RECIT®
GADOR
Grupo farmacoterapéutico: simpaticomiméticos de acción central. Código ATC: N06BA09.
Composición.
Cada cápsula de RECIT® 10 contiene: Atomoxetina base (equivalente Atomoxetina clorhidrato 11,43 mg) 10 mg. Excipientes: Dimeticona, Almidón pregelatinizado c.s. Componentes de la cápsula: Gelatina y Dióxido de titanio Cada cápsula de RECIT® 18 contiene: Atomoxetina base (equivalente Atomoxetina clorhidrato 20,57 mg) 18 mg. Excipientes: Dimeticona, Almidón pregelatinizado c.s. Componentes de la cápsula: Gelatina, Dióxido de titanio y Óxido de hierro amarillo. Cada cápsula de RECIT® 25 contiene: Atomoxetina base (equivalente Atomoxetina clorhidrato 28,57 mg) 25 mg. Excipientes: Dimeticona, Almidón pregelatinizado c.s. Componentes de la cápsula: Gelatina, Dióxido de titanio, FD&C Azul N°2 y Óxido de hierro negro. Cada cápsula de RECIT® 40 contiene: Atomoxetina base (equivalente Atomoxetina clorhidrato 45,71 mg) 40 mg. Excipientes: Dimeticona, Almidón pregelatinizado c.s. Componentes de la cápsula: Gelatina, Dióxido de titanio, FD&C Azul N°2 y Óxido de hierro negro. Cada cápsula de RECIT® 60 contiene: Atomoxetina base (equivalente Atomoxetina clorhidrato 68,56 mg) 60 mg. Excipientes: Dimeticona, Almidón pregelatinizado c.s. Componentes de la cápsula: Gelatina, Dióxido de titanio, FD&C Azul N°2, Óxido de hierro negro y Óxido de hierro amarillo.
Farmacología.
Mecanismo de acción: RECIT® (clorhidrato de atomoxetina) es un inhibidor selectivo de la recaptación de norepinefrina. Su efecto terapéutico en el TDAH se considera relacionado con su potente inhibición del transportador presináptico de norepinefrina, con afinidad mínima por otros receptores noradrenérgicos u otros transportadores o receptores de neurotransmisores. Farmacodinámica: En estudios ex vivo de captación y depleción de neurotransmisores, se encontró que atomoxetina inhibe selectivamente al transportador presináptico de norepinefrina sin afectar directamente a los transportadores de serotonina o dopamina. Atomoxetina presenta afinidad mínima por otros sistemas de receptores. La atomoxetina se oxida principalmente a 4-hidroxiatomoxetina, que es un potente inhibidor del transportador presináptico de norepinefrina. Seguridad cardiovascular: Se estudió la seguridad y tolerabilidad de regímenes de aumento gradual de múltiples dosis de 60 a 150 mg/día de atomoxetina en 16 adultos sanos (10 sujetos MP (metabolizadores potentes) y 6 sujetos MD (metabolizadores débiles)). Ninguno de los intervalos medios o individuales del QTc(F) superó el límite superior normal para cada sexo. El grupo MP no presentó cambios estadísticamente significativos en el intervalo QTc(F) medio en comparación con el tratamiento placebo. No se observaron cambios estadísticamente significativos en el QTc(F) 1 hora después de la dosis (durante la concentración plasmática máxima) en el grupo MD. El grupo MD presentó un aumento estadísticamente significativo en el intervalo QTc(F) medio medido en el plazo 0 (durante la concentración plasmática mínima) el último día de los regímenes de dosis de 60 y 75 mg de atomoxetina dos veces por día, en comparación con placebo. La mayor prolongación media fue de aproximadamente 17 mseg con el nivel de dosis de 60 mg dos veces por día, con una duración media del intervalo de 417,2 mseg. Con el nivel de dosis de 75 mg de atomoxetina dos veces por día, la mayor prolongación media fue de 15 mseg y la duración media del inter valo fue de 414,9 mseg. La dosis de 60 mg dos veces por día y de 75 mg dos veces por día corresponde a 1,4 - 2,24 mg/kg/día y 1,75 - 2,8 mg/kg/día respectivamente. Los ECG iniciales obtenidos durante la selección de los pacientes pediátricos en los estudios clínicos de atomoxetina se revisaron buscando casos de prolongación del QT. Usando un método de corrección fundamentado en los datos de los ECG iniciales, se presentaron 32/3902 casos (0,8%) con QTc(D) > 450 mseg y 5/3902 casos (0,1%) con QTc(D) > 500 mseg. En un metaanálisis de los datos del ECG de pacientes que recibieron atomoxetina en estudios clínicos pediátricos, no se observó relación entre los cambios desde el QTc(D) inicial al final, y la dosis indicada de atomoxetina, ni entre los cambios desde el QTc(D) inicial al momento de la exposición máxima prevista, y la dosis indicada de atomoxetina. En general, los datos no sugieren una relación significativa entre la concentración plasmática de atomoxetina y la duración del intervalo QT corregido por frecuencia cardíaca, en el rango de dosis recomendado. Sin embargo, dado que no existe un requerimiento de selección previa de pacientes con TDAH en cuanto al estado de metabolización por CYP2D6 antes de iniciar el tratamiento con atomoxetina, es importante usar la menor dosis efectiva para minimizar los potenciales efectos adversos cardíacos. Farmacocinética: La atomoxetina se absorbe bien después de la administración oral y los alimentos la afectan en forma mínima. Se elimina principalmente por metabolismo oxidativo a través de la vía enzimática del citocromo P450 2D6 (CYP2D6) y posterior glucuronización. La atomoxetina tiene una vida media de aproximadamente 5 horas. Una fracción de la población (aproximadamente el 7% de los caucásicos y el 2% de los afroamericanos) son metabolizadores débiles (MD) de los medicamentos metabolizados por la CYP2D6. Estos individuos presentan una reducción de la actividad en esta vía, lo que resulta en un ABC 10 veces mayor, concentración plasmática máxima 5 veces mayor y eliminación más lenta (vida media plasmática de 21,6 horas) de atomoxetina en comparación con las personas con actividad normal [metabolizadores potentes (MP)]. Los medicamentos que inhiben la CYP2D6 tal como fluoxetina, paroxetina y quinidina, causan un aumento similar en la exposición. La farmacocinética de atomoxetina se evaluó en más de 400 niños y adolescentes en estudios clínicos seleccionados mediante una estrategia poblacional. Los datos farmacocinéticos individuales de dosis únicas y en estado de equilibrio también se obtuvieron en niños, adolescentes y adultos. Cuando se normalizaron las dosis en base a mg/kg, se observaron valores similares de vida media, Cmáx y ABC en niños, adolescentes y adultos. La depuración y el volumen de distribución después del ajuste por peso corporal también fueron similares. La farmacocinética de atomoxetina es proporcional a la dosis dentro del rango terapéutico; por ende, la administración de RECIT® una o dos veces por día se prevé que resulte en la misma exposición sistémica (ABC) durante un período de 24 horas. Los resultados de los análisis de eficacia muestran que la administración una vez por día de RECIT® es eficaz para el tratamiento del TDAH. Las cápsulas de 80 mg de RECIT® son bioequivalentes a 2 cápsulas de 40 mg. Las cápsulas de 100 mg de RECIT® son bioequivalentes a la combinación de una cápsula de 40 mg y una de 60 mg. Absorción: La atomoxetina se absorbe rápidamente después de la administración oral, con una biodisponibilidad absoluta de aproximadamente el 63% en metabolizadores potentes (MP) y del 94% en metabolizadores débiles (MD). La concentración plasmática máxima media (Cmáx) se alcanza aproximadamente 1 a 2 horas después de la administración. RECIT® se puede administrar con o sin alimentos. En estudios clínicos con niños y adolescentes, la administración de RECIT® con alimentos resultó en una Cmáx 9% menor. La administración de RECIT® con un alimento con alto contenido graso en adultos, no afectó el grado de absorción oral de atomoxetina (ABC), pero disminuyó la velocidad de absorción, lo que resultó en una Cmáx 37% menor y una Tmáx demorada en 3 horas. Distribución: el volumen de distribución en estado de equilibrio después de la administración intravenosa, fue de aproximadamente 0,85 L/kg, lo que indica que atomoxetina se distribuye principalmente en el agua corporal total. En niños y adolescentes, el volumen de distribución aumentó en forma aproximadamente proporcional a los aumentos de peso corporal. El volumen de distribución es similar a través del rango de peso del paciente después de la normalización por peso corporal. En concentraciones terapéuticas, el 98% de atomoxetina en plasma se encuentra unido a las proteínas, principalmente a la albúmina. Metabolismo: la atomoxetina sufre una biotransformación principalmente a través de la vía enzimática del citocromo P450 2D6 (CYP2D6). Las personas con actividad reducida de la vía CYP2D6 (MD) presentan mayores concentraciones plasmáticas de atomoxetina en comparación con las personas con actividad normal (MP). Para los MD, el ABC de atomoxetina en estado de equilibrio es aproximadamente 10 veces mayor y la Css,máx es aproximadamente el quíntuple de aquella en los MP. La administración concomitante de RECIT® con inhibidores potentes de la CYP2D6 tal como fluoxetina, paroxetina o quinidina, resulta en un aumento sustancial en la exposición plasmática a atomoxetina, y puede ser necesario un ajuste de la dosis (véase interacciones farmacológicas). En pacientes MP tratados con inhibidores potentes de la CYP2D6 tal como fluoxetina y paroxetina, el ABC de atomoxetina es aproximadamente 6 a 8 veces mayor y la Css,máx aproximadamente el triple o el cuádruple de aquella con atomoxetina solamente. Los estudios in vitro sugieren que la administración concomitante de inhibidores del citocromo P450 a los MD, no aumentará la concentración plasmática de atomoxetina. Atomoxetina no inhibe ni induce la vía del CYP2D6. El principal metabolito oxidativo que se forma independientemente del estado CYP2D6 es la 4-hidroxiatomoxetina, que es rápidamente glucuronizada. La 4-hidroxiatomoxetina es equipotente a la atomoxetina en cuanto a inhibición del transportador de norepinefrina, pero circula en el plasma en una concentración mucho menor (1% de la concentración de atomoxetina en los MP y 0,1% de la concentración de atomoxetina en los MD). La 4-hidroxiatomoxetina se forma principalmente por la CYP2D6. En individuos con ausencia de actividad del CYP2D6 (metabolizadores débiles), la 4-hidroxiatomoxetina se forma por varias otras enzimas del citocromo P450, pero a velocidad más lenta. La N-desmetil-atomoxetina se forma por la CYP2C19 y otras enzimas del citocromo P450, pero presenta una actividad farmacológica mucho menor a la de atomoxetina, y en menor concentración plasmática (5% de la concentración de atomoxetina en MP y 45% de la concentración de atomoxetina en MD). Eliminación: la vida media de eliminación media de atomoxetina después de la administración oral, es de 5,2 horas y 21,6 horas en sujetos MP y MD respectivamente. La vida media de eliminación de 4-hidroxiatomoxetina es similar a la de la N-desmetil atomoxetina (6 a 8 horas) en sujetos MP, mientras que la vida media de la N-desmetil atomoxetina es mucho mayor en los sujetos MD (34 a 40 horas). La atomoxetina se excreta principalmente como 4-hidroxiatomoxetina-O-glucurónico, principalmente en la orina (más del 80% de la dosis) y en menor grado en las heces (menos del 17% de la dosis). Solo una pequeña fracción (menos del 3%) de la dosis de RECIT® se excreta como atomoxetina sin modificar, lo que indica una importante biotransformación. Poblaciones y afecciones especiales: Pediatría: la farmacocinética de atomoxetina en niños y adolescentes es similar a la de los adultos. No se evaluó la farmacocinética en niños menores de 6 años de edad. Mayores: no se evaluó en forma sistemática la farmacocinética de atomoxetina en la población mayor. Sexo: el sexo no modifica la disposición de atomoxetina. Raza: el origen étnico no modifica la disposición de atomoxetina. Insuficiencia hepática: dosis únicas de RECIT® administradas a sujetos MP con insuficiencia hepática moderada a grave (clase Child-Pugh B y C), resultaron en un aumento de la exposición a atomoxetina, reducción de la depuración de atomoxetina y prolongación de la vida media del medicamento original, en comparación con sujetos sanos. Se recomienda el ajuste de la dosis en pacientes con insuficiencia hepática moderada o grave (véase Dosificacion). Insuficiencia renal: dosis únicas de RECIT® administradas a sujetos MP con enfermedad renal en etapa terminal, resultaron en una mayor exposición a atomoxetina (ABC) que en sujetos sanos (aproximadamente un aumento del 65%), pero no se observó diferencia cuando se corrigió la exposición por dosis en mg/kg. Por lo tanto, RECIT® se puede administrar a pacientes con TDAH con enfermedad renal en etapa terminal o grados menores de insuficiencia renal usando un régimen de dosis normal. Polimorfismo genético: existen dos fenotipos principales asociados con la CYP2D6: metabolizadores potentes que comprenden > 90% de la población, y metabolizadores débiles. Aproximadamente el 7% de la población caucásica y el 2% de la población negra son metabolizadores débiles por la CYP2D6. Pacientes con enfermedades concomitantes: Pacientes pediátricos con TDAH y tics comórbidos: El estudio LYAS inscribió pacientes que cumplían los criterios DSM-IV para TDAH y trastorno de Tourette comórbido, o tics motores crónicos. RECIT® cumplió el objetivo principal del estudio de no inferioridad con placebo con respecto al empeoramiento de los tics, y presentó un efecto beneficioso en la reducción de la gravedad de los tics. RECIT® también fue marcadamente superior a placebo en la reducción de síntomas de TDAH, según lo evaluado por la calificación ADHDRS-IV-Parent:Inv total (p = 0,002). Pacientes con TDAH y trastorno de ansiedad comórbido Estudio LYBP: pacientes pediátricos (n = 176, edad 8 - 17) que cumplían los criterios DSM-IV para TDAH y al menos uno de los criterios de trastorno de ansiedad del trastorno de ansiedad de separación, trastorno de ansiedad generalizado o fobia social, se aleatorizaron en proporción 1:1 en un estudio de 12 semanas, doble ciego controlado con placebo. El plan de análisis estadístico especificó que los pacientes con respuesta (es decir, reducción > 25% en la calificación total de la escala pediátrica de calificación de ansiedad [Pediatric Anxiety Rating Scale - PARS]) durante un período de introducción ciego con placebo, se excluyeran de los análisis principales de eficacia. Sin embargo, todos los pacientes incluidos con una calificación inicial y al menos una posterior a la inicial, se incluyeron en los análisis de eficacia restantes. La dosis inicial de RECIT® fue de 0,8 mg/kg/día, aumentando hasta una dosis terapéutica de 1,2 mg/kg/día (mediana de la dosis 1,3 mg/kg/día ± 0,3 mg/kg/día). RECIT® fue marcadamente superior al placebo para la reducción de los síntomas de TDAH según lo evaluado por la calificación total del ADHDRS-IV-Parent:Inv (p < 0,001) y la calificación total media PARS mejoró significativamente con RECIT® en relación con el placebo (p = 0,011). Posteriormente se definió empeoramiento de la ansiedad como un aumento del 25% en el PARS o ansiedad informada por el paciente como evento adverso. Fundamentado en estos criterios, los pacientes con RECIT® no presentaron empeoramiento de la ansiedad en relación con el grupo placebo. De los 158 pacientes que completaron la introducción doble ciego con placebo, 26 (16%) suspendieron el estudio. Estudio LYDQ: pacientes adultos (n = 442, edad 18 - 65 años) que cumplían los criterios DSM-IV para TDAH adulto y trastorno de ansiedad social (de los cuales el 23% también presentaban trastorno de ansiedad generalizado) se aleatorizaron en un estudio de 16 semanas, doble ciego controlado con placebo. El plan de análisis estadístico especificó que los pacientes con respuesta (es decir, disminución > 25% en los síntomas de ansiedad social según medición por la escala de ansiedad social de Liebowitz) durante el período de introducción ciego con placebo, se excluyeran de los análisis principales de eficacia. Sin embargo, todos los pacientes inscritos con calificación inicial y al menos una posterior a la inicial, se incluyeron en los análisis de eficacia restantes. RECIT® se inició con 40 mg/día hasta una dosis máxima de 100 mg/día (dosis diaria media 83 mg/día ± 19,5 mg/día). RECIT® fue marcadamente superior al placebo en la reducción de los síntomas de TDAH según lo evaluado mediante la escala de calificación de TDAH en adultos de Conners (p < 0,001) y la calificación total media LSAS mejoró significativamente con RECIT® en relación al placebo (p 0,01). Posteriormente se definió empeoramiento de la ansiedad como el aumento del 25% en LSAS, o ansiedad informada por el paciente como evento adverso. Fundamentado en estos criterios, los pacientes con RECIT® no presentaron empeoramiento de la ansiedad en relación al grupo placebo. De los 436 pacientes que completaron la introducción doble ciego con placebo, 172 (39%) suspendieron el estudio. Toxicología: Toxicidad aguda: La mediana de la dosis letal oral de clorhidrato de atomoxetina en animales se estimó en 25 mg/kg para gatos, > 37,5 mg/kg para perros y 190 mg/kg en ratas y ratones. Los signos premonitorios de toxicidad luego de dosis orales únicas de atomoxetina en animales, incluyeron midriasis y reducción del refl ejo pupilar a la luz, heces mucosas, salivación, vómitos, ataxia, temblores, mioclonías erráticas y convulsiones. Toxicidad a largo plazo: Se efectuaron estudios de toxicidad de hasta 1 año en ratas y perros adultos para evaluar la potencial toxicidad crónica de atomoxetina. No se observó una toxicidad importante en los órganos blancos principales en perros que recibieron dosis orales de hasta 16 mg/kg/día o en ratas que recibieron atomoxetina en la dieta en dosis medias ponderadas en el tiempo de hasta 47 mg/kg/día. Estas dosis son aproximadamente 6 - 7 veces la dosis oral diaria máxima recomendada en niños y aproximadamente 4 - 5 veces la dosis oral diaria máxima recomendad en adultos en base a mg/m². Se observaron efectos hepáticos leves, caracterizados por color moteado y pálido del hígado, aumento del peso hepático relativo, vacuolación hepatocelular y niveles séricos levemente aumentados de ALT en ratas macho que recibieron dosis medias ponderadas en el tiempo ≥14 mg/kg/día. No se observaron efectos hepáticos en perros. En éstos se observaron signos clínicos de midriasis, reducción del reflejo pupilar a la luz, emesis y temblores, y estos efectos fueron mínimos en perros adultos que recibieron ≤8 mg/kg/día. Se observaron signos clínicos similares en perros jóvenes (8 semanas de edad) que recibieron atomoxetina oral durante 1 mes. Se efectuaron estudios de toxicidad para evaluar los potenciales efectos de atomoxetina sobre el crecimiento, neurocomportamiento y desarrollo sexual, como así también el desempeño reproductivo en ratas que recibieron dosis desde el período posnatal precoz hasta la primera juventud. No se observaron efectos importantes sobre el crecimiento óseo, morfología cerebral, neurocomportamiento ni desempeño reproductivo en ratas que recibieron clorhidrato de atomoxetina desde los 10 días de edad hasta la adultez, con dosis orales de hasta 50 mg/kg, aproximadamente 7 veces la dosis oral diaria máxima recomendada en niños en base a mg/m². No se observaron efectos sobre la anatomía ni morfología de los órganos reproductores masculinos ni femeninos. Se observó un aumento menor, relacionado con la dosis, del tiempo de inicio de permeabilidad vaginal y separación del prepucio (1 a 3 días en comparación con los controles), pero todos los animales alcanzaron la madurez sexual y no se observaron malformaciones en la vagina ni prepucio/glande del pene. En ratas macho, la espermatogénesis fue normal. Se observó una leve disminución del número de espermatozoides mantenido en el epidídimo, pero sin efecto sobre la fertilidad. Tanto en ratas macho como en hembras, no se observó efecto sobre el apareamiento ni sobre el índice de fertilidad, tampoco efectos sobre la implantación embrionaria ni viabilidad de embriones, ni efectos sobre las pruebas de aprendizaje y memoria. Se desconoce el significado de estos hallazgos para los seres humanos. Carcinogenicidad: El clorhidrato de atomoxetina no fue carcinógeno en ratas ni ratones cuando se administró en la dieta durante 2 años con dosis medias ponderadas por tiempo de hasta 47 y 458 mg/kg/día respectivamente. La dosis máxima usada en ratas es aproximadamente 8 y 5 veces la dosis máxima en seres humanos para niños y adultos respectivamente, en base a mg/m². Se calculó que los niveles plasmáticos (ABC) de atomoxetina con esta dosis en ratas son 1,8 veces (metabolizad ores potentes) o 0,2 veces (metabolizadores débiles) de aquellas en seres humanos que reciben la dosis máxima. La mayor dosis usada en ratones es aproximadamente 39 y 26 veces la dosis máxima en seres humanos para niños y adultos, respectivamente, en base a mg/m². Mutagenicidad: El clorhidrato de atomoxetina fue negativo en una batería de estudios de genotoxicidad que incluyó un ensayo de mutación de punto reverso (prueba de Ames), un ensayo in vitro en linfoma de ratón, una prueba de aberración cromosómica en células de ovario de hámster chino, una prueba de síntesis no programada de ADN en hepatocitos de rata y una prueba in vivo de micronúcleo en ratones. Sin embargo, se observó un leve aumento en el porcentaje de células de ovario de hámster chino con diplocromosis, lo que sugiere endorreduplicación (aberración numérica). El metabolito clorhidrato de N-desmetilatomoxetina fue negativo en la prueba de Ames, ensayo en linfoma de ratón y prueba de síntesis no programada de ADN. Teratogenicidad: No se encontró evidencia de teratogenicidad ni retardo en el desarrollo fetal asociados al medicamento, en conejos o ratas que recibieron clorhidrato de atomoxetina durante la organogénesis en dosis orales de hasta 100 mg/kg/día y 150 mg/kg/día (al menos 20 veces la dosis diaria oral máxima recomendada en niños y 13 veces la dosis oral diaria máxima recomendada en adultos, sobre una base de mg/m²). En un estudio de fertilidad en ratas, se observó disminución del peso y supervivencia de las crías, predominantemente durante la primera semana posparto luego de dosis maternas promedio ponderada por tiempo de atomoxetina con la dieta de hasta 23 mg/kg/día o mayores. No se observaron efectos adversos en los grupos sobrevivientes. El clorhidrato de atomoxetina no alteró la fertilidad cuando se administró a ratas desde los 10 días de edad hasta la adultez en dosis orales de hasta 50 mg/kg/día (hasta 7 veces la dosis oral diaria máxima recomendada en niños y 4 veces la dosis oral diaria máxima recomendada en adultos, en base a mg/m²). Además, no se observó evidencia de alteración de la fertilidad en ninguno de los dos estudios de fertilidad en ratas adultas con clorhidrato de atomoxetina en la dieta en dosis promedio ponderadas por tiempo de hasta 57 mg/kg/día (hasta 8 veces la dosis oral diaria máxima recomendada en niños y 5 veces la dosis oral diaria máxima recomendada en adultos, en base a mg/m²). Atomoxetina no afectó el parto en las ratas. Deterioro de la fertilidad: El clorhidrato de atomoxetina no afectó la fertilidad en ratas que recibieron dosis en la dieta de hasta 57 mg/kg/día, lo que es aproximadamente 6 veces la dosis máxima en seres humanos en base a mg/m². Crecimiento y desarrollo del neurocomportamiento/sexual en ratas: Se efectuó un estudio en ratas jóvenes para evaluar los efectos de atomoxetina sobre el crecimiento y desarrollo del neurocomportamiento y sexual. Las ratas se trataron con 1, 10 o 50 mg/kg/día (aproximadamente 0,2; 2 y 8 veces respectivamente, la dosis máxima en seres humanos en base a mg/m²) de atomoxetina administrada por sonda desde el período posnatal precoz (día 10 de edad) hasta la adultez. Se observó una leve demora en el inicio de la permeabilidad vaginal (todas las dosis) y separación del prepucio (10 y 50 mg/kg), leve disminución del peso del epidídimo y número de espermatozoides (10 y 50 mg/kg) y leve disminución de los cuerpos lúteos (50 mg/kg), pero no se observaron efectos sobre la fertilidad ni desempeño reproductivo. Se observó una leve demora en la erupción de incisivos con 50 mg/kg. Se observó un leve aumento de la actividad motora al día 15 (machos con 10 y 50 mg/kg y hembras con 50 mg/kg) y al día 30 (hembras con 50 mg/kg) pero no al día 60 de edad. No se observaron efectos sobre las pruebas de aprendizaje y memoria. Se desconoce la importancia de estos hallazgos para los seres humanos.
Indicaciones.
RECIT® (clorhidrato de atomoxetina) está indicado para el tratamiento del trastorno de déficit de atención e hiperactividad (TDAH) en niños mayores de 6 años de edad, adolescentes y adultos. El diagnóstico de TDAH (DSM IV) implica la presencia de síntomas hiperactivos - impulsivos o de falta de atención que causan deterioro y se presentan antes de la edad de 7 años. Los síntomas deben ser persistentes, más graves que lo habitualmente observado en individuos con un nivel de desarrollo comparable, deben causar deterioro clínicamente significativo, por ej. en el desempeño social, académico u ocupacional, y debe estar presente en 2 o más ámbitos, por ej. escuela (o trabajo) y en el hogar. Los síntomas no deben poder explicarse por otro trastorno mental. Para el tipo de déficit de atención, debieron persistir al menos 6 de los siguientes síntomas durante al menos 6 meses: falta de atención a los detalles / errores por falta de cuidado, falta de atención sostenida, pobre escucha, fracaso en seguir tareas, mala organización, renuncia a realizar tareas que requieren esfuerzo mental sostenido, pérdida de objetos, distracción fácil, olvidos. Para el tipo hiperactivo impulsivo, debieron persistir al menos 6 de los siguientes síntomas durante al menos 6 meses: movimiento en exceso de manos o pies o imposibilidad de mantenerse inmóvil, dificultad para permanecer sentado, corre o trepa en situaciones que es inapropiado hacerlo, dificultad para participar en tareas tranquilas, comportamiento como si estuviera siempre "en marcha", habla excesiva, hablar sin pensar, no puede esperar su turno, intrusivo. Para un diagnóstico de tipo combinado, deben encontrarse criterios tanto de falta de atención como de hiper actividad impulsividad. Consideraciones especiales de diagnóstico: Se desconoce la etiología específica del TDAH y no existe una prueba diagnóstica única. El diagnóstico adecuado requiere el uso no solo de recursos médicos sino también psicológicos, educacionales y sociales especiales. El aprendizaje puede o no estar deteriorado. El diagnóstico se debe fundamentar en antecedentes y evaluación completa del paciente y no únicamente en la presencia del número necesario de características según el DSM IV. Necesidad de un programa integral de tratamiento: RECIT® está indicado como parte integral de un programa total de tratamiento para el TDAH que puede incluir otras medidas (psicológicas, educacionales y sociales) para los pacientes con este síndrome. No todos los pacientes tienen indicación de tratamiento farmacológico para este síndrome. El tratamiento farmacológico no está destinado para usarse en pacientes que presentan síntomas secundarios a factores ambientales y/u otros trastornos psiquiátricos primarios, incluyendo psicosis. Es esencial una ubicación educacional adecuada en niños y adolescentes con este diagnóstico y con frecuencia la intervención psicosocial es útil. Cuando las medidas correctivas son insuficientes por sí solas, la decisión de indicar tratamiento farmacológico dependerá de la evaluación del médico de la cronicidad y gravedad de los síntomas del paciente. Pediatría ( < 6 años de edad): No se estableció la seguridad y eficacia de RECIT® en pacientes pediátricos menores a 6 años de edad.
Dosificación.
Consideraciones de la dosis: RECIT® se debe administrar comenzando con la menor dosis posible. Luego se debe individualizar la dosis y ajustar lentamente hasta la menor dosis efectiva, dado que la respuesta individual de los pacientes a RECIT® presenta gran variación. RECIT® no se debe usar en pacientes con enfermedad cardiovascular sintomática y en general no se debe usar en pacientes con alteraciones cardíacas estructurales conocidas (véase Contraindicaciones y Advertencias). Niños: teóricamente existe el potencial farmacológico en todos los medicamentos para el TDAH de aumentar el riesgo de muerte súbita/cardíaca. Aunque falta la confirmación del aumento del riesgo de eventos adversos cardíacos por el tratamiento con medicamentos para el TDAH, el médico tratante debe considerar este riesgo potencial. Todos los medicamentos con efectos simpaticomiméticos indicados para el manejo del TDAH se deben usar con precaución en pacientes que: a) están involucrados en ejercicios o actividades extenuantes; b) usan estimulantes; o c) presentan antecedentes familiares de muerte súbita/cardíaca. Antes de iniciar el tratamiento con medicamentos simpaticomiméticos, se deben obtener los antecedentes personales y familiares (incluyendo la evaluación de antecedentes familiares de muerte súbita o arritmia ventricular) y un examen físico para evaluar la presencia de enfermedad cardíaca. En pacientes con factores de riesgo relevantes y fundamentado en el juicio clínico, puede requerirse mayor evaluación cardiovascular (por ej. electrocardiograma y ecocardiograma). Los pacientes que desarrollan síntomas tal como dolor torácico al esfuerzo, síncope inexplicado u otros síntomas sugestivos de enfermedad cardíaca durante el tratamiento del TDAH, deben efectuar una evaluación cardíaca de inmediato. Los pacientes considerados con necesidad de extender el tratamiento con RECIT® deben efectuar una evaluación periódica del estado cardiovascular (véase Advertencias). Se recomienda un riguroso monitoreo clínico en cuanto a ideación suicida u otros indicadores de potencial comportamiento suicida, en pacientes de cualquier edad. Esto incluye el monitoreo de cambios emocionales y del comportamiento de tipo agitación y empeoramiento clínico (véase Advertencias, Potencial asociación con la ocurrencia de cambios del comportamiento y emocionales incluyendo daño autoinfligido). RECIT® (clorhidrato de atomoxetina) está indicado para administración oral y se puede ingerir con o sin alimentos, como dosis diaria única por la mañana o en dosis divididas por la mañana y por la noche. En general los síntomas iniciales de mejoría del TDAH se observan dentro de 1 a 4 semanas desde el inicio del tratamiento. RECIT® no empeora los tics en pacientes pediátricos y se puede usar en pacientes con TDAH y tics motores comórbidos o con diagnóstico de trastorno de Tourette. RECIT® no empeora la ansiedad en pacientes pediátricos ni en adultos y se puede usar en pacientes con TDAH y trastornos comórbidos de ansiedad. (Véase Propiedades Farmacológicas, poblaciones y afecciones especiales, pacientes con enfermedades concomitantes). Si el paciente saltea una dosis, la debe ingerir lo antes posible; sin embargo, no debe ingerir más de la cantidad total diaria indicada de RECIT® en cualquier período de 24 horas. RECIT® se puede suspender sin disminución paulatina de la dosis. Dosis recomendada y ajuste de dosis: Niños (6 años o más) y adolescentes hasta 70 kg de peso corporal: No superar la dosis inicial recomendada ni el incremento posterior de la dosis de RECIT®. Un incremento más rápido de la dosis se puede asociar con una tasa aumentada de somnolencia y molestias del sistema digestivo. No superar la dosis diaria total máxima recomendada de 1,4 mg/kg o 100 mg, lo que sea menor. No se demostró beneficio adicional con dosis mayores a 1,2 mg/kg/día. No se evaluó en forma sistemática la seguridad de dosis únicas mayores a 1,8 mg/kg/día y dosis diarias totales superiores a los 1,8 mg/kg y por lo tanto no se deben administrar por los potenciales efectos adversos (véase Propiedades Farmacológicas, seguridad cardiovascular y Sobredosis). RECIT® se debe inicial con una dosis diaria total de aproximadamente 0,5 mg/kg (etapa 1) durante 7 a 14 días. Fundamentado en la tolerabilidad, la dosis se debe aumentar en forma sucesiva hasta aproximadamente 0,8 mg/kg/día (etapa 2) durante 7 a 14 días y luego a aproximadamente 1,2 mg/kg/día (etapa 3). La tabla 1 suministra la dosis diaria correspondiente de RECIT® para cada paso del ajuste de dosis, de acuerdo con el peso corporal. Después de un mínimo de 30 días, la dosis de mantenimiento se debe reevaluar y ajustar de acuerdo con la respuesta clínica. La dosis diaria total en niños y adolescentes de hasta 70 kg no debe superar los 1,4 mg/kg o los 100 mg, lo que sea menor. Dado que la menor concentración disponible en las cápsulas es de 10 mg, el niño debe pesar al menos 20 kg al momento de iniciar el tratamiento. Se deben administrar solamente cápsulas enteras.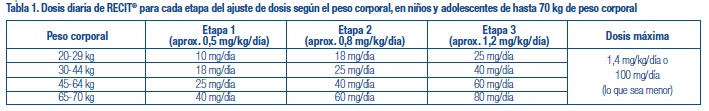
Niños y adolescentes de más de 70 kg de peso corporal, y adultos: No superar la dosis inicial recomendada y los incrementos posteriores de la dosis de RECIT®. No superar la dosis diaria total máxima recomendada de 100 mg. La seguridad de las dosis únicas superiores a 120 mg y las dosis diarias totales superiores a los 150 mg no se evaluaron en forma sistemática y por lo tanto no se deben administrar debido a los potenciales efectos adversos (véase Propiedades Farmacológicas, seguridad cardiovascular y Sobredosis). RECIT® se debe iniciar con una dosis total diaria de 40 mg/día (etapa 1) durante 7 a 14 días. Fundamentado en la tolerabilidad, la dosis se debe aumentar sucesivamente a 60 mg/día (etapa 2) durante 7 a 14 días, y luego a 80 mg/día (etapa 3). Después de 2 a 4 semanas adicionales, la dosis diaria total se puede aumentar hasta un máximo de 100 mg, en pacientes que no lograron una respuesta óptima. La dosis diaria total máxima recomendada en niños y adolescentes de más de 70 kg y adultos, es de 100 mg. Ajuste de dosis para poblaciones especiales: Insuficiencia hepática: La depuración de atomoxetina puede estar reducida en pacientes con TDAH con insuficiencia hepática. En pacientes con insuficiencia hepática moderada (clase Child-Pugh B), la dosis inicial y terapéutica se debe reducir al 50% de la dosis normal. En pacientes con insuficiencia hepática grave (clase Child-Pugh C), la dosis inicial y la dosis terapéutica se debe reducir al 25% de la dosis normal. Insuficiencia renal: Los sujetos con enfermedad renal en etapa terminal presentan una mayor exposición sistémica a atomoxetina que los sujetos sanos (aumento de aproximadamente 65%), pero no se observó diferencia cuando la exposición se corrigió por dosis en mg/kg. Por lo tanto, RECIT® se puede administrar a pacientes con TDAH con enfermedad renal en etapa terminal o grados menores de insuficiencia renal usando el régimen de dosis habitual. Atomoxetina puede exacerbar la hipertensión en pacientes con enfermedad renal en etapa terminal. Ajuste de la dosis para usar con un inhibidor potente de la CYP2D6: En niños ≥6 años de edad) y adolescentes de hasta 70 kg de peso corporal que reciben inhibidores potentes de la CYP2D6, por ej. Paroxetina, fluoxetina y quinidina, RECIT® se debe iniciar con 0,5 mg/kg/día y solamente aumentar al siguiente nivel de dosis si los síntomas no mejoran después de 14 días y la dosis previa se tolera bien. En niños ≥6 años de edad) y adolescentes de más de 70 kg de peso corporal y adultos que reciben inhibidores potentes de la CYP2D6, por ej. Paroxetina, fluoxetina y quinidina, RECIT® se debe iniciar con 40 mg/día y solamente aumentar al siguiente nivel de dosis si los síntomas no mejoran después de 14 días y la dosis previa se tolera bien. Mantenimiento / tratamiento extendido: El tratamiento farmacológico del TDAH puede requerir períodos extensos. Se estudió la eficacia de RECIT® para el mantenimiento de respuesta a los síntomas durante el tratamiento prolongado en niños y adolescentes, en un estudio de 18 meses (3 meses de tratamiento agudo abierto seguido de hasta 15 meses de tratamiento de mantenimiento controlado con placebo). Los resultados de este estudio sugieren que atomoxetina puede ser beneficioso para el tratamiento prolongado del TDAH. Muy pocos pacientes completaron el estudio como para permitir una evaluación adecuada del perfil de seguridad a largo plazo de RECIT® en este estudio. La seguridad a largo plazo de RECIT® se demostró en estudios clínicos en doble ciego y abiertos de al menos 24 meses. El médico que decida usar RECIT® durante períodos prolongados debe reevaluar en forma periódica la utilidad a largo plazo del medicamento para cada paciente individual (véase Indicaciones).
Contraindicaciones.
Hipersensibilidad: RECIT® (clorhidrato de atomoxetina) está contraindicado en pacientes con hipersensibilidad conocida a atomoxetina o a otros componentes del producto (véase Advertencias). Inhibidores de la monoaminooxidasa: RECIT® no se debe tomar con inhibidores de la monoaminooxidasa (IMAO) o dentro de las 2 semanas de la suspensión de IMAO. El tratamiento con IMAO no se debe iniciar dentro de las 2 semanas desde la suspensión de RECIT®. Con otros medicamentos que afectan la concentración de monoaminas cerebrales, se recibieron informes de reacciones graves y algunas veces fatales (incluyendo hipertermia, rigidez, mioclonías, inestabilidad autonómica con posible fl uctuación rápida de los signos vitales y cambios del estado mental que incluye agitación extrema que progresa a delirio y coma) cuando se ingieren en combinación con IMAO. Se presentaron algunos casos con características semejantes al síndrome neuroléptico maligno. Dichas reacciones pueden ocurrir cuando estos medicamentos se administran en forma concurrente o próximamente cercanos. Feocromocitoma: RECIT® no se debe usar en pacientes con feocromocitoma o antecedentes de feocromocitoma. Se informaron reacciones graves incluyendo aumento de la presión arterial y taquiarritmia, en pacientes con feocromocitoma o antecedentes de feocromocitoma que recibieron RECIT®. Glaucoma de ángulo estrecho: en estudios clínicos, RECIT® se asoció al aumento del riesgo de midriasis y por lo tanto, no se recomienda su uso en pacientes con glaucoma de ángulo estrecho. Enfermedad cardiovascular sintomática. Trastornos cardiovasculares graves: RECIT® no se debe usar en pacientes con trastornos cardiovasculares graves cuya condición se espera que empeore si experimentan aumento de la presión arterial o de la frecuencia cardíaca que podrían ser clínicamente importantes. Hipertensión moderada a grave. Arteriosclerosis avanzada. Hipertiroidismo no controlado.
Reacciones adversas.
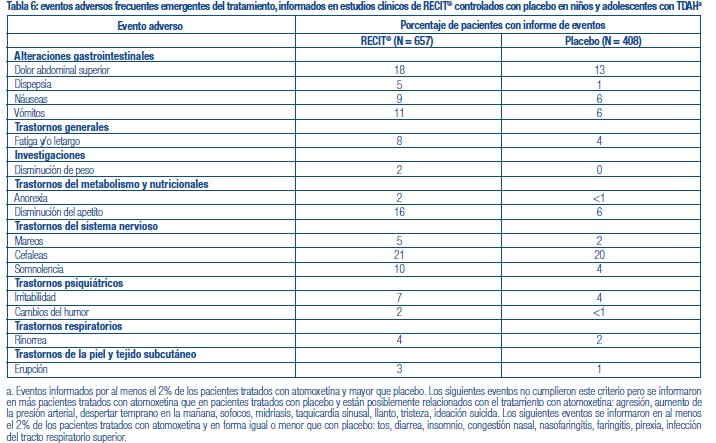
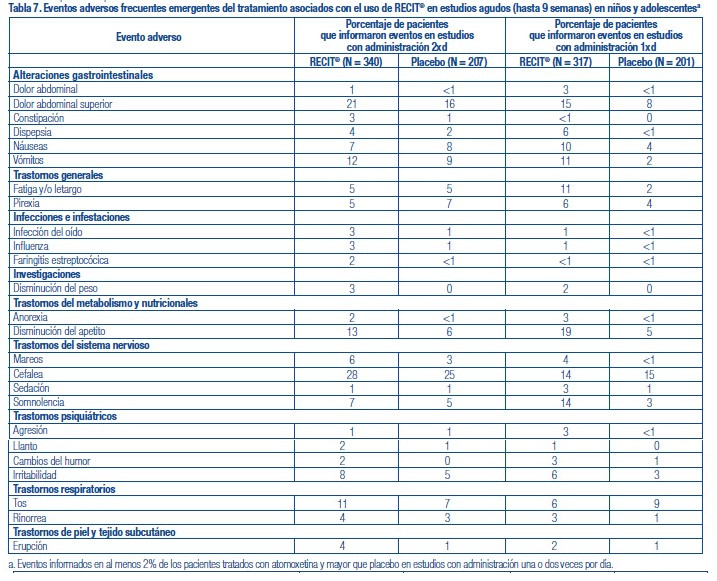
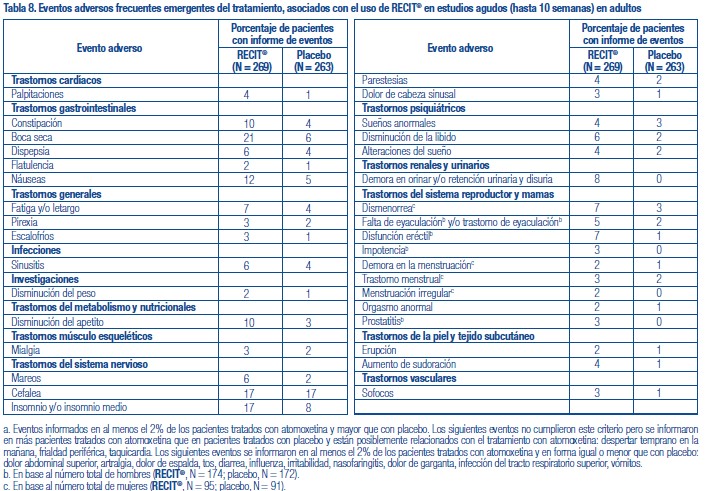
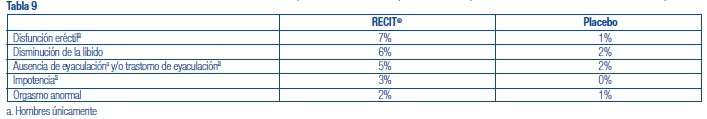
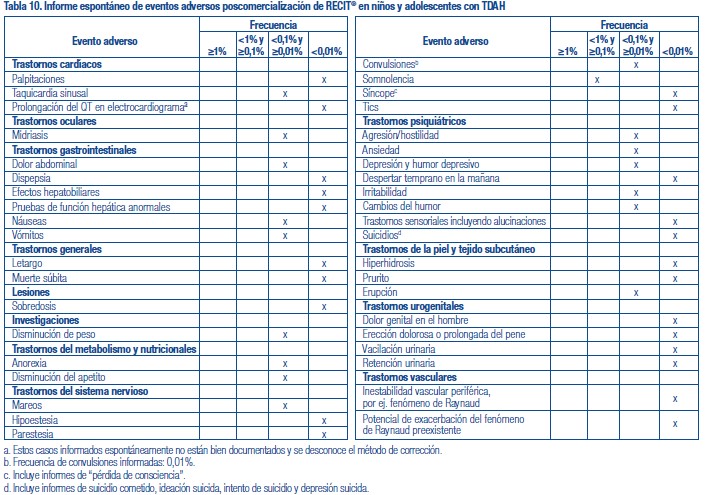
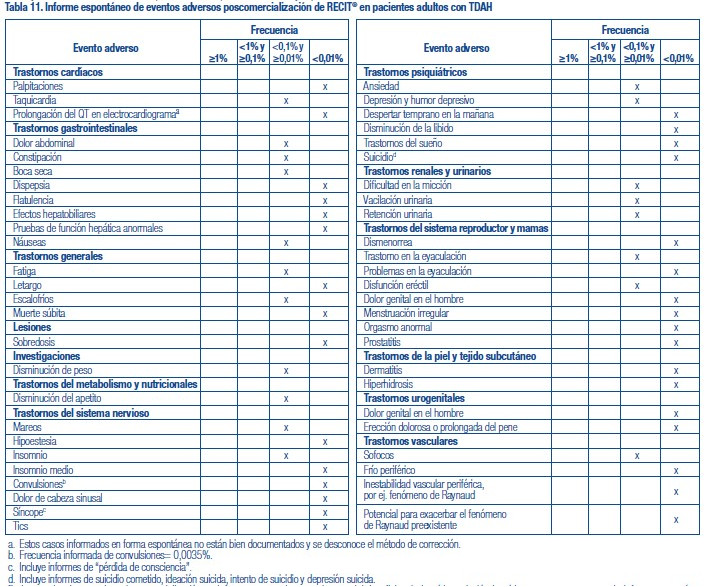
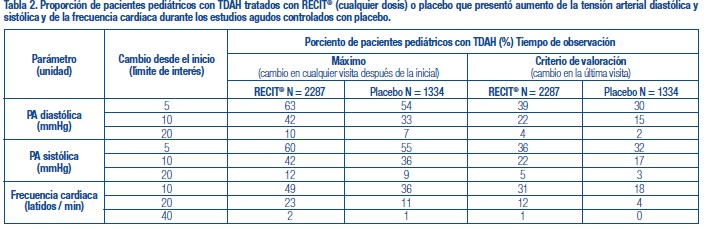
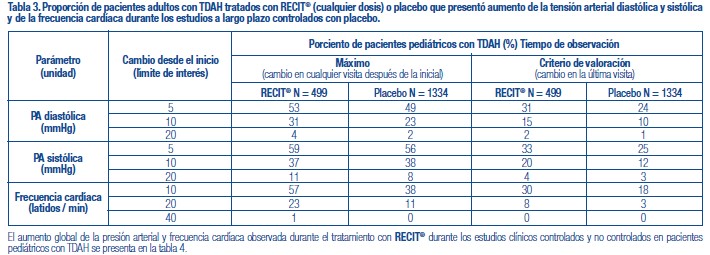
Se administró RECIT® a 3262 pacientes niños o adolescentes con TDAH y a 471 adultos con TDAH en estudios clínicos. Durante los estudios clínicos de TDAH, 1409 pacientes (1236 pediátricos y 173 adultos) se trataron durante más de 1 año y 1940 pacientes (1704 pediátricos y 236 adultos) se trataron durante más de 6 meses. Los datos en las siguientes tablas y texto no se pueden usar para predecir la incidencia de efectos adversos en el curso de la práctica médica habitual, donde las características de