RYBREVANT
JANSSEN CILAG
Anticuerpo monoclonal. Antineoplásico.
Composición.
Cada frasco de RYBREVANT® contiene: Amivantamab 350 mg (50 mg/ml). Excipientes: L-histidina, L-histidina clorhidrato monohidrato, sacarosa, polisorbato 80, L-metionina, EDTA disódico dihidrato, agua para inyectable.
Farmacología.
Mecanismo de acción Amivantamab es un anticuerpo biespecífico que se une a los dominios extracelulares de EGFR y MET. En estudios in vitro e in vivo, amivantamab fue capaz de alterar las funciones de señalización de EGFR y MET mediante el bloqueo de la unión del ligando y, en los modelos de mutación del exón 20, la degradación de EGFR y MET. La presencia de EGFR y MET en la superficie de las células tumorales también permite dirigir estas células para su destrucción por parte de células efectoras inmunitarias, como las células citotóxicas naturales y los macrófagos, a través de mecanismos de citotoxicidad celular dependiente de anticuerpos (ADCC) y de trogocitosis, respectivamente. Farmacodinamia: La relación exposición-respuesta y el curso temporal de la respuesta farmacodinámica de amivantamab no se han caracterizado completamente en pacientes con NSCLC con mutaciones de inserción en el exón 20 del EGFR. Farmacocinética: La exposición a amivantamab aumentó proporcionalmente en un intervalo de dosis de 350 a 1750 mg (de 0,25 a 1,25 veces la dosis máxima recomendada aprobada). El estado estacionario de las concentraciones de amivantamab se alcanzó en la novena infusión. El cociente de acumulación en estado estable fue de 2,4. Distribución: El volumen de distribución medio (± SD) de amivantamab es de 5,13 (± 1,78) L. Eliminación: El clearance medio (± SD) de amivantamab es de 360 (± 144) ml/día y la vida media terminal es de 11,3 (± 4,53) días. Poblaciones específicas: No se observaron diferencias clínicamente significativas en la farmacocinética de amivantamab basado en la edad (rango: 32-87 años), el sexo, la raza, el clearance de creatinina (CLcr 29 a 276 ml/min) o la insuficiencia hepática leve [(bilirrubina total ≤ULN y AST > ULN) o (ULN < bilirrubina total ≤ 1,5 veces ULN)]. No se ha estudiado la farmacocinética de amivantamab en pacientes con insuficiencia renal grave (CLcr 15 a 29 ml/min) o en pacientes con insuficiencia hepática moderada (bilirrubina total de 1,5 a 3 veces el ULN) a grave (bilirrubina total > 3 veces el ULN). Peso corporal: El aumento del peso corporal incrementó el volumen de distribución y el clearance de amivantamab. Las exposiciones de amivantamab son un 30-40% menor en los pacientes que pesaban ≥80 kg en comparación con los pacientes con peso corporal < 80 kg a la misma dosis. Las exposiciones de amivantamab fueron comparables entre los pacientes que pesaban < 80 kg y recibieron una dosis de 1050 mg y los pacientes que pesaban ≥80 kg y recibieron una dosis de 1400 mg. Toxicología preclínica: Carcinogénesis, mutagénesis, deterioro de la fertilidad: No se han realizado estudios para evaluar el potencial de amivantamab en cuanto a carcinogenicidad o genotoxicidad. No se han realizado estudios de fertilidad para evaluar los efectos potenciales de amivantamab. En estudios toxicológicos de dosis repetidas de 6 semanas y 3 meses en monos, no hubo efectos notables en los órganos reproductores masculinos y femeninos. Estudios clínicos: La eficacia de RYBREVANT® se evaluó en pacientes con NSCLC localmente avanzado o metastásico con mutaciones de inserción en el exón 20 del EGFR en un ensayo clínico multicéntrico, abierto y de múltiples cohortes (CHRYSALIS, NCT02609776). El estudio incluyó a pacientes con NSCLC localmente avanzado o metastásico con mutaciones de inserción en el exón 20 del EGFR cuya enfermedad había experimentado progresión durante o después de la quimioterapia basada en platino. Los pacientes con metástasis cerebrales no tratadas y los pacientes con antecedentes de EPI que requirieran tratamiento con esteroides por tiempo prolongado u otros agentes inmunosupresores en los últimos 2 años no fueron elegibles para el estudio. En la población de eficacia, el estado de la mutación en el exón 20 del EGFR se determinó mediante pruebas locales prospectivas utilizando muestras de tejido (94%) y/o plasma (6%). De los 81 pacientes con mutaciones de inserción en el exón 20 del EGFR, las muestras de plasma del 96% de los pacientes se analizaron retrospectivamente utilizando Guardant360® CDx. Mientras que en el 76% de los pacientes se identificó una mutación de inserción en el exón 20 del EGFR en una muestra de plasma, en el 20% no se la identificó, y el 3,7% no tenía muestras de plasma para el análisis. Los pacientes recibieron RYBREVANT® en dosis de 1050 mg (para pacientes con peso corporal inicial < 80 kg) o 1400 mg (para pacientes con peso corporal inicial ≥80 kg) una vez a la semana durante 4 semanas, y después cada 2 semanas hasta progresión de la enfermedad o toxicidad inaceptable. La principal medida de resultado de la eficacia fue la tasa de respuesta global (ORR) de acuerdo con los Criterios de Evaluación de la Respuesta en Tumores Sólidos (RECIST v1.1) evaluados mediante una Revisión Central Independiente Ciega (BICR). Una medida de resultado de la eficacia adicional fue la duración de la respuesta (DOR) evaluada mediante una BICR. La población de eficacia incluyó a 81 pacientes con NSCLC con mutación de inserción en el exón 20 del EGFR con enfermedad medible que habían sido tratados previamente con quimioterapia basada en platino. La mediana de la edad fue de 62 (rango: 42 a 84) años, el 59% eran mujeres; el 49% eran asiáticos, el 37% eran blancos, el 2,5% eran negros; el 74% tenían un peso corporal inicial < 80 kg; el 95% presentaban adenocarcinoma; y el 46% habían recibido inmunoterapia previa. La mediana del número de terapias previas fue de 2 (rango: 1 a 7). Al inicio del estudio, el 67% tenía un estado de rendimiento del Grupo Cooperativo de Oncología Oriental (ECOG) de 1; el 53% nunca había fumado; todos los pacientes tenían enfermedad metastásica; y el 22% tenía metástasis cerebrales tratadas previamente. Los resultados de eficacia se resumen en la Tabla 8.
Indicaciones.
RYBREVANT® está indicado para el tratamiento de pacientes adultos con cáncer pulmonar de células no pequeñas (NSCLC, por sus siglas en inglés) localmente avanzado o metastásico con mutaciones de inserción en el exón 20 del receptor del factor de crecimiento epidérmico (EGFR), detectadas mediante un test validado (ver "Posología y administración, Selección de pacientes"), cuya enfermedad ha progresado durante o después de la quimioterapia basada en platino.
Dosificación.
Selección de pacientes: Seleccionar a los pacientes para el tratamiento con RYBREVANT® en base a la presencia de mutaciones de inserción en el exón 20 del EGFR (ver "Estudios clínicos"). Dosificación recomendada: Las dosis recomendadas de RYBREVANT®, basadas en el peso corporal inicial, se indican en la Tabla 1. Administrar RYBREVANT® semanalmente durante 4 semanas, con la dosis inicial como una infusión dividida en la Semana 1, el Día 1 y el Día 2, y luego administrar cada 2 semanas hasta progresión de la enfermedad o toxicidad inaceptable. Administrar la premedicación antes de cada infusión de RYBREVANT® según sea recomendado (ver "Posología y administración, Premedicaciones recomendadas"). Administrar RYBREVANT® diluido por vía intravenosa según las velocidades de infusión de la Tabla 5 (ver "Posología y administración, Preparación" y "Posología y administración, Administración").
Premedicaciones recomendadas: Antes de la infusión inicial de RYBREVANT® (Semana 1, Días 1 y 2), administrar la premedicación según se describe en la Tabla 2 para reducir el riesgo de reacciones relacionadas con la infusión (ver "Advertencias y precauciones, Reacciones relacionadas con la infusión"):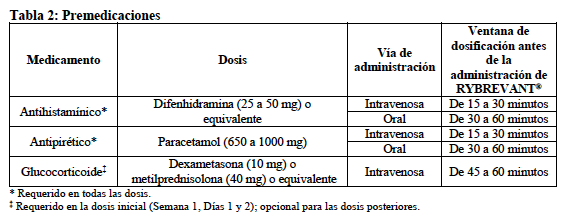
Administrar un antihistamínico y un antipirético antes de todas las infusiones. Se requiere la administración de un glucocorticoide únicamente para las dosis de la Semana 1, Días 1 y 2, y para las infusiones posteriores según sea necesario. Modificaciones de la dosis en caso de reacciones adversas: En la Tabla 3 se enumeran las reducciones de la dosis de RYBREVANT® recomendadas para las reacciones adversas (ver Tabla 4).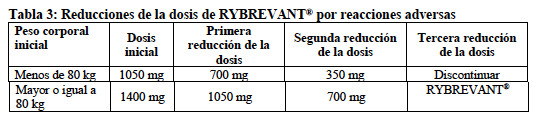
En la Tabla 4 se enumeran las modificaciones de la dosis de RYBREVANT® recomendadas para las reacciones adversas.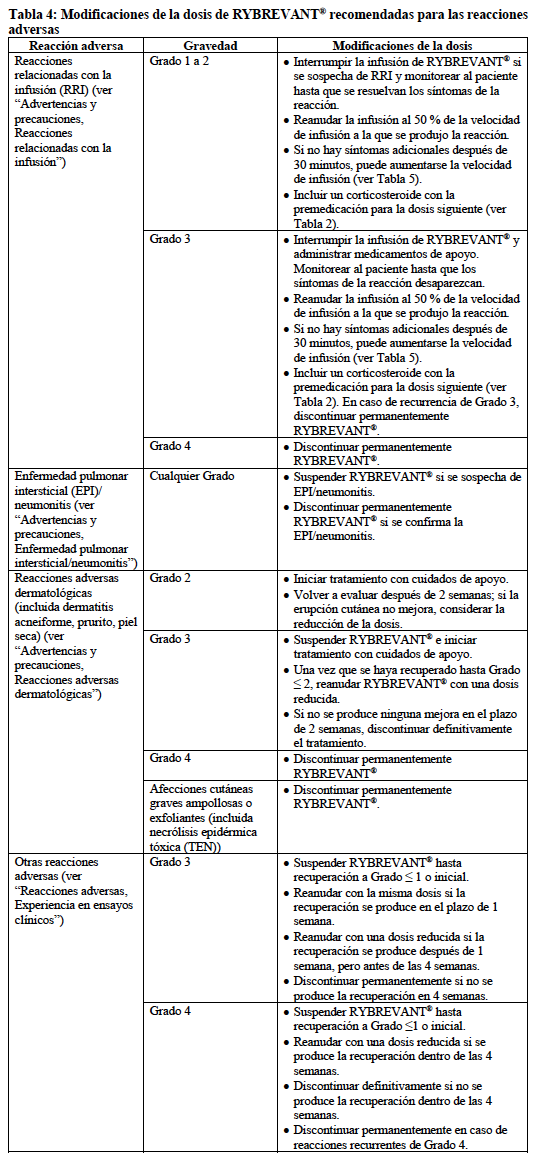
Preparación Diluir y preparar RYBREVANT® para infusión intravenosa antes de su administración. • Comprobar que la solución de RYBREVANT® es incolora a amarillo pálido. Los medicamentos parenterales deben inspeccionarse visualmente en busca de partículas y decoloración antes de su administración, siempre que la solución y el envase lo permitan. No utilizar si hay decoloración o partículas visibles. • Determinar la dosis requerida (1050 mg o 1400 mg) y el número de frascos de RYBREVANT® necesarios basado en el peso inicial del paciente (ver "Posología y administración, Dosificación recomendada"). Cada frasco de RYBREVANT® contiene 350 mg de amivantamab. • Extraer y desechar un volumen de solución de dextrosa al 5% o solución de cloruro de sodio al 0,9% de la bolsa de infusión de 250 ml igual al volumen de RYBREVANT® a añadir (es decir, desechar 7 ml de diluyente de la bolsa de infusión por cada frasco de RYBREVANT®). Utilizar únicamente bolsas de infusión de policloruro de vinilo (PVC), polipropileno (PP), polietileno (PE) o mezcla de poliolefinas (PP+PE). • Extraer 7 ml de RYBREVANT® de cada frasco y añadirlos a la bolsa de infusión. El volumen final en la bolsa de infusión debe ser de 250 ml. Desechar cualquier porción no utilizada que quede en el frasco. • Invertir suavemente la bolsa para mezclar la solución. No agitar. • Las soluciones diluidas deben administrarse dentro de las 10 horas (incluido el tiempo de infusión) a una temperatura ambiente de 15°C a 25°C. Administración Administrar la solución diluida (ver "Posología y administración, Preparación") mediante infusión intravenosa utilizando un equipo de infusión equipado con un regulador de flujo y con un filtro de polietersulfona (PES) en línea, estéril, no pirogénico y de baja unión a proteínas (tamaño de poro de 0,2 micrómetros) cebado únicamente con diluyente. Los equipos de administración deben ser de poliuretano (PU), polibutadieno (PBD) PVC, PP o PE. No infundir RYBREVANT® concomitantemente en la misma vía intravenosa con otros agentes. Administrar RYBREVANT® mediante una vía periférica en la Semana 1 y en la Semana 2, dada la alta incidencia de reacciones relacionadas con la infusión durante el tratamiento inicial (ver "Advertencias y precauciones, Reacciones relacionadas con la infusión"). RYBREVANT® puede administrarse a través de una vía central en las semanas siguientes. Para la infusión inicial, preparar RYBREVANT® lo más cerca posible del momento de la administración para permitir la posibilidad de prolongar el tiempo de infusión en caso de una reacción relacionada con la infusión. Administrar la infusión de RYBREVANT® por vía intravenosa de acuerdo con las velocidades de infusión de la Tabla 5.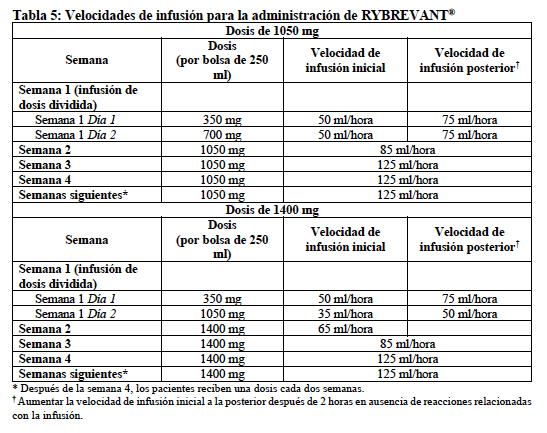
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes.
Reacciones adversas.
Las siguientes reacciones adversas se tratan en otras secciones del prospecto: Reacciones relacionadas con la infusión (ver "Advertencias y precauciones, Reacciones relacionadas con la infusión"). Enfermedad pulmonar intersticial/neumonitis (ver "Advertencias y precauciones, Enfermedad pulmonar intersticial/neumonitis"). Reacciones adversas dermatológicas (ver "Advertencias y precauciones, Reacciones adversas dermatológicas"). Toxicidad ocular (ver "Advertencias y precauciones, Toxicidad ocular"). Experiencia en ensayos clínicos: Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no pueden compararse directamente con las tasas de ensayos clínicos de otro medicamento y podrían no reflejar las tasas observadas en la práctica. La población de seguridad descrita en las "Advertencias y precauciones" refleja la exposición a RYBREVANT® como agente único en el estudio CHRYSALIS en 302 pacientes con NSCLC localmente avanzado o metastásico que recibieron una dosis de 1050 mg (para pacientes < 80 kg) o 1400 mg (para pacientes ≥80 kg) una vez a la semana durante 4 semanas, y después cada 2 semanas. Entre los 302 pacientes que recibieron RYBREVANT®, el 36% estuvo expuesto durante 6 meses o más, y el 12% estuvo expuesto durante más de un año. En la población de seguridad, las reacciones adversas más frecuentes (≥20%) fueron erupción cutánea, reacción relacionada con la infusión, paroniquia, dolor musculoesquelético, disnea, náuseas, edema, tos, fatiga, estomatitis, estreñimiento, vómitos y prurito. Las alteraciones de laboratorio de Grado 3 a 4 más frecuentes (≥2%) fueron disminución de linfocitos, disminución de fosfato, disminución de albúmina, aumento de glucosa, aumento de gamma-glutamil transferasa, disminución de sodio, disminución de potasio y aumento de fosfatasa alcalina. Los datos que se describen a continuación reflejan la exposición a RYBREVANT® a la dosis recomendada en 129 pacientes con NSCLC localmente avanzado o metastásico con mutaciones de inserción del exón 20 del EGFR cuya enfermedad había experimentado progresión con la quimioterapia basada en platino o después de esta. Entre los pacientes que recibieron RYBREVANT®, el 44% estuvo expuesto durante 6 meses o más y el 12% estuvo expuesto durante más de un año. La mediana de la edad fue de 62 años (rango: 36 a 84 años); el 61% eran mujeres; el 55% eran asiáticos, el 35% eran blancos y el 2,3% eran negros; y el 82% tenían un peso corporal inicial < 80 kg. Se produjeron reacciones adversas serias en el 30% de los pacientes que recibieron RYBREVANT®. Las reacciones adversas serias en ≥2% de los pacientes incluyeron embolia pulmonar, neumonitis/EPI, disnea, dolor musculoesquelético, neumonía y debilidad muscular. Se produjeron reacciones adversas mortales en 2 pacientes (1,5%) debido a neumonía y en 1 paciente (0,8%) debido a muerte súbita. Se produjo la discontinuación permanente de RYBREVANT® debido a una reacción adversa en el 11% de los pacientes. Las reacciones adversas que provocaron la interrupción permanente de RYBREVANT® en ≥1% de los pacientes fueron neumonía, RRI, neumonitis/EPI, disnea, derrame pleural y erupción cutánea. Se produjeron interrupciones de la dosis de RYBREVANT® debido a una reacción adversa en el 78% de los pacientes. Se produjeron reacciones relacionadas con la infusión (RRI) que requirieron interrupciones de la infusión en el 59% de los pacientes. Las reacciones adversas que requirieron la interrupción de la dosis en ≥5% de los pacientes incluyeron disnea, náuseas, erupción cutánea, vómitos, fatiga y diarrea. Se produjo reducción de la dosis de RYBREVANT® debido a una reacción adversa en el 15% de los pacientes. Las reacciones adversas que requirieron reducciones de la dosis en ≥2% de los pacientes incluyeron erupción cutánea y paroniquia. Las reacciones adversas más frecuentes (≥20%) fueron erupción cutánea, RRI, paroniquia, dolor musculoesquelético, disnea, náuseas, fatiga, edema, estomatitis, tos, estreñimiento y vómitos. Las alteraciones de laboratorio de Grado 3 a 4 más frecuentes (≥2%) fueron disminución de linfocitos, disminución de albúmina, disminución de fosfato, disminución de potasio, aumento de glucosa, aumento de fosfatasa alcalina, aumento de gamma-glutamil transferasa y disminución de sodio. La Tabla 6 resume las reacciones adversas de CHRYSALIS.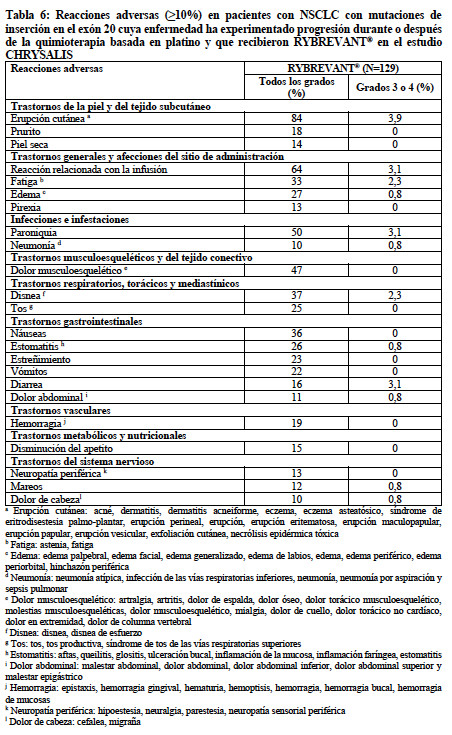
Las reacciones adversas clínicamente relevantes en < 10% de los pacientes que recibieron RYBREVANT® incluyeron toxicidad ocular, EPI/neumonitis y necrólisis epidérmica tóxica (NET). La Tabla 7 resume las alteraciones de laboratorio en el estudio CHRYSALIS.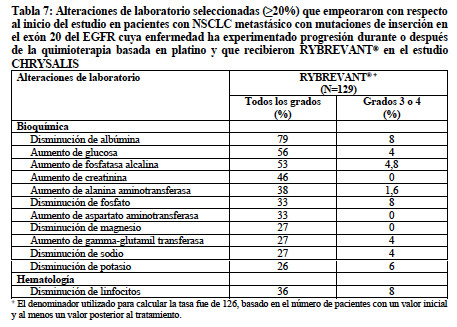
Inmunogenicidad: Como ocurre con todas las proteínas terapéuticas, existe la posibilidad de que se produzca inmunogenicidad. La detección de la formación de anticuerpos depende en gran medida de la sensibilidad y la especificidad del ensayo. Además, la incidencia observada de positividad de anticuerpos (incluidos anticuerpos neutralizantes) en un ensayo puede estar influenciada por varios factores, como la metodología del ensayo, la manipulación de la muestra, el momento de la recolección de la muestra, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos en los estudios descritos a continuación con la incidencia de anticuerpos en otros estudios o con otros productos de amivantamab puede ser engañosa. En el estudio CHRYSALIS, 3 de los 286 (1%) pacientes que fueron tratados con RYBREVANT® y eran evaluables en cuanto a la presencia de anticuerpos antifármaco (ADA), presentaron resultados positivos para anticuerpos anti-amivantamab emergentes del tratamiento (uno a los 27 días, otro a los 59 días y otro a los 168 días después de la primera dosis) con títulos de 1:40 o menos. No hay datos suficientes para evaluar el efecto de los ADA en la farmacocinética, la seguridad o la eficacia de RYBREVANT®. Uso en poblaciones específicas: Embarazo: Resumen de riesgos: Basado en el mecanismo de acción y los hallazgos en modelos animales, RYBREVANT® puede causar daño fetal cuando se administra a una mujer embarazada. No hay datos disponibles sobre el uso de RYBREVANT® en mujeres embarazadas ni datos en animales para evaluar el riesgo de RYBREVANT® en el embarazo. La alteración o el agotamiento del EGFR en modelos animales dio lugar a un deterioro del desarrollo embriofetal, que incluyó efectos en el desarrollo placentario, pulmonar, cardíaco, cutáneo y neural. La ausencia de señalización del EGFR o de MET ha dado lugar a letalidad embrionaria, malformaciones y muerte posnatal en animales (ver "Datos"). Advertir a las mujeres embarazadas del riesgo potencial para el feto. En la población general de Estados Unidos el riesgo de fondo estimado de defectos congénitos mayores y de aborto espontáneo en embarazos clínicamente reconocidos es del 2% al 4% y del 15% al 20%, respectivamente. Datos: Datos en animales: No se han realizado estudios en animales para evaluar los efectos de amivantamab sobre la reproducción y el desarrollo fetal; sin embargo, basado en su mecanismo de acción, RYBREVANT® puede causar daño fetal o anomalías en el desarrollo. En ratones, el EGFR tiene una importancia crítica en los procesos reproductivos y de desarrollo, incluida la implantación del blastocisto, el desarrollo de la placenta y la supervivencia y el desarrollo embriofetal/posnatal. La reducción o eliminación de la señalización del EGFR embriofetal o materno puede impedir la implantación, causar pérdida embriofetal durante varias etapas de la gestación (a través de efectos en el desarrollo de la placenta) y causar anomalías en el desarrollo y muerte temprana en los fetos supervivientes. Se han observado resultados adversos en el desarrollo de múltiples órganos en embriones/neonatos de ratones con señalización del EGFR alterada. Del mismo modo, la inactivación génica de MET o de su ligando HGF fue letal para el embrión debido a graves defectos en el desarrollo de la placenta, y los fetos mostraron defectos en el desarrollo muscular en varios órganos. Se sabe que la IgG1 humana atraviesa la placenta; por lo tanto, amivantamab tiene el potencial de transmitirse de la madre al feto en desarrollo. Lactancia: Resumen de riesgos No hay datos sobre la presencia de amivantamab en la leche humana sobre la producción de leche, o sus efectos en el niño amamantado. Debido a la posibilidad de que se produzcan reacciones adversas serias causadas por RYBREVANT® en niños amamantados, se aconseja a las mujeres que no amamanten durante el tratamiento con RYBREVANT® y en los 3 meses siguientes a la última dosis. Mujeres y hombres en edad fértil RYBREVANT® puede causar daño fetal cuando se administra a una mujer embarazada (ver "Uso en poblaciones específicas, Embarazo"). Pruebas de embarazo: Verificar el estado de embarazo de las mujeres en edad fértil antes de iniciar la administración de RYBREVANT®. Anticoncepción: Mujeres Aconsejar a las mujeres en edad fértil que utilicen métodos anticonceptivos efectivos durante el tratamiento y en los 3 meses siguientes a la última dosis de RYBREVANT®. Uso pediátrico No se ha establecido la seguridad y eficacia de RYBREVANT® en pacientes pediátricos. Uso en pacientes de edad avanzada De los 129 pacientes tratados con RYBREVANT®, el 41% tenía 65 años o más, y el 9% tenía 75 años o más. No se observaron diferencias clínicamente importantes en cuanto a la seguridad o la eficacia entre los pacientes que tenían ≥65 años de edad y los pacientes más jóvenes.
Advertencias.
Reacciones relacionadas con la infusión: RYBREVANT® puede causar reacciones relacionadas con la infusión (RRI); los signos y síntomas de RRI incluyen disnea, rubor, fiebre, escalofríos, náuseas, malestar torácico, hipotensión y vómitos. Basado en la población de seguridad (ver "Reacciones adversas, Experiencia en ensayos clínicos"), se produjeron RRI en el 66% de los pacientes tratados con RYBREVANT®. Entre los pacientes que recibieron el tratamiento el Día 1 de la Semana, el 65% experimentó una RRI, mientras que la incidencia de RRI fue del 3,4% con la infusión del Día 2, del 0,4% con la infusión de la Semana 2 y del 1,1% acumulativamente con las infusiones posteriores. De las RRI notificadas, el 97% fueron de Grado 1-2, el 2,2% de Grado 3 y el 0,4% de Grado 4. La mediana del tiempo hasta la aparición fue de 1 hora (rango de 0,1 a 18 horas) luego del inicio de la infusión. La incidencia de modificaciones de la infusión debido a RRI fue del 62% y el 1,3% de los pacientes interrumpieron permanentemente RYBREVANT® debido a RRI. Premedicar con antihistamínicos, antipiréticos y glucocorticoides e infundir RYBREVANT® según sea recomendado (ver "Posología y administración, Premedicaciones recomendadas"). Administrar RYBREVANT® a través de una vía periférica en la Semana 1 y en la Semana 2 (ver "Posología y administración, Administración"). Monitorear a los pacientes para detectar cualquier signo o síntoma de reacciones a la infusión durante la infusión de RYBREVANT® en un entorno en el que se disponga de medicación y equipo de reanimación cardiopulmonar. Interrumpir la infusión si se sospecha una RRI. Reducir la velocidad de infusión o discontinuar permanentemente RYBREVANT® en función de la gravedad (ver "Posología y administración, Modificaciones de la dosis en caso de reacciones adversas"). Enfermedad pulmonar intersticial/neumonitis: RYBREVANT® puede causar enfermedad pulmonar intersticial (EPI)/neumonitis. En base a la población de seguridad (ver "Reacciones adversas, Experiencia en ensayos clínicos"), se produjo EPI/neumonitis en el 3,3% de los pacientes tratados con RYBREVANT®, y el 0,7% de los pacientes experimentó EPI/neumonitis de Grado 3. Tres pacientes (1%) interrumpieron RYBREVANT® debido a la EPI/neumonitis. Monitorear a los pacientes por si aparecen o empeoran los síntomas indicadores de EPI/neumonitis (por ejemplo, disnea, tos, fiebre). Suspender inmediatamente RYBREVANT® en pacientes con sospecha de EPI/neumonitis y discontinuarlo permanentemente si se confirma la EPI/neumonitis (ver "Posología y administración, Modificaciones de la dosis en caso de reacciones adversas"). Reacciones adversas dermatológicas: RYBREVANT® puede causar erupción cutánea (incluida dermatitis acneiforme), prurito y sequedad de la piel. Basado en la población de seguridad (ver "Reacciones adversas, Experiencia en ensayos clínicos"), se produjo erupción cutánea en el 74 % de los pacientes tratados con RYBREVANT®, incluida erupción cutánea de Grado 3 en el 3,3 % de los pacientes. La mediana del tiempo transcurrido hasta la aparición de la erupción cutánea fue de 14 días (rango: 1 a 276 días). En el 5 % de los pacientes se produjo erupción cutánea que obligó a reducir la dosis, y en el 0,7 % de los pacientes se interrumpió permanentemente el tratamiento con RYBREVANT® debido a la erupción cutánea (ver "Reacciones adversas, Experiencia en ensayos clínicos"). Se produjo necrólisis epidérmica tóxica (NET) en un paciente (0,3%) tratado con RYBREVANT®. Indicar a los pacientes que limiten la exposición al sol durante el tratamiento con RYBREVANT® y en los 2 meses posteriores al mismo. Aconsejar a los pacientes que lleven ropa protectora y utilicen un protector solar de amplio espectro UVA/UVB. Se recomienda una crema emoliente sin alcohol para la piel seca. Si se producen reacciones cutáneas, comenzar la administración de corticosteroides tópicos y antibióticos tópicos y/u orales. En caso de reacciones de Grado 3, añadir esteroides orales y considerar la consulta dermatológica. Derivar inmediatamente a un dermatólogo a los pacientes que presenten erupción cutánea grave, aspecto o distribución atípicos, o que no mejoren en el plazo de 2 semanas. Suspender, reducir la dosis o interrumpir permanentemente RYBREVANT® en función de la gravedad (ver "Posología y administración, Modificaciones de la dosis en caso de reacciones adversas"). Toxicidad ocular: RYBREVANT® puede causar toxicidad ocular, incluida queratitis, síntomas de sequedad ocular, enrojecimiento de la conjuntiva, visión borrosa, deterioro visual, prurito ocular y uveítis. Basado en la población de seguridad (ver "Reacciones adversas, Experiencia en ensayos clínicos"), se produjo queratitis en el 0,7% y uveítis en el 0,3% de los pacientes tratados con RYBREVANT®. Todos los eventos fueron de Grado 1-2. Derivar inmediatamente a un oftalmólogo a los pacientes que presenten síntomas oculares. Suspender, reducir la dosis o interrumpir permanentemente RYBREVANT® en función de la gravedad (ver "Posología y administración, Modificaciones de la dosis en caso de reacciones adversas"). Toxicidad embriofetal: En base a su mecanismo de acción y en los hallazgos en modelos animales, RYBREVANT® puede causar daño fetal cuando se administra a una mujer embarazada. La administración de otras moléculas inhibidoras del EGFR a animales gestantes ha dado lugar a un aumento de la incidencia de alteraciones del desarrollo embriofetal, letalidad embrionaria y aborto. Advertir a las mujeres con potencial reproductivo del riesgo potencial para el feto. Aconsejar a las pacientes con potencial reproductivo que utilicen métodos anticonceptivos efectivos durante el tratamiento y en los 3 meses posteriores a la última dosis de RYBREVANT® (ver "Uso en poblaciones específicas, Embarazo" y "Uso en poblaciones específicas, Mujeres y hombres en edad fértil").
Conservación.
Conservar en heladera a una temperatura de 2°C a 8°C en su envase original para protegerlo de la luz. No congelar.
Sobredosificación.
Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los Centros de Toxicología de: Hospital de Pediatría Dr. Ricardo Gutiérrez - Tel: (011) 4962-6666 / 2247. Hospital A. Posadas - Tel: (011) 4654-6648 y 4658-7777.
Presentación.
RYBREVANT® (amivantamab) es una solución estéril, sin conservantes, de incolora a amarillo pálido para infusión endovenosa. Cada frasco de uso único contiene 350 mg/7 ml (50 mg/ml) de amivantamab. Cada frasco se envasa individualmente en una caja.
Revisión.
28 de junio de 2023.