SINLIP® PREVENT
GADOR
Antagonista de Angiotensina II. Diurético. Hipolipemiante.
Composición.
Cada cápsula dura de SINLIP<<^>® PREVENT contiene 1 comprimido de Rosuvastatina 10 mg + 1 comprimido de Candesartan cilexetil 16 mg - Hidroclorotiazida 12,5 mg. Cada comprimido de Rosuvastatina contiene: Rosuvastatina (como Rosuvastatina cálcica 10,4 mg) 10 mg Excipientes: Ludipress1), Poloxamero 188, Sílica coloidal anhidra, Estearato de magnesio vegetal, Óxido de hierro rojo, Opadry YS 1 7003 blanco2), Opaglos AG 73503) c.s. 1) Compuesto por: Lactosa monohidrato; Povidona K30; Crospovidona. 2) Compuesto por: HPMC 3 cP Methocel E3 LV; HPMC 6 cP Methocel E6 LV; Dióxido de titanio; PEG 400; Polisorbato 80. 3) Compuesto por: Agua purificada; Cera de abejas blanca; Cera carnauba; Polisorbato 20; Ácido sórbico. Cada comprimido recubierto de Candesartan cilexetil / Hidroclorotiazida contiene: Candesartan cilexetil 16 mg Hidroclorotiazida 12,5 mg. Excipientes: Lactosa monohidrato, Almidón 1500, Hidroxipropilcelulosa EFX, Croscaramelosa sódica, Estearato de magnesio vegetal, Polietilenglicol 8000, Óxido de hierro Rojo, Óxido de hierro Amarillo c.s. Componentes de la cápsula: Gelatina; Dióxido de titanio; Verde FD&C N°3; Amarillo FD&C N°6.
Farmacología.
Farmacodinamia: Candesartan cilexetil /Hidroclorotiazida: La angiotensina II es la hormona vasoactiva primaria del sistema renina-angiotensina-aldosterona y desempeña una función importante en la fisiopatología de la hipertensión y otros trastornos cardiovasculares. También desempeña una función importante en la patogénesis de la lesión y de la hipertrofia orgánica. Los principales efectos fisiológicos de la angiotensina II, tales como vasoconstricción, estimulación de aldosterona, regulación de la homeostasis de agua y sal y estimulación del crecimiento celular, son mediados por la vía del receptor tipo 1 (AT,). Candesartán cilexetil es una pro fármaco que se convierte rápidamente en el fármaco Activo, candesartán, por hidrólisis del éster durante la absorción del tracto gastrointestinal. Candesartán es un antagonista de los receptores de angiotensina II, selectivo para receptores AT, con fuerte afinidad y lenta disociación del receptor. No posee actividad agonista. Candesartán no influye en la ECA u otros sistemas enzimáticos por lo general asociados con el uso de inhibidores de la ECA. Debido a que no hay efecto alguno sobre la degradación de las quininas, o en el metabolismo de otras sustancias, tal como la sustancia P, es improbable, que los antagonistas de los receptores de la angiotensina II se asocien con la tos. Candesartán no se une a ni bloquea otros receptores hormonales o canales iónicos de importancia conocida en la regulación cardiovascular. El antagonismo de los receptores AT, da como resultado un aumento en plasma, en relación con la dosis, de los niveles de la renina, angiotensina 1 y la angiotensina II y una disminución de la concentración plasmática de la aldosterona. La Hidroclorotiazida inhibe la reabsorción activa del sodio, principalmente en los túbulos renales distales, y promueve la excreción de sodio, cloruro y agua. La excreción renal de potasio y magnesio aumenta con el aumento de la dosis, mientras que el calcio es reabsorbido en un mayor grado. La hidroclorotiazida disminuye el volumen plasmático y el fluido extracelular y reduce el gasto cardíaco y la presión arterial. Durante la terapia a largo plazo, la reducción de la resistencia periférica contribuye a la reducción de la presión arterial. Candesartán e hidroclorotiazida tienen efectos antihipertensivos aditivos. En pacientes hipertensos, causan una reducción duradera y efectiva en la presión arterial sanguínea sin aumento reflejo en la frecuencia cardíaca. No hay indicación de hipotensión grave o exagerada con la primera dosis o efecto de rebote después de la suspensión del tratamiento. Después de la administración de una dosis única, el inicio del efecto antihipertensivo se presenta generalmente en el lapso de dos horas. Con el tratamiento continuo, la máxima reducción de la presión arterial se alcanza en cuatro semanas y se sostiene durante el tratamiento a largo plazo. La administración una vez al día proporciona una reducción efectiva y suave de la presión arterial, durante 24 horas, con una pequeña diferencia entre efectos máximos y mínimos durante el intervalo de dosificación. Rosuvastatina: Rosuvastatina cálcica es un agente hipolipemiante sintético de administración oral y es un inhibidor selectivo y competitivo de la HMGCoA reductasa, enzima que limita la velocidad de conversión de la coenzima A 3-hidroxi-3-metilglutaril en mevalonato, un precursor del colesterol. El estudio HOPE 3, multicéntrico, a largo plazo, internacional, doble ciego, randomizado, controlado con placebo, con diseño factorial 2x2, incluyó a 12.705 participantes (54% hombres, con una edad media de 65,7 años), se realizó en 228 centros de 21 países y contó con un seguimiento medio de 5,6 años, utilizó la combinación fija de candesartán 16 mg / hidroclorotiazida 12,5 mg / rosuvastatina 10mg y se realizó en pacientes con riesgo cardiovascular intermedio. Se definió como punto final primario de evaluación a: muerte cardiovascular, infarto de miocardio no fatal y accidente cerebrovascular no fatal. Los objetivos secundarios fueron: el desarrollo de insuficiencia cardiaca, paro cardíaco o necesidad de revascularización. Se observó que aquellos pacientes que recibieron candesartán 16 mg / hidroclorotiazida 12.5 mg / rosuvastatina 10 mg tenían menor porcentaje de eventos primarios comparado con placebo (3,6% vs. 5,0%, respectivamente; p=0,005), al igual que eventos secundarios (4,3% vs. 5,9%; p=0,003) y una reducción significativa absoluta de eventos cardiovasculares del 29 % en favor de la terapia combinada (candesartán 16 mg / hidroclorotiazida 12.5 mg / rosuvastatina 10 mg) comparado con placebo. Los eventos adversos mas frecuentes asociados a la terapia combinada comparada con el placebo fueron debilidad muscular y mareos. Farmacocinética: Candesartan cilexetil / Hidroclorotiazida: La administración concomitante de candesartán cilexetil e hidroclorotiazida carece de efectos clínicamente significativos sobre la farmacocinética de ninguno de los medicamentos. Absorción y distribución: Candesartán cilexetil: Después de la administración oral, candesartán cilexetil se convierte en el fármaco activo, candesartán. La biodisponibilidad absoluta de candesartán es aproximadamente del 40% después de una solución oral de candesartán cilexetil. La biodisponibilidad relativa del comprimido comparado con la solución oral es aproximadamente del 34% con una variabilidad muy pequeña. La concentración sérica pico promedio (Cmax) se alcanza 3-4 horas después de la ingestión del comprimido. Las concentraciones laicas de candesartán aumentan linealmente con el incremento de las dosis en el rango de dosis terapéutica. No se han observado diferencias relacionadas con el sexo en la farmacocinética del candesartán. El área bajo la curva de la concentración sérica versus tiempo (ABC) de candesartán no es afectada significativamente por los alimentos. Candesartán se une en alta proporción a las proteínas plasmáticas (más de 99%). El volumen de distribución aparente de candesartán es 0,1 1/kg. Hidroclorotiazida: La Hidroclorotiazida se absorbe rápidamente del tracto gastrointestinal con una biodisponibilidad absoluta de aproximadamente 70%. La ingesta concomitante de alimentos aumenta la absorción en aproximadamente 15%. La biodisponibilidad puede disminuir en pacientes con insuficiencia cardíaca y edema pronunciado. La unión de la hidroclorotiazida a proteínas plasmáticas es aproximadamente 60%. El volumen de distribución aparente es alrededor de 0,8 I/kg. Biotransformación y eliminación: Candesartán cilexetil: Candesartán se elimina en forma inalterada, principalmente por vía urinaria y biliar, y sólo en menor grado se elimina por metabolismo hepático (CYP2C9). Los estudios disponibles no indican efecto alguno sobre CYP2C9 y CYP3A4. En base a datos in vitro, no se espera que se produzca ninguna interacción in vivo con drogas cuyo metabolismo depende de las isoenzimas del citocromo P450, CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, 1CYP2E1 o CYP3A4. La vida media terminal (t1/2) de candesartán es aproximadamente de nueve horas. No hay acumulación después de administrar dosis múltiples. La vida media de candesartán permanece inalterada (aproximadamente 9 horas) después de la administración de candesartán cilexetil en combinación con hidroclorotiazida. No se produce acumulación de candesartán tras la administración repetida de la combinación en comparación con la monoterapia. El clearance plasmático total de candesartán es aproximadamente 0,37 ml/min/kg, con un clearance renal de aproximadamente 0,19 ml/min/kg. La eliminación renal de candesartán cilexetil ocurre tanto por filtración glomerular como secreción tubular activa. Después de una dosis oral de candesartán cilexetil marcado con 14C es excretado en la orina aproximadamente el 26% de la dosis como candesartán y el 7% como un metabolito inactivo, mientras que en las heces es recuperado aproximadamente el 56% de la dosis como candesartán y el 10% como metabolito inactivo. Hidroclorotiazida: La hidroclorotiazida no es metabolizada y se excreta casi completamente como droga inalterada por filtración glomerular y secreción tubular activa. La vida media (t1/2) terminal de hidroclorotiazida es de aproximadamente 8 horas. Alrededor del 70% de una dosis oral es eliminado en la orina dentro de las 48 horas. La vida media de la hidroclorotiazida permanece inalterada (aproximadamente 8 horas) después de la administración de hidroclorotiazida en combinación con candesartán cilexetil. No se produce acumulación adicional de hidroclorotiazida después de dosis repetidas de la combinación en comparación con la monoterapia. Farmacocinética en poblaciones especiales: Candesartán cilexetil: En personas de edad avanzada (mayores de 65 años) la Cmax y el ABC de candesartán aumentan en un 50% y 80% respectivamente en comparación con pacientes jóvenes. Sin embargo, la respuesta de la presión sanguínea y la incidencia de eventos adversos son similares después de, una dosis administrada de candesartan/hidroclorotiazida en pacientes jóvenes y de edad avanzada. En pacientes con deterioro renal leve a moderado, la Cmax y el ABC de candesartán aumentaron durante la administración de dosis repetidas en aproximadamente un 50 a 70% respectivamente, pero la t1/2 terminal del candesartán no fue alterada en comparación con, pacientes con funcionamiento renal normal. Los cambios correspondientes a los pacientes con insuficiencia renal severa fueron de aproximadamente el 50 y 110%, respectivamente. La t1/2 terminal de candesartán fue aproximadamente el doble en pacientes con insuficiencia renal severa. La farmacocinética en pacientes sometidos a hemodiálisis fue similar a los pacientes, con deterioro renal severo. En dos estudios, ambos incluyendo pacientes con insuficiencia hepática de carácter leve a moderado, se produjo un incremento de aproximadamente un 20% en un estudio y de un 80% en el otro estudio en la ABC media del candesartán. No hay experiencia en pacientes con insuficiencia hepática grave. Hidroclorotiazida: La t1/2 terminal de Hidroclorotiazida es prolongada en pacientes con deterioro renal. Rosuvastatina: Absorción: en estudios de farmacología clínica llevados a cabo en seres humanos, las concentraciones plasmáticas máximas de rosuvastatina se alcanzaron 3 a 5 horas después de la administración oral. Tanto la Cmax como el ABC aumentaron en proporción aproximada a la dosis de rosuvastatina. La biodisponibilidad absoluta de la rosuvastatina es de aproximadamente 20%. La administración de rosuvastatina con los alimentos no alteró el ABC de la droga y tampoco difirió luego de la administración diurna o nocturna. Distribución: el volumen medio de distribución en estado de equilibrio dinámico de la rosuvastatina es de alrededor de 134 litros. La rosuvastatina se une en un 88% a las proteínas plasmáticas, principalmente a la albúmina. Esta unión es reversible y no depende de las concentraciones plasmáticas. Metabolismo: rosuvastatina no se metaboliza ampliamente; un 10% de la dosis se recupera como metabolito. El metabolito principal es el N-desmetil rosuvastatina, que se forma principalmente por acción de la enzima 2C9 del citocromo P450, y los estudios in vitro demostraron que el N-desmetil rosuvastatina tiene aproximadamente entre un sexto y la mitad de la actividad inhibitoria del compuesto inalterado sobre la HMG-CoA reductasa. En total, el compuesto sin modificar ejerce más del 90% de la actividad inhibitoria de la HMG-CoA reductasa plasmática activa. Excreción: después de la administración oral, la rosuvastatina y sus metabolitos se excretan principalmente en las heces (90%). La vida media de eliminación (t1/2) de la rosuvastatina es de aproximadamente 19 horas. Después de una dosis endovenosa, aproximadamente el 28% del clearance corporal total se produjo por vía renal, y el 72% por vía hepática. Raza: En un análisis farmacocinético poblacional no se observaron diferencias farmacocinéticas clínicamente significativas entre los grupos de raza blanca, latina y negra. Sin embargo, estudios farmacocinéticos han demostrado un incremento de aproximadamente 2 veces en la mediana de exposición a rosuvastatina en pacientes asiáticos en comparación a controles caucásicos. Género: No se registraron diferencias en las concentraciones plasmáticas de rosuvastatina entre hombres y mujeres. Geriatría: no se registraron diferencias en las concentraciones plasmáticas de rosuvastatina entre la población geriátrica ( > 65 años) y la no geriátrica. Disfunción Renal: El deterioro renal leve a moderado (Clcr > 30ml/min/1,73 m²) no alteró las concentraciones plasmáticas de la rosuvastatina. Sin embargo, las concentraciones plasmáticas de rosuvastatina aumentaron en grado clínicamente significativo (aproximadamente 3 veces) en pacientes con deterioro renal severo (CLcr < 30ml/min/1,73 m²) que no recibían hemodiálisis en comparación con sujetos sanos (CLcr > 80ml/min/1,73 m²). Hemodiálisis: las concentraciones plasmáticas en estado de equilibrio dinámico de rosuvastatina en pacientes bajo hemodiálisis crónica fueron aproximadamente un 50% superiores en comparación con las de voluntarios sanos con función renal normal. Insuficiencia Hepática: en pacientes con hepatopatía alcohólica crónica, las concentraciones de rosuvastatina aumentaron ligeramente. En pacientes con Child-Pugh A, la Cmax y el ABC aumentaron un 60% y 5%, respectivamente, en comparación con pacientes con función hepática normal. En pacientes con Child-Pugh B, la Cmax y el ABC aumentaron un 100% y 21%, respectivamente, en comparación con pacientes con función hepática normal.
Indicaciones.
SINLIP® PREVENT está indicado en prevención cardiovascular primaria como agente reductor del colesterol y la presión arterial en pacientes mayores de 60 años con riesgo cardiovascular intermedio sin enfermedad cardíaca.
Dosificación.
La dosis de SINLIP PREVENT es de 1 cápsula por día. Puede administrarse en cualquier momento del día, junto o alejado de las comidas.
Contraindicaciones.
Hipersensibilidad a los principios activos o excipientes de SINLIP® PREVENT o a las drogas derivadas de la sulfonamida (la hidroclorotiazida es una droga derivada de la sulfonamida). Embarazo y Lactancia. Insuficiencia renal severa (clearance de creatinina < 30 ml/min/1,73 m2). Insuficiencia hepática severa y/o colestasis. Pacientes con hepatopatía activa, que puede incluir elevaciones persistentes e inexplicables de las enzimas hepáticas. Hipokalemia o hipercalcemia refractarias. Gota.
Reacciones adversas.
Las principales reacciones adversas descriptas para la triple combinación en el estudio original publicado HOPE 3 fueron: mialgias, miositis, calambres musculares, mareos, aturdimiento e hipotensión. El estudio HOPE-3 clasifica a las reacciones adversas que promovieron la discontinuación temporaria o permanente de la asociación de la siguiente manera: Temporarias: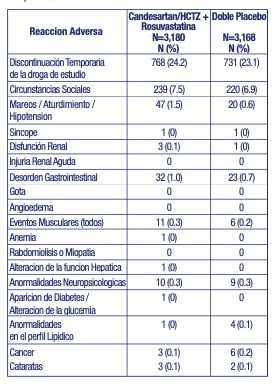
Permanentes: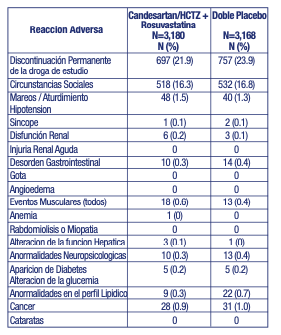
Cabe destacar que no hubo diferencias significativas en el rango permanente de discontinuación por cualquier causa entre la terapia combinada y el doble placebo (26.3% y 28.8%). Notificación de Sospecha de Reacciones Adversas: Es importante notificar la sospecha de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia al siguiente link: http://sistemas.anmat.gov.ar/aplicaciones_net/fvg_eventos_adversos_nuevo/ index.htlm y/o al Departamento de Farmacovigilancia de GADOR S.A. vía email a farmacovigilancia@gador.com o telefónicamente al 0800-220-2273.
Advertencias.
Candesartán cilexetil /Hidroclorotiazida: Insuficiencia renal/ trasplante renal: Cuando candesartán/hidroclorotiazida se utiliza en pacientes con deterioro de la función renal, se recomienda un monitoreo periódico de los niveles de potasio, creatinina y ácido úrico. No existe experiencia con respecto a la administración de candesartán/hidroclorotiazida en pacientes con trasplante renal reciente. Estenosis de la arteria renal: Los inhibidores de la enzima convertidora de angiotensina (ECA) pueden aumentar la urea en sangre y la creatinina sérica en pacientes con estenosis bilateral de la arteria renal o estenosis de la arteria de un solo riñón. Se puede anticipar un efecto similar con los antagonistas de los receptores de angiotensina II. Depleción del volumen intravascular: en pacientes con depleción del volumen intravascular y/o depleción de sodio, puede presentarse hipotensión sintomática por lo tanto, no se recomienda el uso de candesartán/hidroclorotiazida hasta que se corrija esta condición. Anestesia y cirugía: la hipotensión puede ocurrir durante la anestesia y cirugía en pacientes tratados con antagonistas de angiotensina II debido a un bloqueo del sistema renina-angiotensina. Con muy poca frecuencia, la hipotensión puede ser severa de modo que puede justificar el uso de líquidos intravenosos y/o vasopresores. Deterioro hepático: las tiazidas deben usarse con precaución en pacientes con deterioro de la función hepática o enfermedad hepática progresiva, ya que las alteraciones menores del equilibrio electrolítico y de fluidos pueden precipitar el coma hepático. No existe experiencia clínica con candesartán/hidroclorotiazida en pacientes con deterioro hepático. Estenosis de la válvula aorta y mitral (cardiomiopatía hipertrófica obstructiva): se debe tener especial precaución en pacientes con estenosis de la válvula aórtica o mitral hemodinámicamente relevante, o cardiomiopatía hipertrófica obstructiva. Hiperaldosteronismo primario: los pacientes con hiperaldosteronismo primario por lo general no responden a drogas antihipertensivas que actúan a través de la inhibición del sistema renina-angiotensina-aldosterona. Por lo tanto, no se recomienda el uso de candesartán/hidroclorotiazida. Desequilibrio electrolítico: como para cualquier paciente que recibe una terapia diurética, se debe llevar a cabo una determinación periódica de los electrolitos séricos a intervalos apropiados. Las tiazidas, incluyendo la hidroclorotiazida, pueden causar desequilibrio electrolítico o de fluidos (hipercalcemia, hipokalemia, hiponatremia, hipomagnesemia y alcalosis hipoclorémica) también pueden disminuir la excreción urinaria de calcio y pueden causar concentraciones intermitentes y ligeramente aumentadas de calcio sérico. Las tiazidas deben discontinuarse antes de llevar a cabo el análisis para la función paratiroidea. La hidroclorotiazida aumenta, en forma dependiente de la dosis, la excreción urinaria de potasio; lo que puede producir hipokalemia. Este efecto parece ser menos evidente cuando se combina con candesartán cilexetil. El riesgo de hipokalemia puede aumentarse en pacientes con cirrosis hepática, en pacientes que experimentan diuresis rápida, en pacientes con una ingesta oral inadecuada de electrolitos y en pacientes que reciben terapia concomitante con corticosteroides u hormona adrenocorticotrópica (ACTH). El tratamiento con candesartán cilextil puede causar hiperkalemia, especialmente en presencia de insuficiencia cardíaca y/o renal. En base a la experiencia con el uso de otras drogas que afectan el sistema renina-angiotensina-aldosterona, el uso concomitante de candesartán/hidroclorotiazida y diuréticos ahorradores de potasio, suplementos de potasio, sustitutos de sal, u otras drogas que pueden incrementar los niveles de potasio (por ejemplo, heparina sódica) puede producir aumentos en el potasio sérico. Se deben monitorizar los niveles de potasio cuando se estime apropiado. Se mostró que las tiazidas aumentan la excreción urinaria de magnesio, lo cual puede producir hipomagnesemia. Efectos metabólicos y endocrinos: el tratamiento con un diurético tiazídico puede deteriorar la tolerancia a la glucosa. Se puede requerir el ajuste de dosis de drogas antidiabéticas, incluyendo insulina. La diabetes mellitus latente puede manifestarse durante la terapia con tiazidas. Los aumentos en los niveles de colesterol y triglicéridos han sido asociados con la terapia de diuréticos tiazídicos. Los diuréticos tiazídicos aumentan la concentración sérica de ácido úrico y pueden provocar gota en pacientes susceptibles. Fotosensiblidad: se han registrado casos de reacciones de fotosensibilidad durante el uso de diuréticos tiazídicos. Si se produjera una reacción de fotosensiblidad, se recomienda interrumpir el tratamiento. Si la reinstauración del tratamiento es esencial, se recomienda proteger las áreas expuestas al sol o a la radiación UVA artificial. General: en pacientes en los que el tono vascular y la función renal dependen predominantemente de la actividad del sistema renina-angiotensina-aldosterona, el tratamiento con otras drogas que afectan este sistema ha sido asociado con hipotensión aguda, azoemia, oliguria o raramente insuficiencia renal aguda. La posibilidad de efectos similares no puede ser excluida con los antagonistas de los receptores de angiotensina II. Las reacciones de hipersensibilidad a la hidroclorotiazida pueden presentarse en pacientes con o sin una historia de alergia o asma bronquial, pero son más probables en pacientes con tal historia. La exacerbación o activación del lupus eritematoso sistémico ha sido informada con el uso de diuréticos tiazídicos. El efecto antihipertensivo de candesartán/ hidroclorotiazida puede verse potenciado por otros antihipertensivos. Rosuvastatina: Efectos Musculoesqueléticos: con los inhibidores de la HMG-CoA reductasa, incluido rosuvastatina, se informaron casos de miopatía y rabdomiólisis con insuficiencia renal aguda secundaria a mioglobinuria. Estos riesgos pueden presentarse con cualquiera de las dosis, pero aumentan con la dosis más alta (40 mg). Rosuvastatina deberá indicarse con precaución en pacientes con factores predisponentes a la miopatía (por ej. edad > 65 años, hipotiroidismo insatisfactoriamente tratado, disfunción renal). El riesgo de miopatía durante el tratamiento con rosuvastatina podrá verse incrementado con la administración concomitante de otros agentes hipolipemiantes (fibratos o niacina), gemfibrozil, ciclosporina, lopinavir/ritonavir o atazanavir/ ritonavir. También se han reportado casos de miopatía, incluyendo rabdomiolisis, cuando se usan concomitantemente inhibidores de la HMG-CoA reductasa, incluyendo rosuvastatina, con colchicina. Por lo tanto rosuvastatina debe ser usado con precaución cuando se coadministre con colchicina. El tratamiento con rosuvastatina deberá suspenderse si los niveles de creatina-cinasa se elevan sensiblemente o ante el diagnóstico o sospecha de miopatía. Rosuvastatina también deberá interrumpirse momentáneamente en aquellos pacientes que presenten signos o síntomas serios y agudos indicativos de miopatía o predisponentes al desarrollo de insuficiencia renal secundaria a rabdomiólisis (por ej. infección, hipotensión, deshidratación, cirugía mayor, traumatismos, trastornos metabólicos, endocrinos y electrolíticos severos, o convulsiones no controladas). Existen reportes de miopatía necrotizante inmunomediada (MNIM), una miopatía autoinmune, asociada al uso de estatinas. Se caracteriza por: debilidad muscular proximal y elevación sérica de creatina-cinasa, que persiste a pesar de la discontinuación del tratamiento con estatinas; biopsia muscular con miopatía necrotizante sin inflamación significativa; mejoría con agentes inmunosupresores. Se deberá advertir a todos los pacientes que informen inmediatamente sobre cualquier dolor, sensibilidad o debilidad muscular inexplicable, particularmente si cursa con malestar general o fiebre. Anormalidades y control de las enzimas hepáticas: se recomienda controlar las enzimas hepáticas antes de iniciar el tratamiento y si ocurren signos y síntomas de injuria hepática. Con los inhibidores de la HMG-CoA reductasa, incluido rosuvastatina, se informó de aumentos de las aminotransferasas séricas (AST [TGO] o ALT [TGP]). En la mayoría de los casos, las elevaciones fueron transitorias y se resolvieron o mejoraron durante la continuación de la terapéutica o después de una breve interrupción de la misma. Se informaron dos casos de ictericia, cuya relación con el tratamiento con rosuvastatina no pudo determinarse, los cuales se resolvieron después de la suspensión de la terapéutica. No se informaron casos de insuficiencia hepática o de enfermedad hepática irreversible en los estudios clínicos analizados. Según un análisis combinado de estudios controlados contra placebo, el 1,1% de los pacientes tratados con rosuvastatina presentaron aumentos de las aminotransferasas séricas > 3 veces el límite superior del rango normal (LSN) vs. el 0,5% de los pacientes que recibieron placebo. Rosuvastatina deberá emplearse con precaución en pacientes que consumen grandes cantidades de alcohol y/o presentan antecedentes de enfermedad hepática crónica. La enfermedad hepática activa, que puede incluir elevaciones persistentes e inexplicables de las aminotransferasas, constituye una contraindicación para el empleo de rosuvastatina. Coadministración con anticoagulantes cumarínicos: se deberá tener precaución cuando se administren anticoagulantes junto con rosuvastatina debido a la potenciación de los anticoagulantes cumarínicos en prolongar el tiempo de protrombina/RIN. En pacientes que reciben este tipo de anticoagulantes y rosuvastatina en forma concomitante, se deberá determinar el RIN antes de comenzar el tratamiento con rosuvastatina y con suficiente frecuencia durante las primeras etapas del mismo para detectar cualquier alteración significativa en el RIN. Proteinuria y hematuria: en estudios clínicos con rosuvastatina, se informó que los pacientes tratados con esta droga presentaban hematuria microscópica y proteinuria positiva en tiras reactivas. Estos hallazgos fueron más frecuentes en pacientes que recibían 40 mg en comparación con dosis más bajas de rosuvastatina o de inhibidores de la HMG-CoA reductasa comparativos, si bien fueron generalmente transitorios y no se vieron asociados con un empeoramiento de la función renal. Aunque se desconoce la significación clínica de este hallazgo, se deberá considerar una reducción en la dosis en aquellos pacientes tratados con rosuvastatina que presenten proteinuria y/o hematuria persistente de causa desconocida durante los análisis de orina periódicos. Efectos endocrinos: con los inhibidores de la HMGCoA reductasa, incluido rosuvastatina, se informaron aumentos en la HbA1c y en los niveles de glucemia en ayunas. Si bien los estudios clínicos demostraron que rosuvastatina administrado en monoterapia no reduce la concentración plasmática basal de cortisol ni daña la reserva suprarrenal, se deberá tener precaución cuando se coadministre rosuvastatina con agentes que puedan disminuir los niveles o la actividad de las hormonas esteroideas endógenas, tales como el ketoconazol, la espironolactona y la cimetidina. Mutagénesis, carcinogénesis y fertilidad: en un estudio de carcinogénesis de 104 semanas de duración realizado en ratas con dosis orales de 2, 20, 60 u 80 mg/kg/día administradas por sonda, la incidencia de pólipos uterinos estromales se vio significativamente aumentada en las hembras con 80 mg/kg/ día a una exposición sistémica 20 veces superior a la exposición de seres humanos con 40 mg/día en base al ABC. Con dosis menores no se observó una mayor incidencia de pólipos. En un estudio de carcinogénesis de 107 semanas de duración realizado en ratones que recibieron dosis orales de 10, 60, 200 mg/kg/día por sonda, se observó una mayor incidencia de adenoma/carcinoma hepatocelular con 200 mg/kg/día a exposiciones sistémicas 20 veces superiores a la exposición humana con 40 mg/día en base al ABC. No se observó una mayor incidencia de tumores hepatocelulares con dosis menores. La rosuvastatina no resultó mutagénica ni clastogénica con o sin activación metabólica en la prueba de Ames con Salmonella typhimurium y Escherichia coli, el ensayo en linfoma de ratón, y el ensayo de aberraciones cromosómicas en células pulmonares de hamsters chinos. La rosuvastatina fue negativa en el ensayo de micronúcleos de ratón in vivo. En estudios de fertilidad en ratas con dosis orales de 5, 15, 50 mg/kg/día administradas por sonda, los machos fueron tratados durante 9 semanas antes y durante el apareamiento y las hembras fueron tratadas 2 semanas antes y durante el apareamiento hasta el día 7 de gestación. No se observó ningún efecto adverso sobre la fertilidad con 50 mg/kg/día (exposiciones sistémicas de hasta 10 veces la exposición humana con 40 mg/día en base al ABC). Se observaron células espermáticas gigantes en los testículos de perros tratados con 30 mg/kg/día de rosuvastatina durante un mes. Se observaron células espermáticas gigantes en monos que recibieron 30 mg/kg/ día durante 6 meses, además de la vacuolación del epitelio tubular seminífero. En el perro, las exposiciones fueron de 20 veces y en el mono de 10 veces la exposición humana con 40 mg/día en base al área de superficie corporal. Se han observado hallazgos similares con otros agentes de esta clase. Uso en Pediatría: SINLIP® PREVENT no ha sido estudiado ni empleado en pediatría por lo tanto no debe ser indicado ni administrado en niños. Embarazo y Lactancia: SINLIP® PREVENT no debe ser administrado a embarazadas o a mujeres que estén amamantando. Uso en Geriatría: SINLIP® PREVENT puede ser utilizado en pacientes mayores de 60 años.
Interacciones.
Candesartan cilexetil /Hidroclorotiazida: No se ha identificado ninguna interacción medicamentosa clínicamente significativa para candesartán cilexetil. Los compuestos que fueron investigados en estudios de farmacocinética clínica incluyen, warfarina, digoxina, anticonceptivos orales (es decir, etinilestradiol/ levonorgestrel), glibenclamida y nifedipina. Se puede esperar que el efecto reductor de potasio de la hidroclorotiazida sea potenciado por otras drogas asociadas con la pérdida de potasio y la hipokalemia (por ejemplo, otros diuréticos caliuréticos, laxantes, anfotericina, carbenoxolona, penicilina G sódica, derivados del ácido salicílico, esteroides, ACTH). En base a la experiencia con el uso de otras drogas que afectan el sistema renina-angiotensina-aldosterona, el uso concomitante de candesartan/hidroclorotiazida con diuréticos ahorradores de potasio, suplementos de potasio o sustitutos de sal, u otras drogas que pueden incrementar los niveles de potasio sérico (por ejemplo, heparina sódica) puede producir aumentos del potasio sérico. Se deberán monitorizar los niveles de potasio cuando se considere apropiado. La hipokalemia y la hipomagnesemia inducidas por diuréticos predisponen a los efectos cardiotóxicos potenciales de glicósidos digitálicos y antiarrítmicos. Se recomienda el monitoreo periódico del potasio sérico cuando candesartan/hidroclorotiazida se administra con dichas drogas, así como con los siguientes medicamentos, que podrían inducir torsades de pointes: Antiarrítmicos de clase la (por ej. quinidina, hidroquinidina, disopiramida). Antiarrítmicos de clase Ill (por ej. amiodarona, sotalol, dofetilida, ibutilida)• Algunos antipsicóticos (por ej. tioridazina, clorpromazina, levomepromazina, trifluoperazina, ciamemazina, sulpirida, sultoprida, amisulprida, tiaprida, pimozida, haloperidol, droperidol). Otros (por ej. bepridil, cisaprida, difemanilo, eritromicina IV, halofantrina, ketanserina, mizolastina, pentamidina, esparfloxacina, terfenadina, vincamina IV). Se han reportado aumentos reversibles en las concentraciones séricas de litio y la consecuente toxicidad durante la administración concomitante de éste con inhibidores de la ECA o hidroclorotiazida. También se ha registrado un efecto similar con los ARA-ll. No se recomienda el uso de candesartán e hidroclorotiazida con litio. Si se demuestra que el uso de dicha combinación es necesario, se recomienda un cuidadoso control de los niveles séricos de litio. Puede disminuir el efecto antihipertensivo cuando se administran de forma concomitante ARA-ll y antiinflamatorios no esteroideos (AINEs) (como inhibidores selectivos de la COX-2, ácido acetilsalicílico ( > 3 g/día) y AlNEs no selectivos). Al igual que ocurre con los inhibidores de la ECA, el uso concomitante de ARA-ll y AINEs, puede provocar un aumento del riesgo de empeoramiento de la función renal, incluyendo una posible insuficiencia renal aguda, y un aumento del potasio sérico, especialmente en pacientes con trastornos previos de la función renal. La combinación debe administrarse con precaución, especialmente en pacientes ancianos. Los pacientes deben estar adecuadamente hidratados y se evaluará la necesidad de controlar la función renal tras el inicio del tratamiento concomitante y posteriormente, de forma periódica. Los AINEs amortiguan el efecto diurético, natriurético y antihipertensivo de hidroclorotiazida. La absorción de la hidroclorotiazida es reducida por el colestipol o colestiramina. El efecto sobre los relajantes no despolarizantes del músculo esquelético (por ejemplo, la tubocurarina) puede ser potenciado por la hidroclorotiazida. Los diuréticos tiazídicos pueden aumentar los niveles de calcio sérico debido a la disminución de la excreción. Si deben prescribirse suplementos de calcio o Vitamina D, los niveles de calcio sérico deben ser monitoreados y la dosis debe ser ajustada en forma adecuada. Las tiazidas pueden aumentar el efecto hiperglucémico de los betabloqueantes y el diazóxido. Los agentes anticolinérgicos (por ejemplo, atropina, biperiden) pueden aumentar la biodisponibilidad de los diuréticos tipo tiazida disminuyendo la motilidad gastrointestinal y la frecuencia del vaciamiento gástrico. Las tiazidas pueden aumentar el riesgo de efectos adversos causados por la amantadina. Las tiazidas pueden reducir la excreción renal de las drogas citotóxicas (por ejemplo,ciclofosfamida, metotrexato) y potenciar sus efectos mielosupresores. La hipotensión postural puede agravarse por la ingesta simultánea de alcohol, barbituratos o anestésicos. La metformina debe emplearse con precaución debido al riesgo de acidosis láctica inducida por el posible fallo de la función renal asociado a hidroclorotiazida. El tratamiento con un diurético tiazídico puede deteriorar la tolerancia a la glucosa. Se puede requerir el ajuste de dosis de las drogas antidiabéticas, incluyendo insulina. La hidroclorotiazida puede provocar que la respuesta arterial a las aminas presoras (por ejemplo, adrenalina) disminuya pero no lo suficiente como para excluir un efecto presor. La hidroclorotiazida puede aumentar el riesgo de insuficiencia renal aguda especialmente con dosis altas de medios de contraste iodados. El tratamiento concomitante con ciclosporina puede aumentar el riesgo de hiperuricemia y complicaciones de tipo gota. El tratamiento concomitante con baclofeno, amifostina, antidepresivos tricíclicos o neurolépticos puede producir a un aumento del efecto antihipertensivo que puede inducir hipotensión. Rosuvastatina: Ciclosporina: La ciclosporina aumentó significativamente la exposición a la rosuvastatina. Por lo tanto, en pacientes que reciben ciclosporina, el tratamiento con rosuvastatina deberá limitarse a 5 mg una vez al día. Gemfibrozil: El gemfibrozil aumentó significativamente la exposición a la rosuvastatina. Por lo tanto, deberá evitarse la coadministración de rosuvastatina y gemfibrozil. Si se debiera emplear ambos agentes en forma concomitante, no exceder la dosis de 10mg de rosuvastatina una vez al día. Inhibidores de la Proteasa: La coadministración de rosuvastatina con ciertos inhibidores de la proteasa administrados en combinación con ritonavir ejerce distintos efectos sobre la exposición a la rosuvastatina. Las combinaciones de inhibidores de la proteasa lopinavir/ritonavir y atazanavir/ritonavir aumentan la exposición a la rosuvastatina (ABC) hasta tres veces. Con estas combinaciones, la dosis de rosuvastatina deberá limitarse a 10mg. Las combinaciones de tipranavir/ritonavir o de fosamprenavir/ ritonavir alteran poco o no alteran la exposición a la rosuvastatina. Se deberá tener especial precaución cuando la rosuvastatina se coadministre con inhibidores de la proteasa en combinación con ritonavir. Anticoagulantes Cumarínicos: rosuvastatina aumenta significativamente el RIN en pacientes que reciben anticoagulantes cumarínicos. Por lo tanto, se deberá tener precaución cuando se administren anticoagulantes cumarínicos junto con rosuvastaina. En pacientes que reciben anticoagulantes cumarínicos y rosuvastatina en forma concomitante, se deberá determinar el RIN antes de comenzar el tratamiento con rosuvastatina y con suficiente frecuencia durante las primeras etapas del mismo para detectar cualquier alteración significativa del RIN. Niacina: el riesgo de efectos musculoesqueléticos puede verse aumentado cuando la rosuvastatina se emplea en combinación con niacina; se deberá considerar una reducción en la dosis de rosuvastatina en este contexto. Fenofibrato: cuando rosuvastatina se coadministró con fenofibrato no se observó un aumento clínicamente significativo en el ABC de la rosuvastatina ni del fenofibrato. El beneficio sobre los niveles lipídicos derivado del empleo combinado de rosuvastatina y fibratos deberá evaluarse cuidadosamente contra los riesgos potenciales de esta combinación. Colchicina: Se han reportado casos de miopatía, incluyendo rabdomiolisis, cuando se usan concomitantemente inhibidores de la HMG-CoA reductasa, incluyendo rosuvastatina, con colchicina. Por lo tanto rosuvastatina debe ser usado con precaución cuando se coadministre con colchicina.
Conservación.
Conservar en su envase original a temperatura ambiente (entre 15°C y 30°C).
Sobredosificación.
Síntomas: En base a las consideraciones farmacológicas