PROLIA
ADIUM
Grupo farmacoterapéutico: Medicamentos para tratar las enfermedades óseas. Otros fármacos que modifican la estructura y la mineralización ósea, código ATC: M05BX04.
Composición.
Cada jeringa prellenada de PROLIA solución inyectable contiene: Denosumab* 60 mg; Sorbitol 47 mg; Acetato de sodio1 1 mg; Polisorbato 20 0,1 mg; Agua para inyectable c.s.p. 1 ml. 1 Por agregado de Hidróxido de sodio para ajustar el pH a 5,2. *Denosumab es un anticuerpo monoclonal IgG2 humano producido en una línea celular de mamíferos (CHO) mediante tecnología de ADN recombinante.
Indicaciones.
Tratamiento de la osteoporosis en mujeres post menopáusicas con riesgo elevado de fracturas. PROLIA reduce significativamente el riesgo de fracturas vertebrales, no vertebrales y de cadera. Tratamiento de la pérdida ósea asociada con la supresión hormonal en hombres con cáncer de próstata con riesgo elevado de fracturas. En hombres con cáncer de próstata que reciben supresión hormonal, PROLIA reduce significativamente el riesgo de fracturas vertebrales.
Dosificación.
Posología: La dosis recomendada de PROLIA es de 60 mg administrados en una única inyección subcutánea una vez cada 6 meses en el muslo, el abdomen o la parte posterior del brazo. Los pacientes deben tomar suplementos adecuados de calcio y vitamina D. Pacientes con insuficiencia renal: No se requieren ajustes de dosis en pacientes con insuficiencia renal. Pacientes con insuficiencia hepática: No se ha estudiado la seguridad y la eficacia de denosumab en pacientes con insuficiencia hepática. Pacientes de edad avanzada (edad ≥ 65): No se requieren ajustes de dosis en pacientes de edad avanzada. Población pediátrica: PROLIA no está recomendado en pacientes pediátricos (edad < 18) ya que no se ha establecido la seguridad y la eficacia de PROLIA en estos pacientes. La inhibición del RANK/ligando del RANK (RANKL) en estudios con animales se ha asociado con la inhibición del crecimiento óseo y con la falta de aparición de la dentición. Forma de administración: La administración debe realizarla una persona que haya recibido la formación adecuada en técnicas de inyección. Vía subcutánea.
Contraindicaciones.
Hipocalcemia. Hipersensibilidad al principio activo o a alguno de los excipientes.
Reacciones adversas.
Resumen tabulado de reacciones adversas: Se evaluó la seguridad de PROLIA en 10.534 mujeres post menopáusicas con osteoporosis (hasta 5 años de duración) y pacientes con cáncer de mama o próstata que recibían tratamiento de deprivación hormonal en estudios clínicos de fase II y III controlados con placebo. Para clasificar las reacciones adversas notificadas en estos estudios clínicos de fase II y III, se utilizó la convención siguiente (Ver Tabla 3): Muy frecuentes (≥1/10); frecuentes (≥1/100 a < 1/10); poco frecuentes (≥1/1.000 a < 1/100); raras (≥1/10.000 a ≤1/1.000) y muy raras ( < 1/10.000), basándose en tasas de eventos al cabo de 1 año. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia y clasificación de órganos.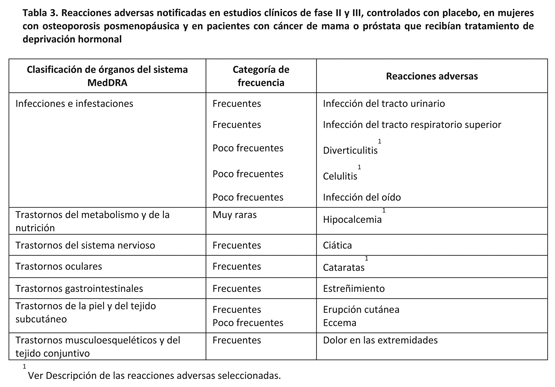
En un análisis combinado de datos de todos los estudios de fase II y fase III controlados con placebo, se notificó síndrome pseudogripal en una proporción de 0,006 por paciente-año para denosumab y de 0,003 por paciente-año para el grupo placebo. Aunque esta distribución desigual se identificó en el análisis combinado, no se identificó en el análisis estratificado que se utilizó para calcular las reacciones adversas incluidas en la Tabla 3. Este desequilibrio no se ha observado en estudios individuales. Descripción de las reacciones adversas seleccionadas: Hipocalcemia: En dos estudios clínicos de fase III controlados con placebo en mujeres post menopáusicas con osteoporosis, aproximadamente el 0,05% (2 de 4.050) de las pacientes presentaron una disminución de los niveles de calcio sérico (menos de 1,88 mmol/l) tras la administración de PROLIA. No se notificaron disminuciones de los niveles de calcio sérico (menos de 1,88 mmol/l) en los dos estudios clínicos de fase III controlados con placebo en pacientes que recibían tratamiento de deprivación hormonal. Infecciones cutáneas: En estudios clínicos de fase III controlados con placebo, la incidencia global de infecciones cutáneas fue similar en el grupo placebo y en el de PROLIA en mujeres post menopáusicas con osteoporosis (placebo [1,2%, 50 de 4.041] frente a PROLIA [1,5%, 59 de 4.050]) y en pacientes con cáncer de mama o próstata que recibían tratamiento de deprivación hormonal (placebo [1,7%, 14 de 845] frente a PROLIA [1,4%, 12 de 860]). Se notificaron infecciones cutáneas que provocaron la hospitalización en el 0,1% (3 de 4.041) de las mujeres post menopáusicas con osteoporosis que recibían placebo, en comparación con el 0,4% (16 de 4.050) de las mujeres que recibían PROLIA. Estos casos fueron principalmente celulitis. Las infecciones cutáneas notificadas como reacciones adversas graves fueron similares en el grupo placebo (0,6%, 5 de 845) y en el de PROLIA (0,6%, 5 de 860) en los estudios de cáncer de mama y próstata. Osteonecrosis del maxilar: En el programa de desarrollo clínico en osteoporosis (8710 pacientes tratados ≥ 1 año), la ONM se notificó raramente con PROLIA. Cataratas: En un único estudio clínico de fase III, controlado con placebo, en pacientes con cáncer de próstata sometidos a tratamiento de deprivación androgénica, se observó una distribución desigual del número de casos de cataratas como reacción adversa (4,7% denosumab, 1,2% placebo). En mujeres post menopáusicas con osteoporosis o en mujeres con cáncer de mama no metastásico tratadas con inhibidores de la aromatasa, no se observó esa diferencia. Diverticulitis: En un único estudio clínico de fase III, controlado con placebo, en pacientes con cáncer de próstata sometidos a tratamiento de deprivación androgénica se observó una distribución desigual del número de casos de diverticulitis como reacción adversa (1,2% denosumab, 0% placebo). La incidencia de diverticulitis fue comparable entre ambos grupos de tratamiento en mujeres post menopáusicas con osteoporosis y en mujeres con cáncer de mama no metastásico tratadas con inhibidores de la aromatasa. Otras poblaciones especiales: En los estudios clínicos, los pacientes con insuficiencia renal grave (clearance de creatinina < 30 ml/min) o en diálisis presentaron un mayor riesgo de desarrollar hipocalcemia si no tomaban suplementos de calcio. Es importante que los pacientes con insuficiencia renal grave o en diálisis tomen una cantidad adecuada de calcio y vitamina D.
Advertencias.
Suplementos de calcio y vitamina D: Es importante que todos los pacientes reciban un aporte adecuado de calcio y vitamina D. Precauciones de uso: La hipocalcemia debe corregirse mediante el aporte adecuado de calcio y vitamina D antes de iniciar el tratamiento. Los pacientes con insuficiencia renal grave (clearance de creatinina < 30 ml/min) o en diálisis presentan un riesgo más alto de desarrollar hipocalcemia. Se recomienda la monitorización clínica de los niveles de calcio en pacientes con predisposición a la hipocalcemia. Los pacientes que reciban PROLIA pueden presentar infecciones cutáneas (principalmente celulitis) que requieran hospitalización. Debe recomendarse a los pacientes que soliciten asistencia médica inmediata, si presentan signos o síntomas de celulitis. Se han notificado casos de osteonecrosis maxilar (ONM) en pacientes tratados con denosumab o con bisfosfonatos, otra clase de fármacos antirresortivos. La mayoría de casos se han producido en pacientes con cáncer; sin embargo, algunos se han observado en pacientes con osteoporosis. En raras ocasiones, se han notificado casos de ONM en estudios clínicos en pacientes que recibían una dosis de 60 mg de denosumab cada 6 meses para la osteoporosis. Se ha notificado casos de ONM en estudios clínicos realizados en pacientes con cáncer avanzado tratados con denosumab a una dosis mensual de 120 mg. Los factores de riesgo conocidos de la ONM incluyen el diagnóstico de cáncer con lesiones óseas, los tratamientos concomitantes (p. ej., quimioterapia, medicamentos biológicos antiangiogénicos, corticosteroides, radioterapia de cabeza y cuello), una higiene bucal deficiente, extracciones dentales y comorbilidades (p. ej., enfermedad dental preexistente, anemia, coagulopatía, infección) y tratamiento previo con bisfosfonatos. En pacientes con factores de riesgo concomitantes se debe considerar la realización de una revisión dental con un tratamiento odontológico preventivo apropiado antes de iniciar el tratamiento con PROLIA. Durante el tratamiento, si es posible, estos pacientes deben evitar procedimientos dentales invasivos. Durante el tratamiento con PROLIA debe mantenerse una buena práctica de higiene bucal. En los pacientes que desarrollen ONM durante el tratamiento con PROLIA, la cirugía dental puede empeorar su estado. Si se produce ONM durante el tratamiento con PROLIA, siga el criterio clínico y elabore una pauta de tratamiento para cada paciente basada en la evaluación del beneficio/riesgo a nivel individual. La cubierta de la aguja de la jeringa prellenada contiene caucho natural (un derivado del látex), que puede causar reacciones alérgicas. Advertencias sobre los excipientes: Los pacientes con problemas hereditarios raros de intolerancia a la fructosa no deben utilizar PROLIA. Este medicamento contiene menos de 1 mmol (23 mg) de sodio por 60 mg, por lo que se considera esencialmente exento de sodio. Interacciones: No se ha realizado estudios de interacciones. No hay datos clínicos sobre la administración conjunta de denosumab y tratamiento hormonal sustitutivo (estrógenos), sin embargo la posibilidad de interacción farmacodinámica se considera muy baja. En mujeres post menopáusicas con osteoporosis, no se modificó la farmacocinética y farmacodinámica de denosumab con el tratamiento previo con alendronato, según los datos de un estudio de transición (de alendronato a denosumab). Fertilidad, embarazo y lactancia: Embarazo: No se dispone de datos adecuados sobre el uso de PROLIA en mujeres embarazadas. Los estudios en animales no evidencian efectos perjudiciales directos o indirectos relativos a la toxicidad reproductiva. En ratones manipulados genéticamente en los que se inactivó el RANKL mediante la eliminación de genes (ratones knockout), los estudios indican que la ausencia del RANKL (la diana de denosumab) podría interferir en el desarrollo de los ganglios linfáticos del feto y podría causar un trastorno post natal de la dentición y el crecimiento óseo. No está recomendado el uso de PROLIA en mujeres embarazadas. Lactancia: Se desconoce si denosumab se excreta en la leche materna. Los estudios en ratones knockout indican que la ausencia del RANKL durante el embarazo puede interferir en la maduración de las glándulas mamarias alterando la lactancia posparto. La decisión entre no amamantar o no seguir el tratamiento con PROLIA debe tomarse teniendo en cuenta las ventajas de la lactancia para el recién nacido/lactante y las ventajas del tratamiento con PROLIA para la mujer. Fertilidad: No hay datos disponibles del efecto de denosumab sobre la fertilidad humana. Los estudios en animales no evidencian efectos perjudiciales directos o indirectos relativos a la fertilidad.
Conservación.
Conservar en heladera (entre 2°C y 8°C). No congelar. No agitar excesivamente. Conservar la jeringa prellenada en su envase para protegerla de la luz. PROLIA puede conservarse a temperatura ambiente (hasta 25°C) durante un máximo de 30 días en el envase original. Una vez fuera de la heladera, PROLIA debe utilizarse dentro del plazo de 30 días. Precauciones especiales de eliminación y otras manipulaciones: La solución de PROLIA debe examinarse antes de su administración. No inyecte la solución si contiene partículas, si está turbia o descolorida. No agitar excesivamente. Para evitar molestias en la zona de inyección, deje que la jeringa prellenada alcance la temperatura ambiente (hasta 25°C) antes de inyectarla y realice la inyección lentamente. Inyecte todo el contenido de la jeringa prellenada o el vial. Deseche cualquier resto de medicamento de la jeringa prellenada. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
Sobredosificación.
No hay experiencia de sobredosis en los estudios clínicos. PROLIA se ha administrado en estudios clínicos utilizando dosis de hasta 180 mg cada 4 semanas (dosis acumuladas de hasta 1.080 mg durante 6 meses) y no se han observado reacciones adversas adicionales. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Ricardo Gutiérrez (011) 4962-6666/2247 o al Hospital A. Posadas (011) 4654-6648/4658-7777. Optativamente a otros Centros de Intoxicaciones.
Presentación.
PROLIA se presenta en jeringa prellenada conteniendo 1 ml de solución.
Nota.
Es información de prescribir abreviada.