Perjeta®
ROCHE
Pertuzumab.
Agente antineoplásico, anticuerpo monoclonal, inhibidor de la dimerización de HER2.
Composición.
Cada vial de 20 ml, con 14 ml de concentrado para solución para infusión, contiene 420 mg de pertuzumab (30 mg/ml), en un excipiente compuesto por L-histidina 43,5 mg, ácido acético glacial 9,2 mg, sacarosa 575,1 mg, polisorbato 20: 2,8 mg y agua para inyectables c.s.p. 14 ml. Un vial contiene 420 mg de pertuzumab en total. La concentración de la solución final de Perjeta debe ser aproximadamente de 3,02 mg/ml (840 mg/278 ml) para la dosis inicial y de 1,59 mg/ml (420 mg/264 ml) para la dosis de mantenimiento (véase Precauciones especiales de eliminación y otras manipulaciones).
Farmacología.
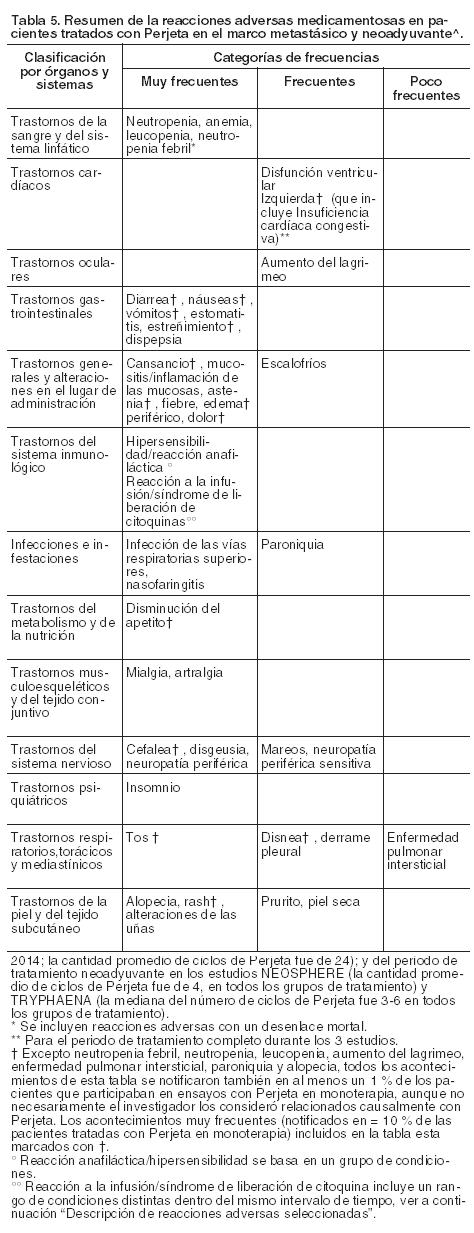
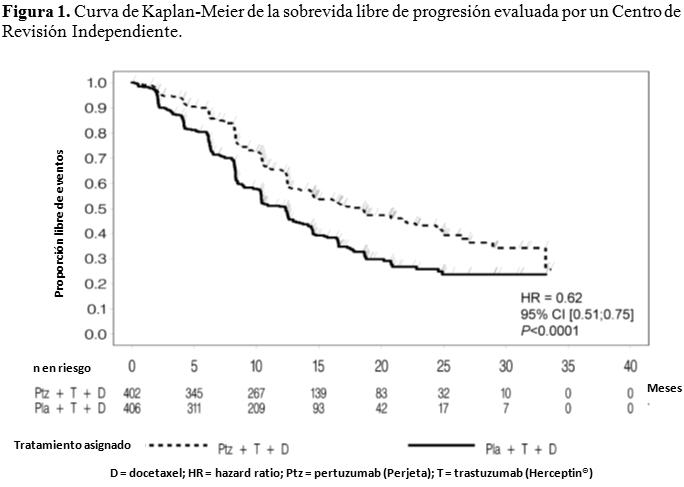
Código ATC: L01XC13. Grupo farmacoterapéutico: Agente antineoplásico, anticuerpo monoclonal, inhibidor de la dimerización de HER2. Propiedades farmacodinámicas: Mecanismo de acción: Perjeta es un anticuerpo monoclonal recombinante humanizado que se dirige específicamente al dominio de dimerización extracelular (subdominio II) de la proteína del receptor 2 del factor de crecimiento epidérmico humano (HER2) y, de este modo, bloquea la heterodimerización dependiente de ligando de HER2 con otros miembros de la familia HER, entre ellos EGFR, HER3 y HER4. Como consecuencia, Perjeta inhibe la señalización intracelular iniciada por ligando a través de dos vías de señalización principales, la proteína quinasa activada por mitógeno (MAP) y la fosfoinositida 3-quinasa (PI3K). La inhibición de estas vías de señalización puede inducir la detención del crecimiento celular y apoptosis, respectivamente. Además, Perjeta interviene en la citotoxicidad celular dependiente de anticuerpo (ADCC). Mientras que Perjeta en monoterapia inhibió la proliferación de las células tumorales humanas, su administración como tratamiento combinado con Herceptin® aumentó significativamente la actividad antitumoral en los modelos de xenoinjertos que sobreexpresan HER2. Eficacia y seguridad clínica: La eficacia de Perjeta en el tratamiento del cáncer de mama HER2 positivo se demostró en un ensayo comparativo aleatorizado Fase III en pacientes con cáncer de mama metastásico y en tres estudios Fase II (uno de rama única en pacientes con cáncer de mama metastásico y dos comparativos aleatorizados en los que se administró tratamiento en neoadyuvancia). Cáncer de mama metastásico: Perjeta en combinación con Herceptin® y docetaxel: WO20698 (CLEOPATRA): es un ensayo clínico multicéntrico, aleatorizado, doble-ciego, controlado con placebo, Fase III que incorporó a 808 pacientes con cáncer de mama HER2 positivo metastásico o locamente recurrente irresecable, que no habían recibido tratamiento previo anti-HER2 o quimioterapia para su enfermedad metastásica. Los pacientes con factores de riesgo cardíacos de importancia clínica no se incluyeron (véase Precauciones). Debido a la exclusión de pacientes con metástasis cerebrales no existen datos disponibles de la acción de Perjeta sobre las metástasis cerebrales. Se dispone de escasa información en pacientes con enfermedad localmente recidivante irresecable. Las muestras de tumores de mama requerían mostrar sobreexpresión de HER2 definida como una puntuación de 3+ por inmunohistoquímica (IHQ) o un cociente ≥ 2,0 por hibridación in situ (HIS) determinado en un laboratorio central. Los pacientes fueron aleatorizados 1:1 para recibir placebo + Herceptin® + docetaxel o Perjeta + Herceptin® + docetaxel. La distribución al azar se estratificó por estado de tratamiento previo (de novo o terapia adyuvante previa / neoadyuvante) y por región geográfica (Europa, América del Norte, América del Sur y Asia). Se les requirió a los pacientes con terapia adyuvante previa o neoadyuvante tener un intervalo libre de enfermedad de por lo menos 12 meses antes de la inscripción en el ensayo. Se administró una dosis estándar de Perjeta y Herceptin® en una pauta cada 3 semanas. Los pacientes recibieron tratamiento con Perjeta y Herceptin® hasta la progresión de la enfermedad, retiro de consentimiento o toxicidad no controlable. Docetaxel se administró en una dosis inicial de 75 mg/m2 como infusión intravenosa cada 3 semanas durante al menos 6 ciclos. La dosis de docetaxel podía incrementarse a 100 mg/m2 según el criterio del Investigador si se observaba buena tolerancia de la dosis inicial. Al momento de realizar el análisis primario de eficacia, el grupo al que se le administró placebo había recibido un promedio de 16,2 ciclos del tratamiento del estudio, mientras que el tratado con Perjeta un promedio de 19,9 ciclos. El criterio de valoración primario del estudio fue la sobrevida libre de progresión, de acuerdo con la evaluación de un Centro de Revisión Independiente y definida como el período de tiempo transcurrido desde la distribución aleatoria de los pacientes hasta la fecha de la progresión de la enfermedad o muerte (por cualquier causa) que hubiere ocurrido en las 18 semanas posteriores a la última evaluación tumoral. Los criterios de valoración secundarios de eficacia fueron la sobrevida global, la sobrevida libre de progresión (evaluada por el Investigador), la tasa de respuesta objetiva, la duración de la respuesta, y el tiempo transcurrido hasta la progresión de los síntomas, según el Cuestionario para la calidad de vida (FACT B). Las características demográficas fueron uniformes (la mediana de la edad fue de 54 años, la mayoría de los pacientes eran caucásicos [59%] y todos eran mujeres, con excepción de 2). Aproximadamente la mitad de los pacientes de cada grupo de tratamiento había desarrollado enfermedad positiva para receptores hormonales (definida como receptor de estrógenos positivo y/o receptor de progesterona positivo) y alrededor de la mitad de las pacientes de cada grupo de tratamiento habían recibido tratamiento adyuvante o neoadyuvante previo (192 pacientes [47,3%] en el grupo tratado con placebo comparado con 184 [45,8%] en el de Perjeta. A la mayoría se le había administrado previamente tratamiento con antraciclinas y un 11% de todas las pacientes fueron tratadas anteriormente con Herceptin®. Un total de 43% de pacientes de ambos grupos de tratamiento había recibido radioterapia previa. La FEVI mediana de los pacientes al inicio fue de 65,5% (rango 50% - 88%) en ambos grupos. Al momento de realizar el análisis primario de sobrevida libre de progresión, un total de 242 pacientes (59%) en el grupo tratado con placebo y 191 (47,5%) en el tratado con Perjeta había presentado enfermedad progresiva confirmada por un Centro de Revisión Independiente o había fallecido dentro de las 18 semanas de su última evaluación tumoral. Al momento del análisis primario el estudio CLEOPATRA demostró una mejoría estadísticamente significativa de la sobrevida libre de progresión (hazard ratio [HR] = 0,62, IC 95% = 0,51, 0,75, p < 0,0001) evaluada por un Centro de Revisión Independiente en el grupo tratado con Perjeta comparado con el de placebo, y un incremento en la mediana de sobrevida libre de progresión de 6,1 meses (mediana de sobrevida libre de progresión de 12,4 meses en el grupo tratado con placebo en comparación con 18,5 meses en el de Perjeta) (véase Figura 1). Los resultados de la sobrevida libre de progresión evaluada por el Investigador fueron comparables con aquellos observados en la sobrevida libre de progresión evaluada por el Centro de Revisión Independiente (la mediana de sobrevida libre de progresión fue de 12,4 meses para el grupo placebo comparada con 18,5 meses para el de Perjeta) (véase Tabla 1). Se observaron resultados consistentes en todos los subgrupos preespecificados de pacientes, incluso en los subgrupos basados sobre factores de estratificación por región geográfica y tratamiento de novo para cáncer de mama metastásico o adyuvante/neoadyuvante previo (véase Figura 2). Un análisis exploratorio adicional posterior mostró que en los pacientes que habían recibido trastuzumab previamente (n=88), el hazard ratio para la SLP valorada por el CRI fue de 0,62 (IC 95% 0,35; 1,07) comparado con 0,60 (IC 95% 0,43; 0,83) en aquellos que habían recibido tratamiento previo que no incluía trastuzumab (n=288). Los resultados de eficacia del ensayo CLEOPATRA están resumidos en la Tabla 1.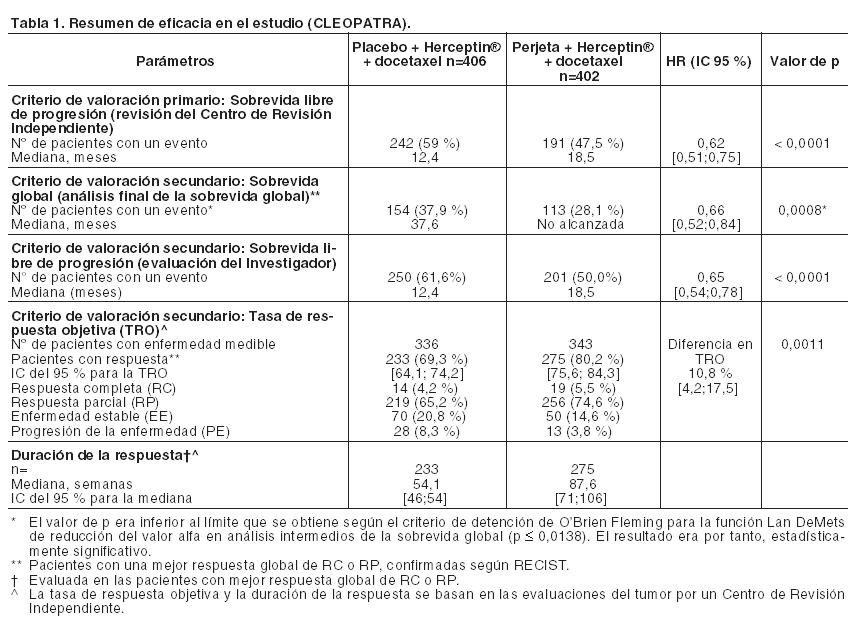
En el análisis primario de eficacia un análisis provisional de la sobrevida global mostró una fuerte tendencia sugerente de un beneficio de sobrevida a favor del grupo tratado con Perjeta. Un análisis provisional de sobrevida global realizado un año después del análisis primario de eficacia, demostró una ventaja en la sobrevida global estadísticamente significativa a favor del grupo tratado con Perjeta (HR 0,66, p = 0,0008 log rank test). La mediana de tiempo hasta la muerte fue de 37,6 meses en el grupo tratado con placebo, pero no se había alcanzado aún en el tratado con Perjeta. El análisis final de sobrevida global se realizó cuando 389 pacientes habían fallecido (221 del grupo tratado con placebo y 168 del tratado con Perjeta). El beneficio estadísticamente significativo de la sobrevida global en favor del grupo tratado con Perjeta se mantuvo (HR 0,68, p = 0,0002 log rank test). La mediana de tiempo hasta la muerte fue de 40,8 meses en el grupo tratado con placebo y de 56,5 meses en el de Perjeta (véanse Tabla 1 y Figura 3).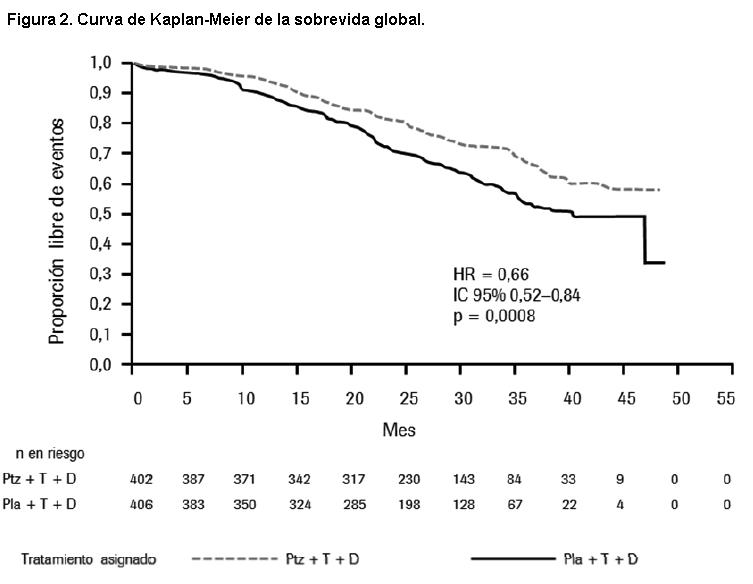
No se registraron diferencias estadísticamente significativas entre los dos grupos de tratamiento en la Calidad de Vida Relacionada con la Salud según lo evaluado por el tiempo de progresión de los síntomas sobre la subescala FACT-B TOI-PFB, definida como una reducción de 5 puntos en el puntaje de la subescala (HR = 0,97, IC 95% = 0,81; 1,16). En un análisis exploratorio, los pacientes tratados con Perjeta en combinación con Herceptin® y docetaxel experimentaron un riesgo menor de progresión de los síntomas sobre la subescala FACT-B de cáncer de mama (definida como una reducción de 2 puntos en el puntaje de la subescala) comparado con aquéllos tratados con Herceptin® y docetaxel solo (HR = 0,78, IC 95% = 0,65; 0,94). Información adicional de apoyo procedente de ensayos clínicos: BO17929: Este fue un ensayo con Perjeta, de Fase II, de rama única, no aleatorizado y llevado a cabo en pacientes con cáncer de mama metastásico HER-2 positivo que habían recibido tratamiento previo con Herceptin®. El estudio estaba dividido en 3 cohortes. Cohortes 1 y 2: 66 pacientes en las cohortes 1 y 2 recibieron como mínimo una dosis de Perjeta y Herceptin® (todas las poblaciones y todos los pacientes habían sido tratados para enfermedad metastásica; a la mitad se administró tratamiento en segunda línea para sus metástasis, mientras que el 35% lo recibió en tercera línea y más allá de ésta. Además, 71% había recibido quimioterapia neoadyuvante). Al momento del análisis primario, la mediana de duración del tratamiento en estudio fue de 9 ciclos (27 semanas). La tasa de respuesta objetiva (TRO) y la tasa de beneficio clínico (TBC) al momento del análisis primario se presentan en la Tabla 2. La mediana de sobrevida libre de progresión y el tiempo hasta progresión fueron de 24 semanas. La mediana de tiempo hasta respuesta fue de 11 semanas, y en aquellos pacientes con una respuesta, la mediana de duración de respuesta fue de 25 semanas. Cohorte 3: 29 pacientes recibieron por lo menos un ciclo de Perjeta, de éstos, 12 participaron en la fase de monoterapia con Perjeta, y 17 fueron tratados con Perjeta y Herceptin® luego de la progresión documentada a Perjeta solo. Los 29 pacientes habían progresado a terapia en primera línea en el marco metastásico, y 41,4% también había progresado luego de terapia en segunda línea. A todos los pacientes en la cohorte 3 se administró como mínimo una dosis completa de Perjeta. Los tratados con Perjeta y Herceptin® recibieron una mediana de 12 ciclos totales. La Tabla 2 demuestra que Perjeta como monoterapia tuvo una actividad modesta en los pacientes luego del fracaso con Herceptin® (columna del medio). Estas respuestas ocurrieron en pacientes cuya enfermedad había progresado recientemente a cada anticuerpo cuando se administraron en forma separada. Además, 3 pacientes tuvieron enfermedad estable durante 6 meses o más para una tasa de beneficio clínico total del 35,3%.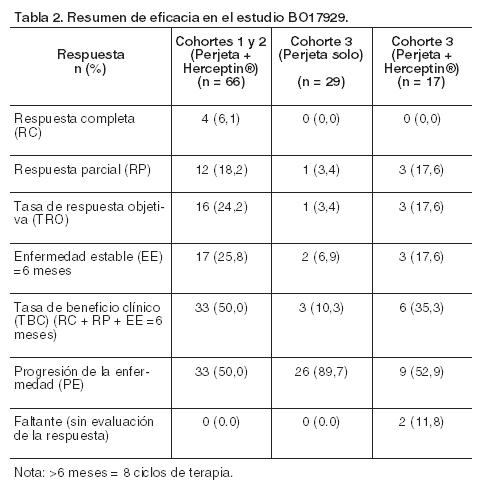
Tratamiento neoadyuvante para el cáncer de mama: En neoadyuvancia, el cáncer de mama localmente avanzado e inflamatorio es considerado de alto riesgo, independientemente del estado del receptor hormonal. En el cáncer de mama en estadio temprano, el tamaño del tumor, el grado, el estado del receptor hormonal y la metástasis en los ganglios linfáticos deben tenerse en cuenta en la evaluación de riesgos. La indicación del tratamiento neoadyuvante del cáncer de mama se basa en la demostración de una mejora en la tasa de respuesta patológica completa, y las tendencias de un aumento en la sobrevida libre de enfermedad que, sin embargo, no establece o mide con precisión un beneficio con respecto a los resultados a largo plazo, tales como la sobrevida global o la sobrevida libre de enfermedad. WO20697 (NEOSPHERE): ensayo de Fase II, multicéntrico, aleatorizado, que incorporó 417 pacientes con cáncer de mama HER2 positivo, operable, localmente avanzado, precoz o inflamatorio diagnosticado de novo (T2-4d; tumor primario 2 cm de diámetro) asignados a terapia neoadyuvante que no habían recibido tratamiento previo con trastuzumab, quimioterapia o radioterapia. Las muestras del tumor de mama debían mostrar sobreexpresión de HER2 definida como IHQ 3+ o cociente de amplificación por FISH ≥ 2,0 determinado en un laboratorio central. Las pacientes con metástasis, cáncer de mama bilateral, factores de riesgo cardíacos clínicamente importantes (véase Precauciones) o FEVI < 55% no fueron incluidas. La mayoría de las pacientes eran menores de 65 años. Los pacientes fueron distribuidos al azar para recibir 1 de los 4 regímenes de tratamiento neoadyuvante antes de la cirugía, como se indica a continuación: Herceptin® más docetaxel, Perjeta más Herceptin® y docetaxel, Perjeta más Herceptin® o Perjeta más docetaxel. La aleatorización fue estratificada por tipo de cáncer de mama (operable, localmente avanzado o inflamatorio) y receptor de estrógeno (RE) o receptor de progesterona (RPg) positivos. Perjeta fue administrado por vía intravenosa en una dosis inicial de 840 mg, seguida por 420 mg cada 3 semanas por 4 ciclos, Herceptin® por vía intravenosa en una dosis inicial de 8 mg/kg, seguida por 6 mg/kg cada 3 semanas por 4 ciclos. Docetaxel fue administrado por vía intravenosa con una dosis inicial de 75 mg/m2 seguida de 75 mg/m2 o 100 mg/m2 (si era tolerada) cada 3 semanas. Luego de la cirugía, todos los pacientes recibieron 3 ciclos de 5-fluorouracilo (600 mg/m2), epirrubicina (90 mg/m2) y ciclofosfamida (600 mg/m2) (FEC) por vía intravenosa cada 3 semanas y Herceptin® por infusión intravenosa cada 3 semanas hasta completar 1 año de tratamiento. Los pacientes en el grupo Perjeta más Herceptin® y docetaxel fueron tratados con docetaxel cada 3 semanas por 4 ciclos antes de recibir FEC previo a la cirugía, de modo que a todos los pacientes se administraron dosis acumuladas equivalentes de agentes quimioterapéuticos y Herceptin®. Los pacientes que sólo recibieron Perjeta más trastuzumab antes de la cirugía, posteriormente, después de ésta, recibieron tanto FEC como docetaxel. El criterio de valoración primario del estudio fue la tasa de respuesta patológica completa (RpC) en la mama (ypT0/is). Los criterios de valoración secundarios de eficacia fueron tasa de respuesta clínica, tasa de cirugía conservadora de la mama (sólo tumores T2-3), sobrevida libre de enfermedad y sobrevida libre de progresión. Tasas de respuesta patológica completa exploratorias adicionales incluyeron el estado ganglionar (ypTO/isNO e ypTONO). Las características demográficas estuvieron equilibradas (la edad promedio fue de 49-50 años; la mayoría fueron caucásicas [71%]) y todas pacientes de sexo femenino. En total, el 7% padecía cáncer de mama inflamatorio; el 32%, cáncer de mama localmente avanzado y el 61%, cáncer de mama operable. Aproximadamente la mitad de las pacientes en cada grupo de tratamiento tenía enfermedad con receptor hormonal positivo (definido como RE positivo y/o RPg positivo). Los resultados de eficacia se resumen en la Tabla 3. Las mejorías estadística y clínicamente significativas en las tasas de respuesta patológica completa (ypTO/is), se observaron en los pacientes tratados con Perjeta más Herceptin® y docetaxel en comparación con aquéllos que recibieron Herceptin® y docetaxel (45,8% comparado con 29,0%, valor de p = 0,0141). Se verificó un patrón consistente de los resultados, independientemente de la definición de RpC. Se considera probable que la diferencia en la tasa de RpC se traduzca en un valor clínicamente significativo en los resultados a largo plazo y está apoyado por la tendencia positiva en SLP (HR 0,69, IC del 95% 0,34, 1,40) y SLE (HR 0,60, IC del 95% 0,28, 1,27). Las tasas de respuesta patológica completa, así como la magnitud de la mejoría con Perjeta (Perjeta más trastuzumab y docetaxel comparado con pacientes que recibían trastuzumab y docetaxel) fueron menores en el subgrupo de pacientes con tumores con receptores hormonales positivos en comparación con aquéllos con receptores hormonales negativos (5,9% a 26,0% y 27,3% a 63,2%, respectivamente). Las tasas de RpC fueron similares en las pacientes con enfermedad operable que en las que tenían enfermedad localmente avanzada. Hubo muy pocas pacientes con cáncer de mama inflamatorio para establecer conclusiones firmes, pero la tasa de RpC fue mayor en aquellas que recibieron Perjeta más trastuzumab y docetaxel.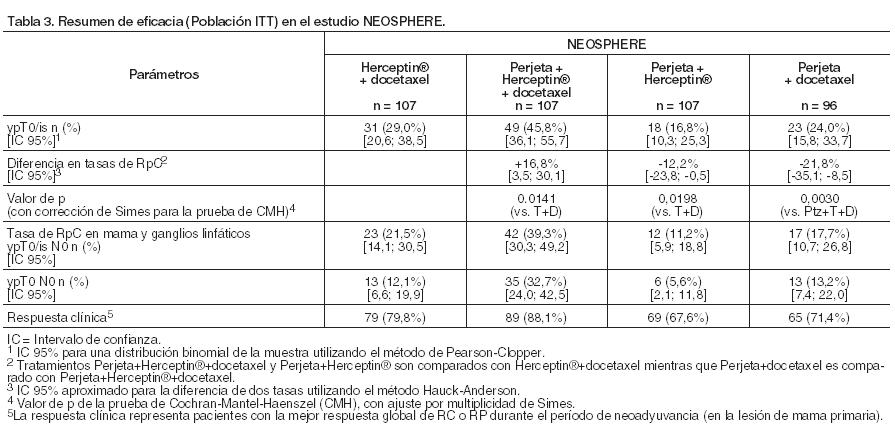
BO22280 (TRYPHAENA): es un estudio de Fase II, multicéntrico, aleatorizado, llevado a cabo en 225 pacientes con cáncer de mama HER2 positivo, localmente avanzado, operable o inflamatorio (T2-4d; tumor primario 2 cm de diámetro) que anteriormente no hubieran recibido trastuzumab, quimioterapia o radioterapia. Las muestras del tumor de mama debían mostrar sobreexpresión de HER2 definida como IHQ 3+ o cociente de amplificación por FISH ≥2,0 determinado en un laboratorio central. Las pacientes con metástasis, cáncer de mama bilateral, factores de riesgo cardíacos clínicamente importantes (véase Precauciones) o FEVI < 55% no fueron incluidas. La mayoría de las pacientes eran menores de 65 años. Los pacientes fueron distribuidos al azar para recibir 1 de los 3 regímenes de tratamiento neoadyuvante antes de la cirugía, de la siguiente manera: 3 ciclos de FEC seguidos por 3 ciclos de docetaxel, todos en combinación con Perjeta y Herceptin®, 3 ciclos de FEC solo seguidos por 3 ciclos de docetaxel y Herceptin® en combinación con Perjeta administrados simultáneamente, o 6 ciclos de docetaxel, carboplatino y Herceptin® (DCH) más Perjeta. La aleatorización fue estratificada por tipo de cáncer de mama (operable, localmente avanzado o inflamatorio) y receptor RE y/o receptor RPg positivos. Perjeta fue administrado por infusión intravenosa en una dosis inicial de 840 mg, seguida por 420 mg cada 3 semanas y Herceptin® por infusión intravenosa en una dosis inicial de 8 mg/kg, seguida por 6 mg/kg cada 3 semanas. Se administró por vía intravenosa 5-Fluorouracilo (500 mg/m2), epirrubicina (100 mg/m2) y ciclofosfamida (600 mg/m2) cada 3 semanas por 3 ciclos. Docetaxel fue administrado por infusión intravenosa en una dosis de 75 mg/m2 cada 3 semanas, con la posibilidad de aumentarla a 100 mg/m2 según el criterio del Investigador si la dosis inicial era bien tolerada. No obstante, en el grupo Perjeta más DCH, docetaxel fue administrado por infusión intravenosa en una dosis de 75 mg/m2 (sin incrementos de dosis permitidos) y carboplatino (ABC 6) fue administrado por infusión intravenosa cada 3 semanas. Luego de la cirugía, todos los pacientes recibieron Herceptin® por vía intravenosa cada 3 semanas hasta completar 1 año de tratamiento. El criterio de valoración primario de este ensayo fue la seguridad cardíaca durante el período de tratamiento neoadyuvante del estudio. Los criterios de valoración secundarios de eficacia fueron las tasas de respuesta patológica completa en la mama (ypTO/is), sobrevida libre de enfermedad, sobrevida libre de progresión y sobrevida global. Las características demográficas estuvieron equilibradas (la edad promedio fue de 49-50 años; la mayoría de las pacientes fueron caucásicas [77%]) y en su totalidad de sexo femenino. El 6% padecía cáncer de mama inflamatorio; el 25% cáncer de mama localmente avanzado y el 69%, cáncer de mama operable. Aproximadamente, la mitad de las pacientes en cada grupo de tratamiento tenía enfermedad con RE positivo y/o RPg positivo. En comparación con datos publicados de regímenes similares sin pertuzumab, altas tasas de RpC fueron observadas en los tres grupos de tratamiento (véase Tabla 4). Se comprobó un patrón consistente de los resultados independientemente de la definición de RpC. Las tasas de respuesta patológica completa fueron menores en el subgrupo de pacientes con tumores con receptores hormonales positivos en comparación con aquéllos con receptores hormonales negativos (46,2% a 50,0% y 65,0% a 83,8%, respectivamente). Las tasas de RpC fueron similares en las pacientes con enfermedad operable que en las que tenían enfermedad localmente avanzada. Hubo muy pocas pacientes con cáncer de mama inflamatorio para establecer conclusiones firmes.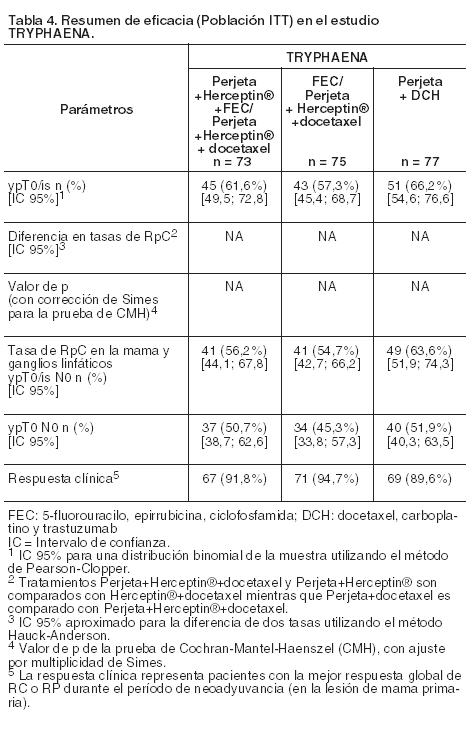
Inmunogenicidad: Los pacientes del ensayo pivotal CLEOPATRA se sometieron a análisis de anticuerpos antiterapéuticos (ATA) contra Perjeta en múltiples intervalos de tiempo. Aproximadamente, el 2,8% (11/386) de los pacientes tratados con Perjeta y el 6,2% (23/372) de los que recibieron placebo dieron positivo para ATA. Ninguno de estos 34 pacientes, tuvo reacciones graves (Grado 4 NCI-CTCAE) a la infusión o de anafilaxia/hipersensibilidad que estuvieran claramente relacionadas con ATA. Sin embargo, las reacciones de hipersensibilidad de Grado 3 relacionadas con ATA detectables ocurrieron en 2 de 366 pacientes tratadas con Perjeta (0,5 %) en ensayos de Fases I y II. Actualmente no existen datos suficientes para evaluar los efectos de ATA en la eficacia de Perjeta en combinación con trastuzumab y docetaxel. Los resultados de los ensayos de inmunogenicidad son altamente dependientes de diversos factores que incluyen estudios de sensibilidad y especificidad, de metodología, manipulación de la muestra, tiempo de la recolección de la muestra, medicación concomitante y enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos contra Perjeta con la incidencia de anticuerpos contra otros productos puede tener carácter engañoso. Población pediátrica: La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar resultados de ensayos realizados con Perjeta en todos los subgrupos de pacientes pediátricos en cáncer de mama (véase Dosificación). Propiedades farmacocinéticas: El análisis de farmacocinética poblacional se realizó en 481 pacientes de diferentes ensayos clínicos (Fases I, II y III), con varios tipos de tumores avanzados que habían recibido Perjeta en monoterapia o en combinación, con un rango de dosis entre 2 y 25 mg/kg administrados en infusión intravenosa de 30-60 minutos cada 3 semanas. Absorción: Perjeta es administrado en infusión intravenosa. No se llevaron a cabo ensayos con otras vías de administración. Distribución: En todos los ensayos clínicos, el volumen de distribución del compartimiento central (Vc) y periférico (Vp) de un paciente típico, fue de 3,11 litros y de 2,46 litros, respectivamente. Biotransformación: El metabolismo de Perjeta no ha sido estudiado directamente. Los anticuerpos se eliminan principalmente por catabolismo. Eliminación: La mediana del clearance de Perjeta fue de 0,235 litros/día y la de la vida media de 18 días. Linealidad/No linealidad: Se obtuvo una farmacocinética lineal con Perjeta dentro del rango de dosis recomendado. Farmacocinética en poblaciones especiales: Pacientes pediátricos: No se han llevado a cabo estudios para investigar la farmacocinética de Perjeta en esta población. Pacientes de edad avanzada: No se han realizado estudios específicos en pacientes de edad avanzada. En un análisis de farmacocinética poblacional, no se observó que la edad tuviera un efecto significativo sobre la farmacocinética de pertuzumab. En el análisis de farmacocinética de población, el 32,5% (n = 143) de los pacientes tenía 65 o más años de edad y el 9,1% (n = 40) 75 o más años de edad. Pacientes con insuficiencia renal: No se ha efectuado un ensayo específico de Perjeta en insuficiencia renal. Sobre la base de los resultados del análisis de farmacocinética poblacional, la exposición de Perjeta en pacientes con insuficiencia renal leve (clearance de creatinina [CLcr] 60 a 90 ml/min, n=200) y con insuficiencia renal moderada (CLcr 30 a 60 ml/min, n=71) fue similar a la de aquéllos con función renal normal (CLcr mayor de 90 ml/min, n=200). No se observó relación entre el clearance y la exposición de Perjeta en el rango de CLcr (27 a 244 ml/min). Otras poblaciones especiales: El análisis farmacocinético de la población no registró diferencias farmacocinéticas según edad, sexo y etnia (japoneses comparados con no japoneses). Los valores iniciales de albúmina y peso corporal magro fueron las covariables más significativas que influyeron sobre el clearance. Este se redujo en los pacientes con concentraciones iniciales de albúmina superiores y se incrementó en aquéllos con mayor peso corporal magro. No obstante, los análisis de sensibilidad realizados con la dosis recomendada y el esquema de administración de Perjeta demostraron que con los valores extremos de estas dos covariables, no se produjo un impacto significativo en la posibilidad de alcanzar el objetivo de las concentraciones en estado estacionario identificadas en los modelos preclínicos de xenoinjertos tumorales. Por lo tanto, de acuerdo con estas covariables, no es necesario ajustar la dosis de Perjeta. Los resultados de farmacocinética de pertuzumab en el estudio NEOSPHERE concordaron con los pronósticos previos del modelo de farmacocinética poblacional. Datos preclínicos sobre seguridad: No se han realizado ensayos de fertilidad en animales para evaluar el efecto de Perjeta. En los estudios de toxicidad con dosis repetidas de hasta 6 meses de duración realizados en macacos cynomolgus, no se observaron reacciones adversas sobre los órganos reproductores masculinos y femeninos. Se han llevado a cabo estudios de toxicología para la reproducción en macacos cynomolgus embarazadas (desde el día de gestación (DG) 19 hasta el DG 50), con dosis iniciales de 30 a 150 mg/kg, seguidas de dosis de 10 a 100 mg/kg, dos veces por semana. En base a la Cmáx, estos niveles de dosis produjeron exposiciones clínicamente relevantes de 2,5 a 20 veces mayor que con la dosis recomendada en seres humanos. La administración intravenosa de Perjeta desde el DG 19 hasta el DG 50 (período de organogénesis) fue embriotóxica, con aumentos dosis- dependiente de las muertes embriofetales entre el DG 25 y el DG 70. Las incidencias de pérdidas embriofetales fueron de 33, 50, y 85% para las hembras de macacos embarazadas, tratadas dos veces por semana con dosis de Perjeta de 10, 30 y 100 mg/kg, respectivamente (en base a la Cmáx son 2,5 a 20 veces mayores que con la dosis recomendada en seres humanos). En la cesárea del DG 100, en todos los grupos que recibieron dosis de Perjeta se identificaron oligohidramnios, disminución relativa del peso de los pulmones y riñones, y evidencia microscópica de hipoplasia renal consecuente con el retraso del desarrollo de los riñones. Además, como consecuencias de las limitaciones en el crecimiento fetal, también se registraron oligohidramnios menos importante, hipoplasia pulmonar (1 de 6 en el grupo de 30 mg/kg y 1 de 2 en el grupo de 100 mg/kg), comunicación interventricular (1 de 6 en el grupo de 30 mg/kg), estrechamiento de la pared del ventrículo (1 de 2 en el grupo de 100 mg/kg) y anomalías menores en el esqueleto externo (3 de 6 en el grupo de 30 mg/kg). Se notificó exposición a pertuzumab en la descendencia de todos los grupos de tratamiento, con valores del 29 al 40% de los niveles de suero materno en el DG 100. En macacos cynomolgus, la administración intravenosa semanal de Perjeta en dosis de hasta 150 mg/kg/dosis se toleró bien en general. Con dosis de 15 mg/kg y superiores, se observó diarrea leve e intermitente asociada con el tratamiento. En un subgrupo de macacos, la administración crónica (7 a 26 dosis semanales) originó episodios de diarrea secretoria grave. La diarrea se controló (a excepción de la eutanasia de un animal, 50 mg/kg/dosis) con tratamiento de soporte, incluyendo terapia de reemplazo de líquido por vía intravenosa.
Indicaciones.
Cáncer de mama metastásico (CMM): Perjeta está indicado en combinación con Herceptin® y docetaxel en pacientes con cáncer de mama HER2 positivo metastásico o localmente recurrente irresecable, que no hayan recibido tratamiento previo anti-HER2 o quimioterapia para la enfermedad metastásica. Tratamiento neoadyuvante para el cáncer de mama: Perjeta está indicado en combinación con Herceptin® y quimioterapia para el tratamiento neoadyuvante de pacientes adultos con cáncer de mama HER2 positivo localmente avanzado, inflamatorio o temprano con alto riesgo de recaída (tumor con un diámetro superior a los dos centímetros o con ganglios linfáticos positivos) como parte de un régimen de tratamiento completo para el cáncer de mama temprano (véanse Farmacología, Propiedades farmacodinámicas y Dosificación). Limitaciones en el uso: No se ha determinado la seguridad de Perjeta como parte de un régimen que contenga doxorrubicina, o administrado por más de 6 ciclos para el cáncer de mama temprano.
Dosificación.
Perjeta está supeditado a prescripción médica limitada y el tratamiento sólo debe iniciarse bajo la supervisión de un médico con experiencia en la administración de medicamentos antineoplásicos. Perjeta debe ser administrado por un profesional sanitario preparado para manejar la anafilaxia y en un lugar donde se disponga inmediatamente del servicio de reanimación. Las pacientes que reciben tratamiento con Perjeta deben presentar tumor HER2 positivo, definido como un índice de 3+ IHQ y/o una relación mayor o igual a 2,0 por HIS evaluada mediante un ensayo validado. Para asegurar resultados exactos y reproducibles, la determinación debe ser realizada en un laboratorio especializado, que pueda asegurar la validación de los procedimientos realizados. Para instrucciones completas sobre la realización e interpretación del ensayo, consulte el prospecto del procedimiento de ensayo HER2 validado. Con el fin de prevenir los errores de medicación, es importante consultar los prospectos de los viales para asegurarse de que el medicamento que está siendo preparado y administrado es Perjeta. El tratamiento con Perjeta sólo debe administrarse bajo la supervisión de un profesional de la salud con experiencia en el tratamiento de pacientes con cáncer. Posología: Cáncer de mama metastásico (CMM): La dosis de carga inicial recomendada de Perjeta es de 840 mg, administrada como infusión intravenosa durante 60 minutos, seguida posteriormente por una dosis de mantenimiento de 420 mg en un período de 30 - 60 minutos cada 3 semanas. Cuando se administre con Perjeta, la dosis de carga inicial recomendada de Herceptin® es de 8 mg/kg de peso corporal, administrada como infusión intravenosa, seguida luego cada 3 semanas de una dosis de mantenimiento de 6 mg/kg de peso corporal. Cuando se administre con Perjeta, la dosis inicial recomendada de docetaxel es de 75 mg/m2, administrada en lo sucesivo en un régimen cada 3 semanas. La dosis de docetaxel puede incrementarse a 100 mg/m2 en los siguientes ciclos si se observa buena tolerancia de la dosis inicial (la dosis de docetaxel no se debe aumentar cuando se utilice en combinación con carboplatino, trastuzumab y Perjeta). Los medicamentos deben administrarse secuencialmente y no mezclarse en la misma bolsa de infusión. Perjeta y Herceptin® se pueden aplicar en cualquier orden. Cuando el paciente vaya a recibir docetaxel, éste debe administrarse después de Perjeta y Herceptin®. Se recomienda un período de observación de 30 a 60 minutos luego de cada infusión de Perjeta y antes del comienzo de la infusión posterior de Herceptin® o docetaxel (véase Precauciones). Las pacientes deben recibir tratamiento con Perjeta hasta la progresión de la enfermedad o toxicidad no controlable. Tratamiento neoadyuvante para el cáncer de mama: Perjeta debe administrarse cada 3 semanas por 3 a 6 ciclos como parte de uno de los siguientes regímenes de tratamiento para el cáncer de mama temprano (véase Farmacología, Propiedades farmacodinámicas): Cuatro ciclos preoperatorios de Perjeta en combinación con Herceptin® y docetaxel seguidos por 3 ciclos posoperatorios de fluorouracilo, epirrubicina y ciclofosfamida (FEC). Tres ciclos preoperatorios de FEC solo seguidos por 3 ciclos preoperatorios de Perjeta en combinación con docetaxel y Herceptin®. Seis ciclos preoperatorios de Perjeta en combinación con docetaxel, carboplatino y trastuzumab (DCH) (no se recomienda incrementar la dosis de docetaxel por encima de 75 mg/m2). Luego de la cirugía, los pacientes deberán continuar recibiendo Herceptin® para completar 1 año de tratamiento. No se dispone de evidencia suficiente para recomendar el uso continuo de Perjeta por más de 6 ciclos para el tratamiento neoadyuvante del cáncer de mama temprano. Tampoco, para aconsejar la administración concomitante de una antraciclina con Perjeta, ni datos de seguridad para respaldar el uso consecutivo de doxorrubicina con Perjeta. Retraso u omisión de dosis: Si el intervalo entre dos infusiones consecutivas es inferior a 6 semanas, la dosis de 420 mg de Perjeta debe administrarse lo antes posible, independientemente de la siguiente dosis prevista. Si el intervalo entre dos infusiones consecutivas es de 6 semanas o más, la dosis de carga inicial de 840 mg de Perjeta debe readministrarse como infusión intravenosa durante 60 minutos, seguida en lo sucesivo por una dosis de mantenimiento de 420 mg administrada durante un período de 30 a 60 minutos, cada 3 semanas. Modificación de la dosis: No se recomienda reducir la dosis de Perjeta. Los pacientes pueden continuar el tratamiento durante períodos de mielosupresión reversible inducida por quimioterapia, pero deben ser monitoreados estrechamente por si existen complicaciones debidas a la neutropenia durante este tiempo. Para mayor información sobre las modificaciones de la dosis de docetaxel, véase el prospecto de envase de este fármaco. No se recomienda reducir la dosis de Herceptin®; véase prospecto de envase del producto. Deberá interrumpirse el tratamiento con Perjeta en caso de discontinuar la administración de Herceptin®. Si se discontinúa el tratamiento con docetaxel, deberá continuarse administrando la combinación Perjeta y Herceptin® hasta la progresión de la enfermedad o toxicidad no controlable en la enfermedad metastásica. Insuficiencia ventricular izquierda: Deberá interrumpirse la dosis de Perjeta y Herceptin® durante por lo menos 3 semanas en caso de: Signos y síntomas que sugieran insuficiencia cardíaca congestiva (Perjeta se debe interrumpir si se confirma insuficiencia cardíaca sintomática). Una disminución en la fracción de eyección del ventrículo izquierdo (FEVI) a un valor inferior a 40%. Una FEVI de 40% - 45% asociada con una reducción ≥ 10% puntos por debajo de los valores previos al inicio del tratamiento. Podrá reiniciarse la dosis de Perjeta y Herceptin® si la FEVI se ha restablecido a > 45% ó a un valor de 40 - 45% asociado con < 10% por debajo de los valores previos al inicio del tratamiento. Si después de una evaluación repetida aproximadamente dentro de 3 semanas no se ha observado una mejoría en la FEVI, o si ha disminuido nuevamente, deberá considerarse seriamente la interrupción de Perjeta y Herceptin®, salvo que los beneficios que produce para el paciente sean superiores a los riesgos derivados (véase Precauciones). Reacciones relacionadas con la infusión: La velocidad de la infusión podrá reducirse o interrumpirse si el paciente experimenta una reacción relacionada con la infusión (véase Reacciones adversas). Esta puede reanudarse si los síntomas disminuyen. El tratamiento con oxígeno, agonistas beta, antihistamínicos, fluidos intravenosos rápidos y antipiréticos pueden también ayudar a aliviar los síntomas. Reacciones de hipersensibilidad/anafilaxia: Deberá interrumpirse de inmediato la infusión si el paciente desarrolla una reacción Grado 4 NCI-CTCAE (anafilaxia), broncospasmo o síndrome de distress respiratorio agudo (véase Precauciones). Poblaciones especiales: Pacientes pediátricos: No se ha establecido la seguridad ni la eficacia de Perjeta en niños y adolescentes menores de 18 años. No existe una recomendación de uso específica para Perjeta en la población pediátrica para la indicación de cáncer de mama metastásico. Pacientes de edad avanzada: Se dispone de pocos datos sobre la seguridad y eficacia de Perjeta en pacientes ≥ 65 años de edad. No se observaron diferencias significativas en la seguridad y eficacia de Perjeta entre pacientes de edad avanzada de 65 a 75 años de edad y adultos < 65 años de edad. No es necesario ajustar la dosis en ancianos ≥ 65 años de edad. Se dispone de escasa información sobre los mayores de 75 años de edad. Pacientes con insuficiencia renal: No es necesario ajustar la dosis de Perjeta en pacientes con insuficiencia renal leve o moderada. No se puede hacer una recomendación de las dosis en aquéllos con insuficiencia renal grave debido a que existen pocos datos de farmacocinética disponibles (véase Farmacología, Propiedades farmacocinéticas). Pacientes con insuficiencia hepática: No se ha estudiado la seguridad y la eficacia de Perjeta en pacientes con insuficiencia hepática. No se puede hacer una recomendación específica de las dosis. Formas de administración: Perjeta debe ser diluido por un profesional de la salud y se administra por vía intravenosa mediante infusión. No debe administrarse en inyección intravenosa rápida o bolo. Para consultar las instrucciones de dilución de Perjeta antes de la administració