hormona de crecimiento puede provocar la inhibición de 11bHSD-1 y una reducción de las concentraciones de cortisol sérico. En pacientes tratados con hormona de crecimiento se puede desenmascarar un hipoadrenalismo central (secundario) no diagnosticado previamente y es posible que requiera tratamiento sustitutivo con glucocorticoides. Asimismo, los pacientes en tratamiento sustitutivo con glucocorticoides por hipoadrenalismo diagnosticado previamente, pueden requerir un incremento de sus dosis de mantenimiento o de refuerzo tras el inicio del tratamiento con hormona de crecimiento. Es necesario monitorear a los pacientes con hipoadrenalismo conocido para detectar una reducción de los niveles séricos de cortisol y/o la necesidad de aumentar la dosis de glucocorticoides; ver Interacción con otros medicamentos y otras formas de interacción. Trastorno en el metabolismo de la glucosa: El tratamiento con hormona de crecimiento puede reducir la sensibilidad a la insulina, sobre todo en dosis altas en pacientes susceptibles y, como consecuencia, se puede producir hiperglucemia en personas con una capacidad inadecuada de secreción de insulina. Como resultado, durante el tratamiento con hormona de crecimiento se puede desenmascarar una intolerancia a la glucosa o diabetes mellitus no diagnosticadas previamente. Por lo tanto, se deben monitorizar periódicamente los niveles de glucosa en todos los pacientes tratados con hormona de crecimiento, sobre todo en aquellos con factores de riesgo de diabetes mellitus, como obesidad o antecedentes familiares de diabetes mellitus. Durante el tratamiento con hormona de crecimiento se debe monitorizar estrechamente a los pacientes con diabetes mellitus tipo 1 o tipo 2 o intolerancia a la glucosa preexistentes. Puede que se necesite ajustar las dosis de medicamentos antihiperglucémicos cuando estos pacientes inicien el tratamiento con hormona de crecimiento. Neoplasias: No hay evidencia de un mayor riesgo de nuevos cánceres primarios en pacientes tratados con hormona de crecimiento. No se ha asociado el tratamiento con hormona de crecimiento a un aumento del índice de recidivas en pacientes en remisión completa de neoplasias malignas o que han sido tratados por tumores benignos. Se debe realizar un estrecho seguimiento de los pacientes que han alcanzado la remisión completa de una neoplasia maligna o que han sido tratados por tumores benignos, por si se produjese una recidiva tras comenzar con el tratamiento con hormona de crecimiento. Se debe interrumpir el tratamiento con hormona de crecimiento si se desarrolla o reaparece un tumor maligno o benigno. Se ha observado un ligero aumento general en las segundas neoplasias en los sobrevivientes a cáncer infantil tratados con hormona de crecimiento, siendo los más frecuentes los tumores intracraneales. El factor de riesgo principal para las neoplasias secundarias parece ser la exposición previa a radiación. Hipertensión intracraneal benigna: Se recomienda realizar una fundoscopía en casos de cefalea intensa o recurrente, síntomas visuales, náuseas y/o vómitos para descartar papiledema. Si se confirma papiledema, se debe considerar un diagnóstico de hipertensión intracraneal benigna y, si es necesario, discontinuar el tratamiento con hormona de crecimiento. Actualmente, no hay evidencia suficiente para orientar la toma de decisiones clínicas en pacientes con hipertensión intracraneal resuelta. Si se reinicia el tratamiento con hormona de crecimiento, es necesario un estrecho monitoreo de síntomas de hipertensión intracraneal. Función tiroidea: La hormona de crecimiento incrementa la conversión extratiroidea de T4 a T3 y puede, por ello, desenmascarar un hipotiroidismo incipiente. Dado que el hipotiroidismo interfiere con la respuesta al tratamiento con hormona de crecimiento, los pacientes deben controlar regularmente su función tiroidea y, cuando esté indicado, recibir un tratamiento sustitutivo con hormona tiroidea; ver Interacción con otros medicamentos y otras formas de interacción y Reacciones adversas. Uso con estrógenos por vía oral: Los estrógenos orales influyen en la respuesta de IGF-I a la hormona de crecimiento, incluido somapacitán. Mujeres en tratamiento con cualquier forma de estrógenos orales (terapia hormonal o anticonceptivos) deben considerar cambiar la vía de administración del estrógeno (por ejemplo, transdérmica, productos hormonales vaginales) o usar otro método anticonceptivo. Si una mujer en tratamiento con estrógenos orales inicia el tratamiento con somapacitán, puede requerir dosis de inicio más altas y un período de ajuste de la dosis más prolongado (ver sección Posología y modo de administración). Si una mujer en tratamiento con somapacitán inicia un tratamiento con estrógenos orales, es posible que necesite aumentar la dosis de somapacitán para mantener los niveles séricos de IGF-I dentro del rango normal adecuado para la edad. Por el contrario, si una mujer en tratamiento con somapacitán suspende el tratamiento con estrógenos orales, es posible que sea necesario reducir la dosis de somapacitán para evitar el exceso de somapacitán y/o reacciones adversas; ver secciones Posología y modo de administración e Interacción con otros medicamentos y otras formas de interacción. Trastornos de la piel y del tejido subcutáneo: Si se administra somapacitán en el mismo sitio durante un largo período de tiempo, podrían ocurrir cambios locales en el tejido subcutáneo como lipohipertrofia, lipoatrofia y lipodistrofia adquirida. Se debe rotar el sitio de inyección para minimizar el riesgo; ver secciones Posología y modo de administración y Reacciones adversas. Anticuerpos: No se han observado anticuerpos contra somapacitán en pacientes adultos con GHD. Pocos pacientes pediátricos con GHD resultaron positivos en el análisis de anticuerpos de unión a somapacitán. Ninguno de estos anticuerpos fue neutralizante y no se observó un impacto en los efectos clínicos. Se debe analizar la presencia de anticuerpos anti-somapacitán en pacientes con una falta de respuesta al tratamiento. Enfermedad crítica aguda: Se ha evaluado el efecto de la hormona de crecimiento en la recuperación en dos estudios controlados con placebo en 522 pacientes adultos en estado crítico que sufrieron complicaciones posteriores a cirugía a corazón abierto, cirugía abdominal, traumatismo accidental múltiple o insuficiencia respiratoria aguda. La mortalidad fue superior en los pacientes tratados con 5,3 u 8 mg de hormona de crecimiento diaria en comparación con los pacientes que recibieron placebo, 42% frente a 19%. De acuerdo con esta información, este tipo de pacientes no debería ser tratado con somapacitán. Dado que no hay información disponible sobre la seguridad del tratamiento sustitutivo con hormona de crecimiento en pacientes en estado crítico agudo, se deben considerar los beneficios del tratamiento continuado en esta situación frente a los riesgos potenciales. La deficiencia de hormona de crecimiento en adultos es una enfermedad crónica y necesita ser tratada, sin embargo, la experiencia es todavía limitada en pacientes mayores de 60 años y en pacientes con más de cinco años en tratamiento para la deficiencia de hormona de crecimiento en adultos. Pancreatitis: Se han notificado pocos casos de pancreatitis durante el tratamiento con otros medicamentos de hormona de crecimiento. Por lo tanto, debe ser considerada en pacientes tratados con somapacitán que desarrollen dolor abdominal sin una explicación. Sodio: Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis; esto es, esencialmente "libre de sodio". Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de Sogroya® sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Interacción con otros medicamentos y otras formas de interacción: Medicamentos metabolizados por el citocromo P450: Los datos de un estudio de interacción realizado en adultos con deficiencia de hormona de crecimiento sugieren que la administración de la hormona de crecimiento puede aumentar el clearance de compuestos que son metabolizados por las isoenzimas del citocromo P450. Puede aumentar especialmente el clearance de los compuestos metabolizados por el citocromo P450 (p. ej. esteroides sexuales, corticoesteroides, anticonvulsivantes y ciclosporina) dando lugar a niveles plasmáticos más bajos de estos compuestos. Se desconoce la relevancia clínica de este hallazgo. Glucocorticoides: La hormona de crecimiento disminuye la conversión de cortisona a cortisol y puede desenmascarar un hipo