SAXENDA®
NOVO NORDISK
Análogo acilado humano del péptido-1 similar al glucagón (GLP-1).
Composición.
Un ml de solución contiene 6 mg de liraglutida*. Una lapicera prellenada contiene 18 mg de liraglutida en 3 ml. *Análogo humano del péptido-1 similar al glucagón (GLP-1) producido por tecnología de ADN recombinante en Saccharomyces cerevisiae. Excipientes: Fosfato disódico dihidrato, Propilenglicol, Fenol, Ácido clorhídrico (para ajuste del pH), Hidróxido de sodio (para ajuste del pH), Agua para inyectables. Forma farmacéutica: Solución inyectable. Solución isotónica (pH = 8,15), clara e incolora o casi incolora.
Farmacología.
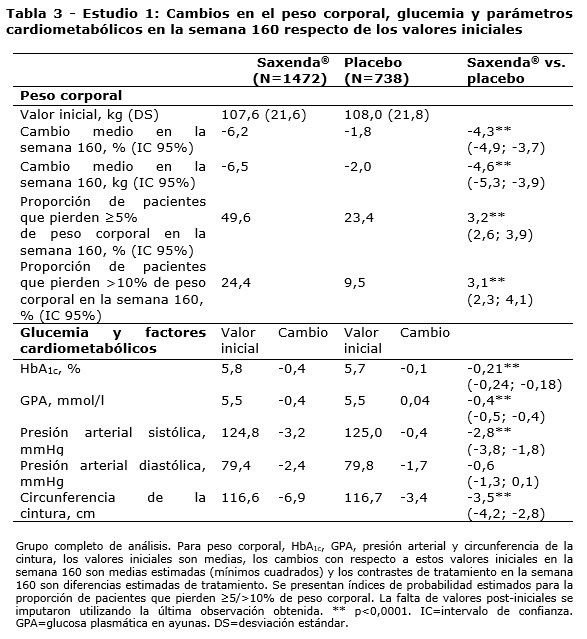
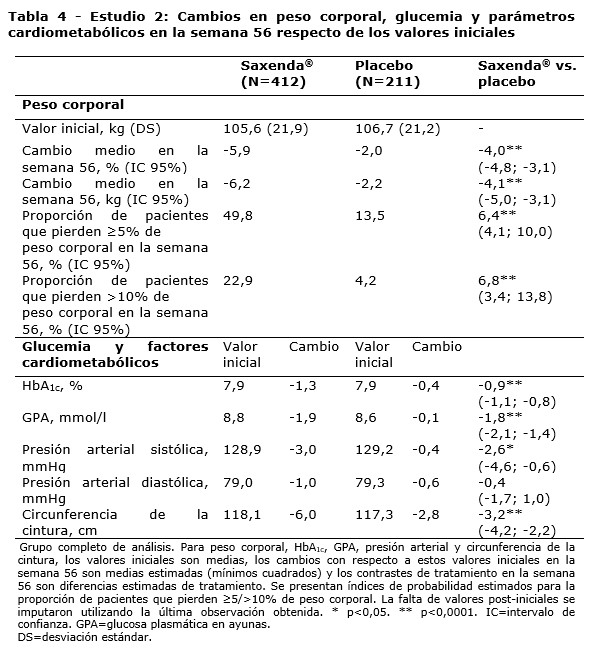
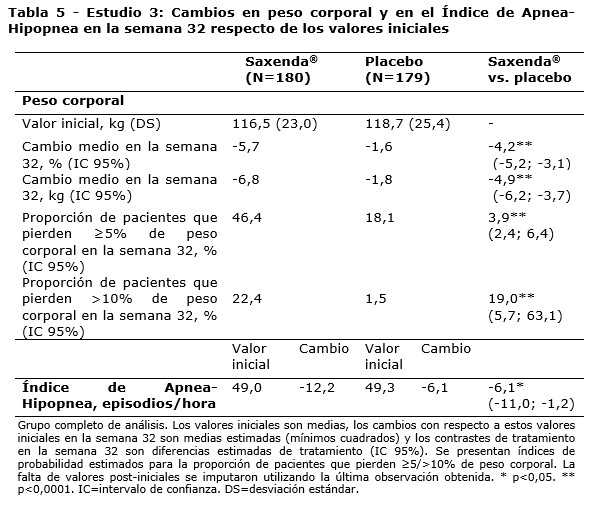
Propiedades farmacodinámicas: Mecanismo de acción: Liraglutida es un análogo acilado humano del péptido-1 similar al glucagón (GLP-1) con un 97% de homología de secuencia de aminoácidos con el GLP-1 humano endógeno. Liraglutida se une al receptor de GLP-1 (GLP-1R) y lo activa. El GLP-1 es un regulador fisiológico del apetito y de la ingesta de alimentos, pero el mecanismo de acción exacto no está completamente claro. En estudios llevados a cabo con animales, la administración periférica de liraglutida condujo a la captación en regiones específicas del cerebro implicadas en la regulación del apetito, donde liraglutida, a través de la activación específica del GLP-1R, incrementó las señales clave de la saciedad y redujo las señales clave del hambre llevando así al descenso del peso corporal. Los receptores de GLP-1 también se expresan en localizaciones específicas en el corazón, la vasculatura, el sistema inmune y los riñones. En los modelos murinos de aterosclerosis, liraglutida evitó la progresión de la placa aórtica y redujo la inflamación en la placa. Además, liraglutida tuvo un efecto beneficioso sobre los lípidos plasmáticos. Liraglutida no redujo el tamaño de la placa de las placas ya establecidas. Datos farmacodinámicos: Liraglutida contribuye a reducir el peso corporal en humanos principalmente a través de la pérdida de masa grasa con reducción relativa de grasa visceral que es mayor que la pérdida de grasa subcutánea. Liraglutida regula el apetito porque aumenta la sensación de plenitud y saciedad, a la vez que reduce la sensación de hambre y el consumo prospectivo de alimentos, lo que conduce a una reducción en la ingesta de alimentos. En comparación con el placebo, liraglutida no incrementa el gasto de energía. Liraglutida estimula la secreción de insulina y disminuye la secreción de glucagón de un modo glucosa-dependiente, lo que reduce la glucosa en ayunas y la glucosa posprandial. El efecto hipoglucemiante es mayor en pacientes con pre-diabetes y diabetes que en los pacientes con normoglucemia. Los estudios clínicos sugieren que liraglutida mejora y mantiene la función de las células beta según HOMA-B y la relación proinsulina/insulina. Datos de eficacia clínica y seguridad: La eficacia y seguridad de liraglutida para controlar el peso en combinación con una menor ingesta de calorías y un aumento de la actividad física se evaluó en cuatro estudios aleatorizados de fase 3, doble ciegos y controlados por placebo en los que participaron un total de 5.358 pacientes adultos. Estudio 1 (SCALE Obesity & Pre-Diabetes - 1839): Un total de 3.731 pacientes con obesidad (IMC ≥30 kg / m²) o con sobrepeso (IMC ≥27 kg / m²) con dislipidemia y/o hipertensión fueron estratificados de acuerdo con el estado de pre-diabetes al momento del screening y el IMC al inicio (≥30 kg/ m² o < 30 kg/m²). Todos los 3.731 pacientes fueron aleatorizados a 56 semanas de tratamiento y los 2.254 pacientes con pre-diabetes al momento del screening fueron aleatorizados a 160 semanas de tratamiento. Ambos períodos de tratamiento fueron seguidos por un período de seguimiento observacional de 12 semanas sin medicamento/placebo. El tratamiento de base para todos los pacientes consistió en la intervención del estilo de vida, en forma de dieta restringida en calorías y recomendación de ejercicio físico. En la semana 56 del estudio 1 se evaluó la pérdida de peso corporal en todos los 3.731 pacientes aleatorizados (2.590 pacientes completaron el estudio). En la semana 160 del estudio 1 se evaluó el tiempo transcurrido hasta el inicio de la diabetes tipo 2 en los 2.254 pacientes aleatorizados con pre-diabetes (1.128 personas completaron el estudio). Estudio 2 (SCALE Diabetes - 1922): Estudio de 56 semanas de duración en el que se evaluó la pérdida de peso corporal en 846 pacientes aleatorizados (de los cuales 628 completaron el estudio) con obesidad y sobrepeso, además de diabetes mellitus tipo 2 insuficientemente controlada (rango de HbA1c 7-10%). El tratamiento de base al comienzo del estudio consistía en dieta y ejercicio solamente o en metformina, una sulfonilurea o una glitazona en monoterapia, o una combinación de estos. Estudio 3 (SCALE Sleep Apnoea - 3970): Estudio de 32 semanas de duración en el que se evaluó la severidad de la apnea del sueño y la pérdida de peso corporal en 359 pacientes aleatorizados (de los cuales 276 completaron el estudio) con obesidad y apnea obstructiva del sueño moderada o severa. Estudio 4 (SCALE Maintenance - 1923): Estudio de 56 semanas de duración en el que se evaluó el mantenimiento y la pérdida de peso corporal en 422 pacientes aleatorizados (de los cuales 305 completaron el estudio) con obesidad y sobrepeso, además de hipertensión o dislipidemia tras experimentar una pérdida de peso anterior del ≥5% como resultado de una dieta baja en calorías. Peso corporal: Se alcanzó una mayor pérdida de peso con liraglutida que con placebo en pacientes con obesidad o sobrepeso en todos los grupos estudiados. En todas las poblaciones del estudio, fue mayor el porcentaje de pacientes que experimentaron una pérdida de peso ≥5% y > 10% con liraglutida en comparación con placebo (tablas 2-4).En la parte de 160 semanas del estudio 1, la pérdida de peso se produjo, principalmente, en el primer año y se mantuvo durante las 160 semanas. En el estudio 4, fue mayor el número de pacientes que mantuvieron la pérdida de peso alcanzada con anterioridad al inicio del tratamiento con liraglutida que con placebo (81,4% y 48,9%, respectivamente). Los datos específicos sobre la pérdida de peso, los pacientes que respondieron al tratamiento, la evolución temporal y la distribución acumulada del cambio de peso (en %) correspondientes a los estudios 1-4 se presentan en las tablas 2-6 y en las figuras 1, 2 y 3. Pérdida de peso después de 12 semanas de tratamiento con liraglutida (3,0 mg): Los pacientes con respuesta temprana se definieron como los pacientes en los que se produjo una pérdida de peso ≥5% tras 12 semanas con la dosis de tratamiento de liraglutida (4 semanas de escalamiento de dosis y 12 semanas con la dosis de tratamiento). En la semana 56 del estudio 1, el 67,5% de los pacientes alcanzaron una pérdida de peso ≥5% después de 12 semanas. En el estudio 2, el 50,4% de los pacientes alcanzaron una pérdida de peso ≥5% después de 12 semanas. Con un tratamiento continuo con liraglutida, se prevé que el 86,2% de estos pacientes con respuesta temprana alcancen una pérdida de peso de ≥5% y que el 51% alcance una pérdida de peso de ≥10% después de 1 año de tratamiento. Se prevé que los pacientes con respuesta temprana que completen 1 año de tratamiento pierdan una media del 11,2% de su peso corporal inicial (9,7% en hombres y 11,6% en mujeres). Para los pacientes que alcanzan una pérdida de peso de < 5% tras 12 semanas con la dosis de tratamiento de liraglutida, la proporción de pacientes que no consiguen alcanzar una pérdida de peso de ≥10% tras 1 año es del 93,4%. Control glucémico: El tratamiento con liraglutida mejora de forma significativa los parámetros glucémicos en subpoblaciones con normoglucemia, pre-diabetes y diabetes mellitus tipo 2. En la semana 56 del estudio 1, desarrollaron menos diabetes mellitus tipo 2 los pacientes tratados con liraglutida que los tratados con placebo (0,2% frente a 1,1%). En comparación con los pacientes tratados con placebo, la pre-diabetes inicial se revirtió en más pacientes (69,2% frente a 32,7%). En la semana 160 del estudio 1, el criterio de valoración primario de eficacia fue la proporción de pacientes con inicio de diabetes mellitus tipo 2 evaluado como tiempo hasta el inicio. A la semana 160, estando en tratamiento, el 3% tratado con Saxenda® y el 11% tratado con placebo fueron diagnosticados con diabetes mellitus tipo 2. El tiempo estimado de aparición de la diabetes mellitus tipo 2 para los pacientes tratados con liraglutida 3,0 mg fue 2,7 veces mayor (con un intervalo de confianza del 95% de [1,9, 3,9]) y la relación de riesgo de desarrollar diabetes mellitus tipo 2 fue de 0,2 para liraglutida versus placebo. Factores de riesgo cardiometabólicos: El tratamiento con liraglutida mejoró de forma significativa la presión arterial sistólica y la circunferencia de la cintura en comparación con placebo (tablas 2, 3 y 4). Índice de Apnea-Hipopnea (IAH): El tratamiento con liraglutida reduce de manera significativa la gravedad de la apnea obstructiva del sueño como indica el cambio con respecto al nivel basal del IAH frente a placebo (ver tabla 5).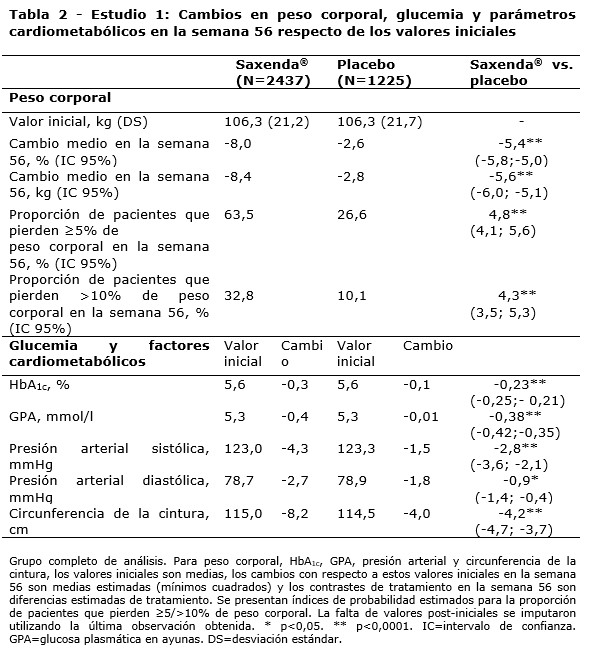
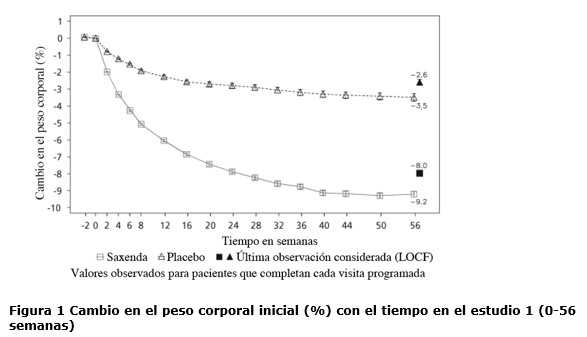
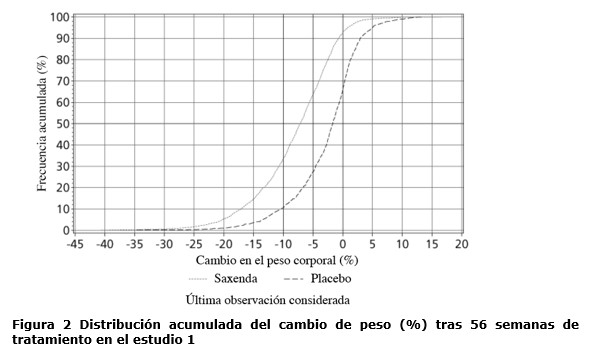

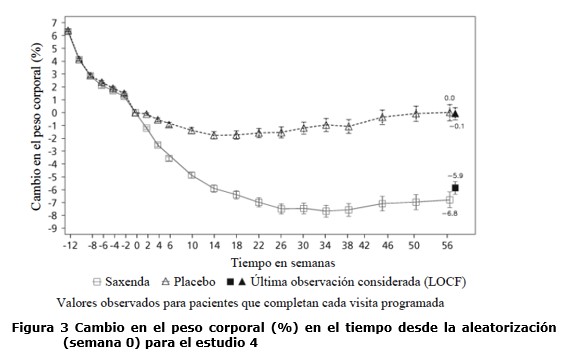
Antes de la semana 0, el tratamiento de los pacientes consistía exclusivamente en una dieta baja en calorías y ejercicio. En la semana 0 los pacientes fueron aleatorizados para recibir Saxenda® o placebo. Inmunogenicidad: De acuerdo con las propiedades potencialmente inmunogénicas de los fármacos que contienen proteínas o péptidos, los pacientes pueden desarrollar anticuerpos anti-liraglutida tras el tratamiento con liraglutida. En los estudios clínicos, el 2,5% de los pacientes tratados con liraglutida desarrolló anticuerpos anti-liraglutida. La formación de anticuerpos no se ha asociado con una reducción en la eficacia de liraglutida. Evaluación cardiovascular: Los eventos adversos cardiovasculares mayores (MACE) fueron adjudicados por un grupo de expertos externo e independiente y definidos como infarto de miocardio no fatal, accidente cerebrovascular no fatal y muerte cardiovascular. En todos los estudios clínicos a largo plazo realizados con Saxenda®, se produjeron 6 MACE en pacientes tratados con liraglutida y 10 MACE en pacientes que recibieron placebo. La relación de riesgo y el IC 95% es 0,33 [0,12; 0,90] para liraglutida en comparación con placebo. En los estudios clínicos de fase 3 se ha observado que liraglutida produce un aumento promedio de la frecuencia cardíaca desde el valor inicial de 2,5 latidos por minuto (el valor varía entre los estudios de 1,6 a 3,6 latidos por minuto). La frecuencia cardíaca alcanzó su valor máximo después de 6 semanas aproximadamente. No se ha determinado el impacto clínico a largo plazo de este aumento promedio de la frecuencia cardíaca. Este cambio en la frecuencia cardíaca fue reversible tras la interrupción del tratamiento con liraglutida (ver sección Precauciones y advertencias especiales de uso). El estudio "Efecto y acción de la liraglutida en la diabetes: evaluación de los resultados de criterios de valoración cardiovasculares" (LEADER), incluyó a 9.340 pacientes con diabetes tipo 2 que no estaba controlada de forma adecuada. La gran mayoría de ellos con enfermedad cardiovascular establecida. Los pacientes fueron asignados aleatoriamente a liraglutida en una dosis diaria de hasta 1,8 mg (4.668) o placebo (4.672), ambos agregados al cuidado estándar. La duración de la exposición fue entre 3,5 y 5 años. La edad promedio fue de 64 años y el IMC promedio fue de 32,5 kg/m². La HbA1c basal media fue 8,7 con una mejora después de 3 años del 1,2% en pacientes asignados a liraglutida y del 0,8% en pacientes asignados a placebo. El criterio de valoración primario fue el tiempo desde la aleatorización hasta la primera aparición de cualquier evento cardiovascular adverso mayor (MACE): muerte cardiovascular, infarto de miocardio no fatal o accidente cerebrovascular no fatal. Liraglutida redujo significativamente la tasa de eventos adversos cardiovasculares mayores (criterio de valoración primario, MACE) en comparación con placebo (3,41 comparado con 3,90 por 100 pacientes-años de observación en los grupos de liraglutida y placebo, respectivamente) con una reducción del riesgo del 13%, HR 0,87, [0,78, 0,97] [IC de 95% ] (p=0,005) (ver figura 4).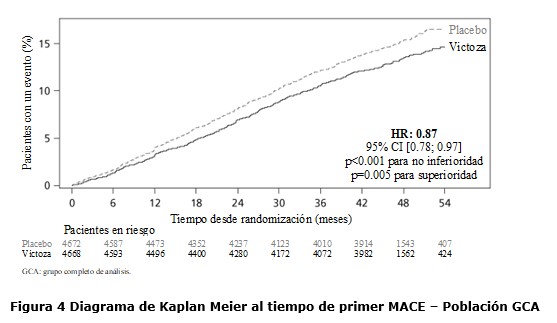
Población pediátrica: En un estudio doble ciego que compara la eficacia y la seguridad de Saxenda® versus placebo en la pérdida de peso en pacientes adolescentes de 12 años o más con obesidad, Saxenda® fue superior al placebo en la reducción de peso (evaluado como puntuación del desvío estandar del IMC) después de 56 semanas de tratamiento (ver tabla 7). Una mayor proporción de pacientes lograron reducciones del IMC de ≥5% y ≥10% con liraglutida que con placebo, así como mayores reducciones en el IMC promedio y el peso corporal (tabla 7). Después de las 26 semanas del período de seguimiento del estudio sin el producto, se observó una recuperación de peso con liraglutida vs placebo (ver tabla 7).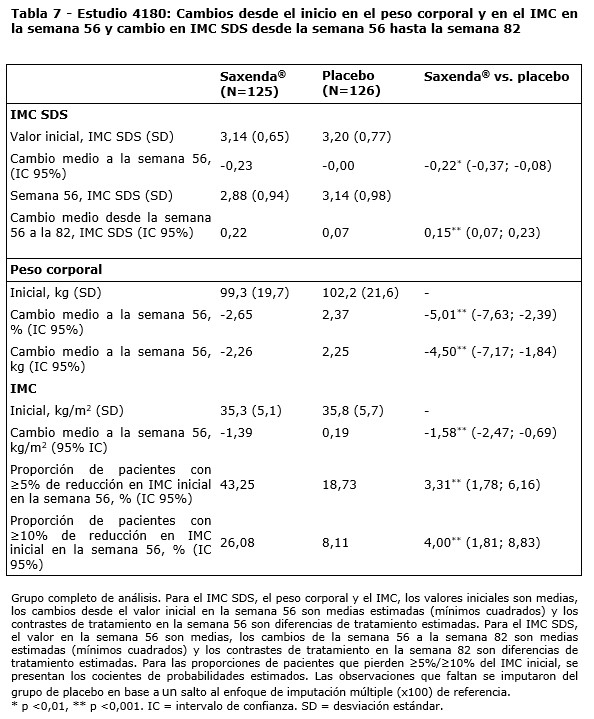
Con base en la tolerabilidad, 103 pacientes (82,4%) aumentaron y permanecieron con una dosis de 3,0 mg, 11 pacientes (8,8%) aumentaron y continuaron con una dosis de 2,4 mg, 4 pacientes (3,2%) aumentaron y continuaron con una dosis de 1,8 mg, 4 pacientes (3,2%) aumentaron y permanecieron con una dosis de 1,2 mg y 3 pacientes (2,4%) permanecieron con una dosis de 0,6 mg. No se encontraron efectos en el crecimiento o el desarrollo puberal luego de 56 semanas de tratamiento. Propiedades farmacocinéticas: Absorción: La absorción de liraglutida tras la administración por vía subcutánea fue lenta, alcanzando su concentración máxima aproximadamente a las 11 horas tras su administración. La media de la concentración en estado de equilibrio de liraglutida (AUCt/24) alcanzó aproximadamente los 31 nmol/l en pacientes con obesidad (IMC 30-40 kg/m2) tras la administración de 3 mg de liraglutida. La exposición a liraglutida aumentó proporcionalmente con la dosis. La biodisponibilidad absoluta de liraglutida tras su administración por vía subcutánea es de aproximadamente un 55%. Distribución: El volumen de distribución medio aparente tras la administración subcutánea es de 20-25 l (para una persona que pesa aproximadamente 100 kg). Liraglutida se une ampliamente a proteínas plasmáticas ( > 98%). Biotransformación: Durante 24 horas tras la administración de una única dosis de [3H]-liraglutida a sujetos sanos, el componente mayoritario en plasma fue liraglutida intacta. Se detectaron dos metabolitos menores en el plasma (≤9% y ≤5% de la exposición a radioactividad plasmática total). Eliminación: Liraglutida se metaboliza endógenamente de un modo similar al de las proteínas grandes sin un órgano específico como ruta principal de eliminación. Tras una dosis de [3H]-liraglutida, no se detectó liraglutida intacta en orina o heces. Únicamente una proporción menor de la radioactividad administrada se excretó en forma de metabolitos relacionados con liraglutida a través de orina o heces (6% y 5% respectivamente). La radioactividad en orina y heces se excretó principalmente durante los primeros 6-8 días y correspondió a tres metabolitos menores respectivamente. El clearance promedio tras la administración por vía subcutánea de liraglutida es de aproximadamente 0,9-1,4 l/h con una vida media de eliminación de aproximadamente 13 horas. Poblaciones especiales: Pacientes de edad avanzada: La edad no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de liraglutida según los resultados de un análisis de datos farmacocinéticos de la población de pacientes obesos y con sobrepeso (entre 18 y 82 años). No es necesario un ajuste de dosis en función de la edad. Sexo: Según los resultados de los análisis farmacocinéticos de la población, las mujeres tienen un clearance de liraglutida ajustado al peso que es inferior en un 24% en comparación con los hombres. Según los datos de respuesta a la exposición, no es necesario un ajuste de dosis en función del sexo del paciente. Origen étnico: El origen étnico no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de liraglutida según los resultados del análisis farmacocinético de la población en el que se incluyeron pacientes con obesidad y con sobrepeso de grupos de población blanca, negra, asiática, hispanoamericana y no hispanoamericana. Peso corporal: La exposición a liraglutida disminuye con un aumento del peso corporal inicial. La dosis diaria de 3,0 mg de liraglutida proporcionó exposiciones sistémicas adecuadas sobre el rango de pesos de 60 a 234 kg cuya respuesta a la exposición se evaluó en los estudios clínicos. No se estudió la exposición a liraglutida en pacientes con un peso corporal superior a los 234 kg. Insuficiencia hepática: Se evaluó la farmacocinética de liraglutida en pacientes con diversos grados de insuficiencia hepática en un estudio de dosis única (0,75 mg). La exposición a liraglutida disminuyó un 13-23% en pacientes con insuficiencia hepática de leve a moderada en comparación con los sujetos sanos. La exposición fue significativamente menor (44%) en pacientes con insuficiencia hepática grave (puntuación Child Pugh > 9). Insuficiencia renal: La exposición a liraglutida disminuyó en pacientes con insuficiencia renal en comparación con los individuos con una función renal normal en un estudio de dosis única (0,75 mg). La exposición a liraglutida disminuyó un 33%, un 14%, un 27% y un 26%, respectivamente, en pacientes con insuficiencia renal leve (clearance de creatinina, CrCl 50-80 ml/min), moderada (CrCl 30-50 ml/min) y severa (CrCl < 30 ml/min) y con enfermedad renal en etapa terminal con necesidad de diálisis. Población pediátrica: Las propiedades farmacocinéticas de liraglutida 3,0 mg se evaluaron en estudios clínicos para pacientes adolescentes de 12 a menos de 18 años de edad con obesidad (134 pacientes, peso corporal 62-178 kg). La exposición a liraglutida en adolescentes (de 12 años a menos de 18 años) fue similar a la de adultos con obesidad. Las propiedades farmacocinéticas también se evaluaron en un estudio de farmacología clínica en la población pediátrica con obesidad de 7-11 años de edad (13 pacientes, peso corporal 54-87 kg) respectivamente. Se encontró que la exposición asociada con 3,0 mg de liraglutida es comparable entre los niños de 7 a 11 años, adolescentes y adultos con obesidad, después de la corrección para el peso corporal. Datos preclínicos de seguridad: Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas o genotoxicidad. Se observaron tumores no letales en células C de tiroides en estudios de carcinogenicidad de dos años de duración en ratas y ratones. En ratas no se ha observado un nivel de efecto adverso no observado (NOAEL). Estos tumores no se observaron en monos tratados durante 20 meses. Estos hallazgos en roedores están provocados por un mecanismo específico no genotóxico mediado por el receptor GLP-1 al que los roedores son especialmente sensibles. La relevancia en humanos es probablemente baja pero no se puede excluir completamente. No se ha detectado ningún otro tumor relacionado con el tratamiento. Los estudios en animales no sugieren efectos perjudiciales directos en términos de fertilidad, pero sí un leve aumento de las muertes embrionarias tempranas a la dosis más alta. La administración de liraglutida durante el periodo intermedio de gestación provocó una reducción en el peso de la madre y en el crecimiento del feto con efectos no claros sobre las costillas en ratas y en la variación esquelética en el conejo. El crecimiento neonatal se redujo en el caso de las ratas durante su exposición a liraglutida y continuó durante el periodo de destete en el grupo de dosis elevada. Se desconoce si la disminución en el crecimiento de las crías se debe a una reducción en la ingesta de leche debido a un efecto directo del GLP-1 o a una reducción de la producción de leche materna a causa de una disminución de la ingesta calórica. En ratas jóvenes, liraglutida causó un retraso en la maduración sexual tanto en machos como en hembras a exposiciones clínicamente relevantes. Estos retrasos no tuvieron impacto en la fertilidad o la capacidad reproductiva de ninguno de los sexos, ni en la capacidad de las hembras de mantener el embarazo.
Indicaciones.
Adultos: Saxenda® está indicado, en combinación con una dieta baja en calorías y un aumento de la actividad física, para controlar el peso en pacientes adultos con un índice de masa corporal (IMC) inicial de ≥30 kg/m² (obesidad), o ≥ 27 kg/m² a < 30 kg/m² (sobrepeso) que presentan al menos una comorbilidad relacionada con el peso, como alteraciones de la glucemia (prediabetes o diabetes mellitus tipo 2), hipertensión, dislipidemia o apnea obstructiva del sueño. El tratamiento con una dosis diaria de 3,0 mg de Saxenda® se debe interrumpir si después de 12 semanas los pacientes no han perdido al menos el 5% de su peso corporal inicial. Adolescentes (≥12 años): Saxenda® puede ser utilizado en combinación con una nutrición saludable y mayor actividad física para el control del peso en pacientes adolescentes a partir de 12 años de edad con: obesidad (IMC correspondiente a ≥30 kg/m2 para adultos por puntos de corte internacionales)*, y peso corporal superior a 60 kg. El tratamiento con Saxenda debe interrumpirse y re-evaluarse si los pacientes no han perdido al menos el 4% de su IMC o puntuación Z para IMC luego de 12 semanas con una dosis diaria de 3,0 mg o la dosis máxima tolerada. * Puntos de corte del IMC del IOTF para la obesidad por sexo entre 12 y 18 años de edad (ver tabla 1):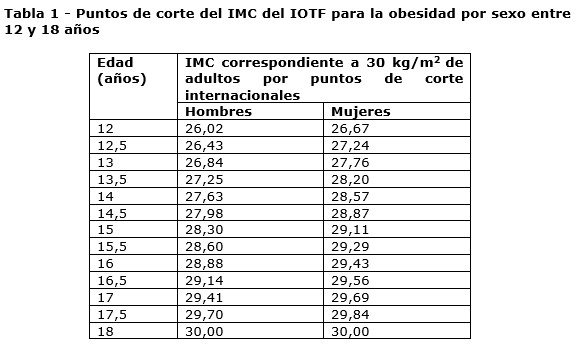
Dosificación.
Posología: Adultos: La dosis inicial es de 0,6 mg una vez al día. La dosis se debe aumentar hasta 3,0 mg una vez al día en incrementos de 0,6 mg en intervalos de al menos una semana para que mejore la tolerancia gastrointestinal (ver tabla 8). Si el paciente no tolera un aumento de la dosis durante dos semanas consecutivas, se debe considerar la discontinuación del tratamiento. No se recomiendan dosis diarias superiores a 3,0 mg.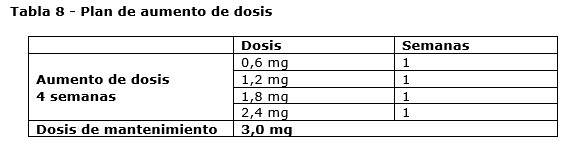
Adolescentes (≥12 años): Para los adolescentes de entre 12 a menos de 18 años de edad se debe aplicar un esquema de aumento de la dosis similar al de los adultos (ver tabla 8). La dosis debe aumentarse hasta que se alcancen 3,0 mg (dosis de mantenimiento) o la dosis máxima tolerada. No se recomiendan dosis diarias superiores a 3,0 mg. Pacientes con diabetes mellitus tipo 2: Saxenda® no se debe utilizar en combinación con otro agonista del receptor de GLP-1. Cuando se inicia el tratamiento con Saxenda®, se debe considerar reducir la dosis de insulina o de secretagogos de la insulina (como sulfonilureas) que se administran de forma concomitante para reducir el riesgo de hipoglucemia. Es necesario realizar automonitoreo de la glucemia para ajustar la dosis de insulina o secretagogos de insulina (ver sección Precauciones y advertencias especiales de uso). Poblaciones especiales: Pacientes de edad avanzada (≥65 años de edad): No es necesario un ajuste de dosis en función de la edad. La experiencia terapéutica en pacientes de 75 años de edad en adelante es limitada, por lo que no se recomienda su uso en estos pacientes (ver secciones Precauciones y advertencias especiales de uso y Propiedades farmacocinéticas). Pacientes con insuficiencia renal: No es necesario un ajuste de dosis en pacientes con insuficiencia renal leve o moderada (clearance de creatinina ≥30 ml/min). No se recomienda utilizar Saxenda® en pacientes con insuficiencia renal severa (clearance de creatinina < 30 ml/min), incluidos los pacientes con enfermedad renal en etapa terminal (ver secciones Precauciones y advertencias especiales de uso, Reacciones adversas y Propiedades farmacocinéticas). Pacientes con insuficiencia hepática: No se recomienda ajustar la dosis en pacientes con insuficiencia hepática leve o moderada. No se recomienda utilizar Saxenda® en pacientes con insuficiencia hepática severa y se debe utilizar con precaución en pacientes con insuficiencia hepática leve o moderada (ver secciones Precauciones y advertencias especiales de uso y Propiedades farmacocinéticas). Población pediátrica: No se requiere ajuste de dosis para adolescentes de 12 años o más. No se ha establecido la seguridad y eficacia de Saxenda® en niños menores de 12 años de edad (ver sección Propiedades farmacodinámicas). Modo de administración: Saxenda® solo se debe administrar por vía subcutánea. No se debe administrar por vía intravenosa o intramuscular. Saxenda® se administra una vez al día en cualquier momento del día, independiente de las comidas. Se debe inyectar en el abdomen, en el muslo o en la parte superior del brazo. Tanto el lugar de inyección como el momento de la administración se pueden modificar sin necesidad de ajustar la dosis. Sin embargo, es preferible que Saxenda® se administre alrededor de la misma hora del día, una vez se haya elegido el momento más conveniente del día para ello. Para consultar más información sobre la administración, ver sección Precauciones especiales de descarte y manipulación. Dosis omitida: Si una dosis es omitida dentro de las 12 horas a partir de la hora de administración habitual, el paciente se debe administrar la dosis lo antes posible. Si quedan menos de 12 horas para la próxima dosis, el paciente no se debe administrar la dosis omitida y debe reanudar el esquema de una vez al día con la siguiente dosis programada. No se debe administrar una dosis adicional o aumentar la dosis para compensar la dosis omitida.
Contraindicaciones.
Hipersensibilidad a liraglutida o a alguno de los excipientes incluidos en la sección Composición.
Reacciones adversas.
Resumen del perfil de seguridad: La seguridad de Saxenda® fue evaluada en 5 estudios clínicos doble ciego, controlados con placebo, en los que han participado 5.813 pacientes adultos con sobrepeso u obesidad con al menos una comorbilidad relacionada con el peso. En general, las reacciones gastrointestinales fueron las reacciones adversas notificadas más frecuentemente durante el tratamiento (67,9%) (ver sección "Descripción de las reacciones adversas seleccionadas"). Tabla de reacciones adversas: En la Tabla 9 se presentan las reacciones adversas notificadas en adultos. Las reacciones adversas figuran en la lista según el sistema de clasificación de órganos y la frecuencia. Las categorías de frecuencia se definen del siguiente modo: muy frecuentes (≥1/10); frecuentes (≥1/100 a < 1/10); poco frecuentes (≥1/1.000 a < 1/100); raras (≥1/10.000 a < 1/1.000); muy raras ( < 1/10.000). Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia.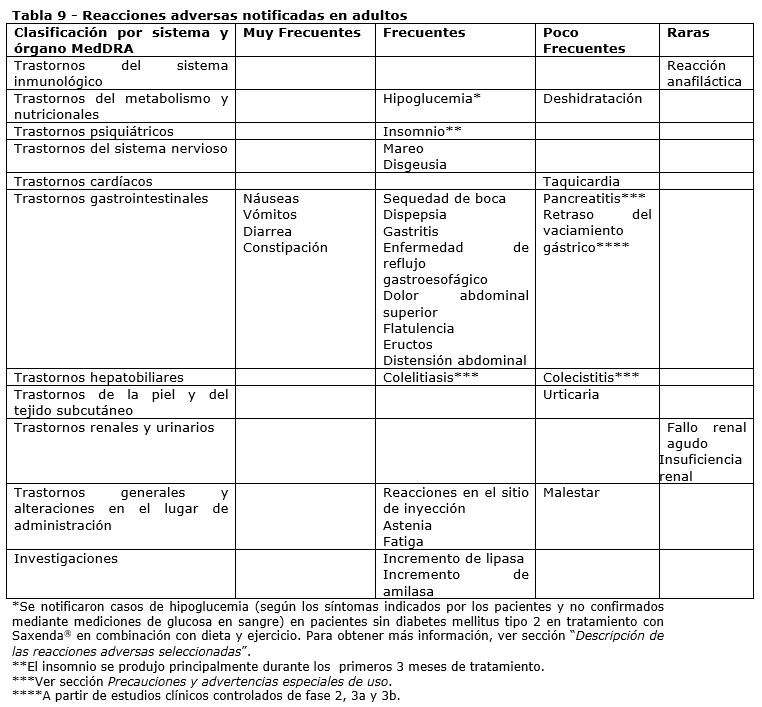
Descripción de las reacciones adversas seleccionadas: Hipoglucemia en pacientes sin diabetes mellitus tipo 2: En estudios clínicos realizados en pacientes con obesidad o con sobrepeso sin diabetes mellitus tipo 2 en tratamiento con Saxenda® en combinación con dieta y ejercicio, no se notificaron episodios hipoglucémicos severos (que requiriesen la asistencia de terceras personas). El 1,6% de los pacientes tratados con Saxenda® y el 1,1% de los pacientes tratados con placebo notificaron que presentaron síntomas de episodios hipoglucémicos; sin embargo, estos episodios no se confirmaron mediante mediciones de glucosa en sangre. La mayoría de los episodios fueron leves. Hipoglucemia en pacientes con diabetes mellitus tipo 2: En un estudio clínico realizado en pacientes con obesidad o con sobrepeso con diabetes mellitus tipo 2 en tratamiento con Saxenda® en combinación con dieta y ejercicio, se notificó hipoglucemia severa (que requirió la asistencia de terceras personas) en el 0,7% de los pacientes tratados con Saxenda® y solo en pacientes tratados de forma concomitante con sulfonilurea. Además, se documentaron casos de hipoglucemia sintomática en el 43,6% de los pacientes tratados con Saxenda® y en el 27,3% de los pacientes tratados con placebo. Entre los pacientes que no se trataron con sulfonilurea de forma concomitante, se documentaron episodios de hipoglucemia sintomática (definidos como glucosa en plasma ≤3,9 mmol/l acompañados de síntomas) en el 15,7% de los pacientes tratados con Saxenda® y el 7,6% de los pacientes tratados con placebo. Hipoglucemia en pacientes con diabetes mellitus tipo 2 tratados con insulina: En un estudio clínico realizado en pacientes con obesidad o sobrepeso con diabetes mellitus tipo 2 en tratamiento con insulina y liraglutida 3,0 mg/día en combinación con dieta y ejercicio y hasta 2 ADOs (Antidiabéticos Orales), se notificó hipoglucemia severa (que requirió la asistencia de terceras personas) en el 1,5% de los pacientes tratados con liraglutida 3,0 mg/día. En este estudio, se documentaron episodios de hipoglucemia sintomática (definidos como glucosa en plasma ≤3,9 mmol/l acompañados de síntomas) en el 47,2% de los pacientes tratados con liraglutida 3,0 mg/día y en el 51,8% de los pacientes tratados con placebo. Entre los pacientes en tratamiento concomitante con sulfonilureas, se documentaron episodios de hipoglucemia sintomática en el 60,9% de los pacientes tratados con liraglutida 3,0 mg/día y en el 60,0% de los pacientes tratados con placebo. Reacciones adversas gastrointestinales: La mayoría de los episodios gastrointestinales fueron de leves a moderados, transitorios y no conllevaron la interrupción del tratamiento. Las reacciones generalmente sucedieron durante las primeras semanas y disminuyeron una vez transcurridos algunos días o semanas de tratamiento continuado. Los pacientes de 65 años de edad en adelante pueden experimentar más efectos gastrointestinales al ser tratados con Saxenda®. Los pacientes con insuficiencia renal leve o moderada (clearance de creatinina ≥30 ml/min) pueden experimentar más efectos gastrointestinales al ser tratados con Saxenda®. Fallo renal agudo: En pacientes tratados con agonistas del receptor de GLP-1 se han notificado casos de fallo renal agudo. La mayoría de los casos notificados se produjeron en pacientes que habían experimentado náuseas, vómitos o diarrea con la consiguiente disminución del volumen (ver sección Precauciones y advertencias especiales de uso). Reacciones alérgicas: Durante la comercialización de liraglutida, se han notificado pocos casos de reacciones anafilácticas con síntomas tales como hipotensión, palpitaciones, disnea y edema. Las reacciones anafilácticas pueden ser potencialmente mortales. Ante la sospecha de una reacción anafiláctica, se debe interrumpir el tratamiento con liraglutida y este no se debe reanudar (ver sección Contraindicaciones). Reacciones en el sitio de inyección: En pacientes tratados con Saxenda® se han notificado reacciones en el lugar de inyección. Estas reacciones fueron por lo general leves y transitorias y la mayoría desaparecieron durante el tratamiento continuado. Taquicardia: En estudios clínicos se notificó taquicardia en el 0,6% de los pacientes tratados con Saxenda® y en el 0,1% de los pacientes tratados con placebo. La mayoría de los episodios fueron leves o moderados. Se trató de episodios aislados que en su mayoría se resolvieron durante el tratamiento continuado con Saxenda®. Población pediátrica: En un estudio clínico realizado en adolescentes de 12 años a menos de 18 años de edad con obesidad, 125 pacientes fueron expuestos a Saxenda® durante 56 semanas. En general, la frecuencia, el tipo y la gravedad de las reacciones adversas en los adolescentes con obesidad fueron comparables a las observadas en la población adulta. Los vómitos ocurrieron con una frecuencia 2 veces mayor en adolescentes en comparación con adultos. El porcentaje de pacientes informando al menos un episodio de hipoglucemia clínicamente significativo fue mayor con liraglutida (1,6%) comparado con placebo (0,8%). No ocurrieron episodios de hipoglucemia severa en el estudio. Reporte de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas.
Precauciones.
Pacientes con insuficiencia cardíaca: No existe experiencia clínica en pacientes con insuficiencia cardíaca congestiva de clase IV según la New York Heart Association (NYHA) y, por lo tanto, el uso de liraglutida no se recomienda en estos pacientes. Poblaciones especiales: No se ha establecido la seguridad y eficacia de liraglutida para controlar el peso en pacientes: de 75 años de edad en adelante, tratados con otros productos para controlar el peso, con obesidad debida a trastornos endocrinos o desórdenes alimentarios o al tratamiento con medicamentos que pueden provocar aumento de peso, con insuficiencia renal severa, con insuficiencia hepática severa. No se recomienda el uso en estos pacientes (ver sección Posología y modo de administración). Como liraglutida no se ha estudiado para controlar el peso en sujetos con insuficiencia hepática leve o moderada, se debe utilizar con precaución en estos pacientes (ver secciones Posología y modo de administración y Propiedades farmacocinéticas). La experiencia en pacientes con enfermedad inflamatoria intestinal y gastroparesia diabética es limitada. No se recomienda el uso de liraglutida en estos pacientes ya que se asocia a reacciones adversas gastrointestinales transitorias, como náuseas, vómitos y diarrea. Pancreatitis: Se ha observado pancreatitis aguda con el uso de agonistas del receptor de GLP-1. Se debe informar a los pacientes de los síntomas característicos de la pancreatitis aguda. Ante la sospecha de pancreatitis, se debe interrumpir el tratamiento con liraglutida, y este no se debe reanudar si se confirma la pancreatitis aguda. Colelitiasis y colecistitis: En estudios clínicos sobre control del peso, se ha observado una mayor tasa de colelitiasis y colecistitis en los pacientes tratados con liraglutida que en los pacientes tratados con placebo. El hecho de que la pérdida importante de peso puede aumentar el riesgo de colelitiasis y, por consiguiente, de colecistitis, solo explicó parcialmente la mayor tasa con liraglutida. La colelitiasis y la colecistitis pueden requerir hospitalización y colecistectomía. Se debe informar a los pacientes de los síntomas característicos de la colelitiasis y la colecistitis. Enfermedad tiroidea: En estudios clínicos llevados a cabo en pacientes con diabetes tipo 2, se han notificado eventos adversos tiroideos, como bocio, especialmente en pacientes con enfermedad tiroidea preexistente. Por lo tanto, liraglutida debe utilizarse con precaución en pacientes con enfermedad tiroidea. Frecuencia cardíaca: En estudios clínicos se ha observado que liraglutida produce un aumento de la frecuencia cardíaca (ver sección Propiedades farmacodinámicas). La frecuencia cardíaca se debe controlar de forma periódica de acuerdo con la práctica clínica habitual. Se debe informar a los pacientes de los síntomas del aumento de la frecuencia cardíaca (palpitaciones o sensación de aceleración del pulso en reposo). El tratamiento con liraglutida se debe discontinuar en pacientes que experimenten un incremento sostenido clínicamente significativo de la frecuencia cardíaca en reposo. Deshidratación: Se han notificado signos y síntomas de deshidratación que incluyen insuficiencia renal y fallo renal agudo en pacientes en tratamiento con agonistas del receptor de GLP-1. Se debe advertir a los pacientes en tratamiento con liraglutida que existe un riesgo potencial de deshidratación relacionado con los eventos adversos gastrointestinales y que tomen precauciones para evitar la pérdida de líquidos. Hipoglucemia en pacientes con diabetes mellitus tipo 2: Los pacientes con diabetes mellitus tipo 2 a los que se les administra liraglutida en combinación con una insulina y/o sulfonilurea podrían presentar un riesgo mayor de hipoglucemia. Es posible disminuir el riesgo de hipoglucemia reduciendo la dosis de insulina y/o sulfonilurea. Población pediátrica: Se han notificado episodios de hipoglucemia clínicamente significativa en adolescentes (≥12 años) tratados con liraglutida. Los pacientes deben estar informados acerca de los síntomas característicos de hipoglucemia y las acciones adecuadas a llevar a cabo. Hiperglucemia en pacientes con diabetes mellitus en tratamiento con insulina: No se debe utilizar Saxenda® como un sustituto de insulina en pacientes con diabetes mellitus. Se ha notificado cetoacidosis diabética en pacientes insulinodependientes después de una interrupción rápida o reducción de la dosis de insulina (ver sección Posología y modo de administración). Excipientes: Saxenda® contiene menos de 1 mmol (23 mg) de sodio por dosis; esto es, esencialmente "exento de sodio". Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de Saxenda® sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Sin embargo, se pueden experimentar mareos principalmente durante los primeros 3 meses de tratamiento con Saxenda®. Si se sufren mareos se debe conducir o utilizar máquinas con precaución. Interacción con otros medicamentos y otras formas de interacción: In vitro, liraglutida ha demostrado un potencial muy bajo de estar implicada en interacciones farmacocinéticas con otras sustancias activas relacionadas con el citocromo P450 (CYP) y la unión a proteínas plasmáticas. El leve retraso en el vaciamiento gástrico asociado a liraglutida puede influir en la absorción de medicamentos orales administrados de forma concomitante. Los estudios de interacción no han demostrado ningún retraso clínicamente significativo en la absorción y, por lo tanto, no se requiere ajuste de dosis. Se han realizado estudios de interacciones con 1,8 mg de liraglutida. El efecto sobre la velocidad de vaciamiento gástrico fue equivalente para 1,8 mg y 3,0 mg de liraglutida (AUC0-300 min de paracetamol). Pocos pacientes tratados con liraglutida notificaron al menos un episod