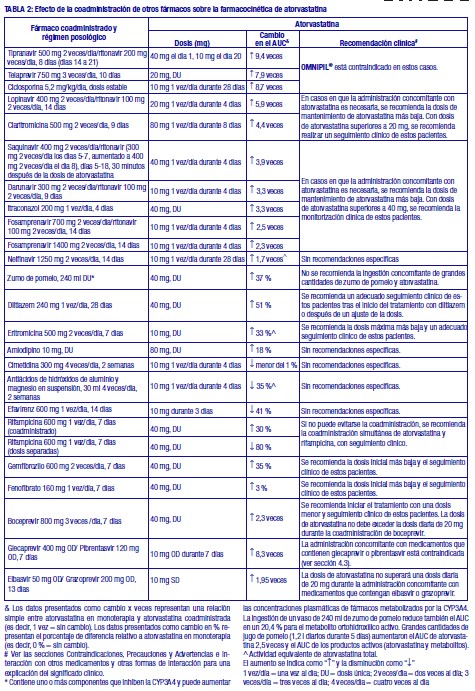
OMNIPIL®
GADOR
Inhibidores de la HMG-CoA reductasa, en combinación.
Composición.
Cada cápsula con minicomprimidos de OMNIPIL® 100/20/2,5 contiene 2 minicomprimidos de Ácido acetilsalicílico, 2 minicomprimidos de Atorvastatina y 1 minicomprimido de Ramipril. Cada 2 minicomprimidos de Ácido acetilsalicílico contienen: Ácido acetilsalicílico (como Ácido acetilsalicílico granular) 100 mg. Excipientes: Almidón glicolato de sodio, Celulosa microcristalina PH 112, Dióxido de silicio coloidal, Talco, Opaglos GS 2 0700 1), Opadry AMB II 88A001840 2) c.s. 1) Compuesto por: Etanol, Goma Laca, Cera carnauba, Cera blanca. 2) Compuesto por: Alcohol polivinílico, Talco, Dióxido de titanio, Glicerol monocapril caprato, Lauril sulfato de sodio. Cada 2 minicomprimidos de Atorvastatina contienen: Atorvastatina (como Atorvastatina cálcica trihidrato 21,7 mg) 20 mg. Excipientes: Lactosa monohidrato, Almidón parcialmente pregelatinizado, Carbonato de calcio liviano, Hidroxipropilcelulosa, Polisorbato 80, Crospovidona, Dióxido de silicio coloidal, Estearato de magnesio, Óxido de hierro amarillo (CI 77492), Óxido de hierro negro (CI 77499), Opaglos GS 2 0700 1), Opadry AMB II 88A001840 2) c.s. 1) Compuesto por: Etanol, Goma Laca, Cera carnauba, Cera blanca. 2) Compuesto por: Alcohol polivinílico, Talco, Dióxido de titanio, Glicerol monocapril caprato, Lauril sulfato de sodio. Cada minicomprimido de Ramipril contiene: Ramipril 2,5 mg. Excipientes: Almidón parcialmente pregelatinizado, Celulosa microcristalina (Avicel PH 112), Hidroxipropilmetilcelulosa, Estearil fumarato de sodio, Óxido de hierro amarillo (CI 77492), Opaglos GS 2 0700 1), Opadry AMB II 88A001840 2) c.s. 1) Compuesto por: Etanol, Goma Laca, Cera carnauba, Cera blanca. 2) Compuesto por: Alcohol polivinílico, Talco, Dióxido de titanio, Glicerol monocapril caprato, Lauril sulfato de sodio. Composición de la cápsula con minicomprimidos: Dióxido de titanio, Gelatina, Colorante amarillo FDyC N°6. Cada cápsula con minicomprimidos de OMNIPIL® 100/20/5 contiene 2 minicomprimidos de Ácido acetilsalicílico, 2 minicomprimidos de Atorvastatina y 1 minicomprimido de Ramipril. Cada 2 minicomprimidos de Ácido acetilsalicílico contienen: Ácido acetilsalicílico (como Ácido acetilsalicílico granular) 100 mg Excipientes: Almidón glicolato de sodio, Celulosa microcristalina PH 112, Dióxido de silicio coloidal, Talco, Opaglos GS 2 0700 1), Opadry AMB II 88A001840 2) c.s. 1) Compuesto por: Etanol, Goma Laca, Cera carnauba, Cera blanca. 2) Compuesto por: Alcohol polivinílico, Talco, Dióxido de titanio, Glicerol monocapril caprato, Lauril sulfato de sodio. Cada 2 minicomprimidos de Atorvastatina contienen: Atorvastatina (como Atorvastatina cálcica trihidrato 21,7 mg) 20 mg. Excipientes: Lactosa monohidrato, Almidón parcialmente pregelatinizado, Carbonato de calcio liviano, Hidroxipropilcelulosa, Polisorbato 80, Crospovidona, Dióxido de silicio coloidal, Estearato de magnesio, Óxido de hierro amarillo (CI 77492), Óxido de hierro negro (CI 77499), Opaglos GS 2 0700 1), Opadry AMB II 88A001840 2) c.s. 1) Compuesto por: Etanol, Goma Laca, Cera carnauba, Cera blanca. 2) Compuesto por: Alcohol polivinílico, Talco, Dióxido de titanio, Glicerol monocapril caprato, Lauril sulfato de sodio. Cada minicomprimido de Ramipril contiene: Ramipril 5 mg. Excipientes: Almidón parcialmente pregelatinizado, Celulosa microcristalina (Avicel PH 112), Hidroxipropilmetilcelulosa, Estearil fumarato de sodio, Óxido de hierro amarillo (CI 77492), Opaglos GS 2 0700 1), Opadry AMB II 88A001840 2) c.s. 1) Compuesto por: Etanol, Goma Laca, Cera carnauba, Cera blanca. 2) Compuesto por: Alcohol polivinílico, Talco, Dióxido de titanio, Glicerol monocapril caprato, Lauril sulfato de sodio. Composición de la cápsula con minicomprimidos: Dióxido de titanio, Gelatina, Colorante rojo FDyC N°40, Colorante FDyC verde N°3, Colorante FDyC amarillo N°10. Cada cápsula con minicomprimidos de OMNIPIL® 100/20/10 contiene 2 minicomprimidos de Ácido acetilsalicílico, 2 minicomprimidos de Atorvastatina y 1 minicomprimido de Ramipril. Cada 2 minicomprimidos de Ácido acetilsalicílico contienen: Ácido acetilsalicílico (como Ácido acetilsalicílico granular) 100 mg. Excipientes: Almidón glicolato de sodio, Celulosa microcristalina PH 112, Dióxido de silicio coloidal, Talco, Opaglos GS 2 0700 1), Opadry AMB II 88A001840 2) c.s. 1) Compuesto por: Etanol, Goma Laca, Cera carnauba, Cera blanca. 2) Compuesto por: Alcohol polivinílico, Talco, Dióxido de titanio, Glicerol monocapril caprato, Lauril sulfato de sodio. Cada 2 minicomprimidos de Atorvastatina contienen: Atorvastatina (como Atorvastatina cálcica trihidrato 21,7 mg) 20 mg. Excipientes: Lactosa monohidrato, Almidón parcialmente pregelatinizado, Carbonato de calcio liviano, Hidroxipropilcelulosa, Polisorbato 80, Crospovidona, Dióxido de silicio coloidal, Estearato de magnesio, Óxido de hierro amarillo (CI 77492), Óxido de hierro negro (CI 77499), Opaglos GS 2 0700 1), Opadry AMB II 88A001840 2) c.s. 1) Compuesto por: Etanol, Goma Laca, Cera carnauba, Cera blanca. 2) Compuesto por: Alcohol polivinílico, Talco, Dióxido de titanio, Glicerol monocapril caprato, Lauril sulfato de sodio. Cada minicomprimido de Ramipril contiene: Ramipril 10 mg. Excipientes: Almidón parcialmente pregelatinizado, Celulosa microcristalina (Avicel PH 112), Hidroxipropilmetilcelulosa, Estearil fumarato de sodio, Óxido de hierro amarillo (CI 77492), Opaglos GS 2 0700 1), Opadry AMB II 88A001840 2) c.s. 1) Compuesto por: Etanol, Goma Laca, Cera carnauba, Cera blanca. 2) Compuesto por: Alcohol polivinílico, Talco, Dióxido de titanio, Glicerol monocapril caprato, Lauril sulfato de sodio. Composición de la cápsula con minicomprimidos: Dióxido de titanio, Gelatina, Colorante rojo FDyC N°3, Colorante FDyC azul N°1.
Farmacología.
Propiedades farmacodinámicas: Ácido acetilsalicílico: El ácido acetilsalicílico inhibe de forma irreversible la agregación plaquetaria. Este efecto en las plaquetas se debe a la acetilación de la ciclooxigenasa, que inhibe de forma irreversible la síntesis de tromboxano A2 (una prostaglandina que favorece la agregación plaquetaria y la vasoconstricción) en las plaquetas. Este efecto es permanente y habitualmente persiste durante los 8 días de vida de la plaqueta. Paradójicamente, el ácido acetilsalicílico también inhibe la síntesis de prostaciclina (una prostaglandina que inhibe la agregación plaquetaria, pero con efectos vasodilatadores) en las células endoteliales de los vasos sanguíneos. Este efecto es transitorio. En cuanto el ácido acetilsalicílico se ha eliminado de la sangre, las células endoteliales nucleadas vuelven a sintetizar prostaciclina. Como consecuencia de ello, una dosis diaria única baja de ácido acetilsalicílico ( < 100 mg/ día) inhibe la síntesis de tromboxano A2 en las plaquetas sin afectar sustancialmente la síntesis de prostaciclina. El ácido acetilsalicílico también pertenece al grupo de antiinflamatorios no esteroideos acídicos con propiedades analgésicas, antipiréticas y antiinflamatorias. Su mecanismo de acción consiste en la inhibición irreversible de las enzimas ciclooxigenasas que participan en la síntesis de prostaglandinas. En dosis altas, el ácido acetilsalicílico se utiliza para el tratamiento del dolor leve a moderado, la elevación de la temperatura corporal y para el tratamiento de las enfermedades inflamatorias agudas y crónicas, como la artritis reumatoide. Atorvastatina: Atorvastatina es un inhibidor selectivo y competitivo de la HMG-CoA reductasa, la enzima limitante responsable de la conversión de 3-hidroxi- 3-metil-glutaril-coenzima A a mevalonato, un precursor de los esteroles, incluido el colesterol. Los triglicéridos y el colesterol se unen en el hígado a lipoproteínas de muy baja densidad (VLDL) y se liberan en el plasma para su distribución a los tejidos periféricos. Las lipoproteínas de baja densidad (LDL) se forman a partir de las VLDL y se catabolizan principalmente a través del receptor con elevada afinidad para las LDL (receptor LDL). Atorvastatina reduce el colesterol plasmático y las concentraciones séricas de lipoproteínas al inhibir la HMG-CoA reductasa y, por tanto, la biosíntesis de colesterol en el hígado, y aumenta el número de receptores LDL en la superficie de las células hepáticas para una mejor captación y catabolismo de las LDL. Atorvastatina disminuye la formación de LDL y el número de partículas LDL. Atorvastatina aumenta de forma profunda y sostenida la actividad de los receptores LDL, junto con un cambio beneficioso en la cantidad de partículas LDL circulantes. Atorvastatina reduce eficazmente el C-LDL en pacientes con hipercolesterolemia familiar homocigota, una población que no suele responder a los fármacos hipolipemiantes. En un estudio de dosis-respuesta, atorvastatina demostró que reduce las concentraciones de C-total (30 % - 46 %), C-LDL (41 % - 61 %), apolipoproteína B (34 % - 50 %) y triglicéridos (14 % - 33 %), a la vez que eleva de manera variable el C-HDL y apolipoproteína A1. Estos resultados se observan tanto en pacientes con hipercolesterolemia familiar heterocigota, formas de hipercolesterolemia no familiar e hiperlipidemia mixta, incluidos pacientes con diabetes mellitus no insulinodependiente. Se ha demostrado que las disminuciones de C-total, C-LDL y apolipoproteína B reducen el riesgo de acontecimientos cardiovasculares y mortalidad cardiovascular. Ramipril: Mecanismo de acción Ramiprilato, el metabolito activo del profármaco ramipril, inhibe la enzima dipeptidilcarboxipeptidasa I (sinónimos: enzima de conversión de la angiotensina; cininasa II). En plasma y tejidos, esta enzima cataliza la transformación de la angiotensina I en la sustancia vasoconstrictora activa angiotensina II y degrada la sustancia vasodilatadora activa bradiquinina. La reducción de la formación de angiotensina II y la inhibición de la degradación de bradiquinina inducen vasodilatación. Dado que la angiotensina II también estimula la liberación de aldosterona, ramiprilato reduce la secreción de aldosterona. La respuesta promedio a la monoterapia con inhibidores de la ECA fue inferior en pacientes hipertensos de raza negra (afrocaribeños) (habitualmente, una población hipertensa hiporreninémica) que en pacientes hipertensos de otras etnias. Efectos farmacodinámicos: Propiedades antihipertensivas: La administración de ramipril reduce sustancialmente la resistencia arterial periférica. En general, el flujo renal y el índice de filtración glomerular no cambian sustancialmente. La administración de ramipril a pacientes hipertensos reduce la presión arterial en bipedestación y en posición supina sin un aumento compensatorio de la frecuencia cardíaca. En la mayoría de los pacientes, el efecto antihipertensivo de una dosis única se inicia de 1 a 2 horas después de la administración oral. El efecto máximo de una dosis única se alcanza de 3 a 6 horas después de la administración oral. El efecto antihipertensivo de una dosis única suele durar unas 24 horas. El efecto antihipertensivo máximo del tratamiento continuado con ramipril suele aparecer al cabo de 3 a 4 semanas. Se ha demostrado que el efecto antihipertensivo se mantiene con un tratamiento a largo plazo hasta de 2 años. La interrupción súbita de ramipril no produce un efecto rebote de aumento excesivo ni rápido de la presión arterial. Insuficiencia cardíaca: Además del tratamiento convencional con diuréticos y glucósidos cardíacos opcionales, se ha demostrado que ramipril es eficaz en pacientes encuadrados en las clases funcionales II-IV de la New-York Heart Association. El fármaco tiene efectos beneficiosos sobre la hemodinámica cardíaca (descenso de las presiones de llenado ventriculares izquierda y derecha, reducción de la resistencia vascular periférica total, aumento del gasto cardíaco y mejora del índice cardíaco). También reduce la activación neuroendocrina. Farmacocinética: Ácido acetilsalicílico: El ácido acetilsalicílico se metaboliza en su principal metabolito activo, ácido salicílico, antes, durante y después de la absorción. Los metabolitos se eliminan básicamente por los riñones. Además del ácido salicílico, los metabolitos principales del ácido acetilsalicílico son el conjugado de glicina de ácido salicílico (ácido salicilúrico), el éter glucurónido y éster del ácido salicílico (glucurónido acilsalicílico y salicilfenólico) y ácido gentísico formado por la oxidación del ácido salicílico y su conjugado de glicina. La absorción del ácido acetilsalicílico tras la administración oral es rápida y completa, en función de la formulación galénica. De hecho, la hidrólisis del residuo acetil del ácido acetilsalicílico tiene lugar, en cierto grado, durante el paso a través de la mucosa gastrointestinal. Las concentraciones plasmáticas máximas se alcanzan al cabo de 10 a 20 minutos (ácido acetilsalicílico) o al cabo de 0,3 a 2 horas (salicilato total). La cinética de eliminación del ácido acetilsalicílico depende en gran medida de la dosis, ya que la capacidad de metabolizar el ácido salicílico es limitada (la semivida de eliminación oscila entre 2 y 30 horas). La semivida de eliminación del ácido acetilsalicílico es de apenas unos minutos; la semivida de eliminación del ácido salicílico es de 2 horas después de la administración de una dosis de 0,5 g de ácido acetilsalicílico, de 4 horas después de la administración de 1 g y aumenta a 20 h tras una dosis única de 5 g. La unión a las proteínas plasmáticas en el ser humano depende de la concentración; se ha descripto que los valores oscilan entre el 49 % a más del 70 % (ácido acetilsalicílico) y del 66 % al 98 % (ácido salicílico). El ácido salicílico se detecta en líquidos corporales y líquido sinovial tras la administración de ácido acetilsalicílico. El ácido salicílico atraviesa la placenta y se excreta en la leche materna. Atorvastatina: Absorción: Atorvastatina se absorbe rápidamente tras la administración oral; La concentración plasmática máxima (Cmáx) se alcanza entre 1 y 2 horas después. El grado de absorción aumenta en proporción a la dosis de atorvastatina. Tras la administración oral, la atorvastatina en comprimidos recubiertos tiene una biodisponibilidad del 95 % al 99 %, en comparación con la solución oral. La biodisponibilidad absoluta de atorvastatina es aproximadamente del 12 % y la disponibilidad sistémica de la actividad inhibidora de la HMG-CoA reductasa es aproximadamente del 30 %. La baja disponibilidad sistémica se atribuye a un aclaramiento presistémico en la mucosa gastrointestinal y/o al metabolismo hepático de primer paso. Distribución: El volumen medio de distribución de atorvastatina es aproximadamente de 381 l. Atorvastatina se une a proteínas plasmáticas en una proporción ≥ 98 %. Biotransformación: Atorvastatina es metabolizada por el citocromo P450 3A4 del citrocromo P450 a derivados ortohidroxilados y parahidroxilados y diversos productos de betaoxidación. Además de por otras vías, estos productos continúan metabolizándose por glucuronidación. In vitro, la inhibición de la HMG-CoA reductasa por los metabolitos ortohidroxilados y parahidroxilados es equivalente a la de atorvastatina. Aproximadamente el 70 % de la actividad inhibidora circulante para la HMG-CoA reductasa se atribuye a los metabolitos activos. Eliminación: Atorvastatina se elimina principalmente en la bilis tras sufrir metabolismo hepático y/o extrahepático. Sin embargo, el fármaco no parece estar sometido a una recirculación enterohepática significativa. La semivida de eliminación plasmática media de atorvastatina en seres humanos es aproximadamente de 14 horas. La semivida de la actividad inhibidora para la HMG-CoA reductasa es aproximadamente de 20 a 30 horas, debido a la contribución de los metabolitos activos. Ramipril: Absorción: Tras la administración oral, ramipril se absorbe rápidamente en el tubo digestivo: las concentraciones plasmáticas máximas del fármaco se alcanzan en 1 hora. En función de la recuperación urinaria, el grado de absorción es como mínimo del 56 %, sin influir significativamente la presencia de alimentos en el tubo digestivo. La biodisponibilidad del metabolito activo ramiprilato tras la administración oral de 2,5 y 5 mg de ramipril es del 45 %. Las concentraciones plasmáticas máximas de ramiprilato, el único metabolito activo de ramipril, se obtienen al cabo de 2 a 4 horas de la ingestión de ramipril. Las concentraciones plasmáticas de ramiprilato en el estado de equilibrio, tras la administración única diaria de las dosis habituales de ramipril se alcanzan alrededor del cuarto día de tratamiento. Distribución: La unión de ramipril a proteínas séricas es aproximadamente del 73 % y la de ramiprilato, aproximadamente del 56 %. Metabolismo: Ramipril se metaboliza casi completamente a ramiprilato, al éster dicetopiperazina, el ácido dicetopiperazínico y los glucurónidos de ramipril y ramiprilato. Eliminación: La excreción de metabolitos es básicamente renal. Las concentraciones plasmáticas de ramiprilato disminuyen de una forma polifásica. Dada su potente unión saturable a la ECA y a la lenta disociación de la enzima, ramiprilato presenta una fase de eliminación terminal prolongada en concentraciones plasmáticas muy bajas. Tras la administración única diaria repetida de ramipril, la semivida eficaz de las concentraciones de ramiprilato fue de 13-17 horas para dosis de 5-10 mg y superior para las dosis más bajas (1,25-2,5 mg). Esta diferencia está relacionada con la capacidad saturable de la enzima para unirse a ramiprilato. Una única dosis oral de ramipril produjo un nivel indetectable de ramipril y su metabolito en la leche materna. No obstante, se desconoce el efecto de dosis repetidas. Pacientes con insuficiencia renal: La excreción renal de ramiprilato se redujo en pacientes con disfunción renal y el aclaramiento renal de ramiprilato es proporcional al aclaramiento de creatinina, lo que resulta en elevaciones de las concentraciones plasmáticas de ramiprilato, que disminuyen más lentamente que en las personas con función renal normal. Pacientes con insuficiencia hepática: En pacientes con disfunción hepática, se retrasó el metabolismo de ramipril a ramiprilato por una disminución de la actividad de las esterasas hepáticas; las concentraciones plasmáticas de ramipril en estos pacientes aumentaron. Sin embargo, las concentraciones máximas de ramiprilato en dichos pacientes no difieren de las observadas en las personas con función hepática normal. Datos preclínicos sobre seguridad: Ácido acetilsalicílico El perfil de seguridad preclínica del ácido acetilsalicílico está bien documentado. En estudios con animales, no se ha demostrado que los salicilatos causen lesiones orgánicas, excepto en el riñón a dosis altas. El ácido acetilsalicílico se ha analizado ampliamente in vitro e in vivo para detectar posibles efectos mutágenos. En su totalidad, los resultados no indican ninguna sospecha de efecto mutágeno. Lo mismo es válido para los estudios en los que se investiga la posibilidad de efectos carcinógenos. En estudios con animales, se han descripto en varias especies los efectos teratógenos de los salicilatos. Se han descripto alteraciones de la implantación, efectos fetotóxicos y embriotóxicos y deterioro de la capacidad de aprendizaje en la descendencia con exposición prenatal. Atorvastatina: En un ensayo in vivo y en una batería de 4 pruebas in vitro, atorvastatina no tuvo efectos mutágenos ni clastogénicos. Atorvastatina no demostró efecto carcinógeno en ratas, pero en altas dosis (de 6 a 11 veces la AUC0-24h alcanzada en seres humanos a las dosis máximas recomendadas) en ratones se observaron adenomas hepatocelulares en los machos y carcinomas hepatocelulares en las hembras. Se dispone de pruebas procedentes de estudios experimentales con animales de que los inhibidores de la HMG-CoA reductasa pueden afectar al desarrollo del embrión o el feto. En ratas, conejos y perros, atorvastatina no afectó a la fertilidad ni tuvo efectos teratógenos; sin embargo, a dosis tóxicas para la madre se observó toxicidad fetal en ratas y conejos. El desarrollo de la progenie en ratas se retrasó y se redujo la supervivencia posnatal con la exposición de las madres a dosis altas de atorvastatina. En ratas, se ha demostrado la transferencia placentaria. Las concentraciones plasmáticas de atorvastatina en ratas son similares a las de la leche. No se sabe si atorvastatina o sus metabolitos se excretan en la leche materna humana. Ramipril: Se ha observado que la administración oral de ramipril carece de toxicidad aguda en roedores y perros. Se han realizado estudios de administración oral crónica en ratas, perros y monos. En las tres especies se han constatado alteraciones de los electrólitos plasmáticos y del hemograma. Como expresión de la actividad farmacodinámica del ramipril, se ha observado un aumento pronunciado del aparato yuxtaglomerular en perros y monos con dosis diarias de 250 mg/kg/día. Ratas, perros y monos toleraron bien las dosis de 2, 2,5 y 8 mg/kg/día, respectivamente, sin efectos perjudiciales. Los estudios sobre toxicología reproductiva en ratas, conejos y monos no demostraron ninguna propiedad teratógena. En ratas, no afectó a la fertilidad de hembras o machos. La administración de ramipril a ratas hembra durante el periodo fetal y la lactancia indujo lesiones renales irreversibles (dilatación de la pelvis renal) en la descendencia con dosis diarias iguales o superiores a 50 mg/kg de peso corporal. Los extensos análisis de mutagenicidad realizados con varios ensayos no han demostrado que ramipril tenga características genotóxicas o mutágenas.
Indicaciones.
OMNIPIL® está indicado en la prevención secundaria de accidentes cardiovasculares, como tratamiento de sustitución en pacientes adultos controlados de forma adecuada con los monocomponentes administrados concomitantemente en dosis terapéuticas equivalentes, es decir, pacientes que han respondido en forma adecuada a las monodrogas administradas concomitantemente en dosis terapéuticas equivalentes.
Dosificación.
Adultos: Los pacientes actualmente controlados con dosis terapéuticas equivalentes de ácido acetilsalicílico, atorvastatina y ramipril pueden cambiar directamente a cápsulas de OMNIPIL®. Se iniciará el tratamiento bajo supervisión médica. Para la prevención cardiovascular, la dosis de mantenimiento de ramipril es de 10 mg una vez al día. Población pediátrica: OMNIPIL® está contraindicado en niños y adolescentes menores de 18 años. Poblaciones especiales: Pacientes con insuficiencia renal: La dosis diaria en pacientes con insuficiencia renal debe basarse en el aclaramiento de creatinina: Si el aclaramiento de creatinina es ≥ 60 ml/min, la dosis máxima diaria de ramipril es de 10 mg; Si el aclaramiento de creatinina está entre 30-60 ml/min, la dosis máxima diaria de ramipril es de 5 mg; En pacientes sometidos a hemodiálisis o con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min), OMNIPIL® está contraindicado. Pacientes con insuficiencia hepática: La dosis máxima diaria de ramipril en estos pacientes es de 2,5 mg. En pacientes con insuficiencia hepática grave, OMNIPIL® está contraindicado. Se recomienda realizar pruebas de función hepática antes de iniciar el tratamiento con atorvastatina y posteriormente de forma periódica. Se deben realizar pruebas de función hepática a los pacientes que desarrollen cualquier síntoma o signo que sugiera lesión hepática. Los pacientes que presenten un aumento en los niveles de transaminasas se deben controlar hasta que esta anomalía(s) quede(n) resuelta(s). En caso de un aumento persistente de las transaminasas 3 veces el valor máximo de normalidad, se recomienda una reducción de la dosis o la retirada de OMNIPIL®. OMNIPIL® debe utilizarse con precaución en pacientes que consuman cantidades importantes de alcohol y/o con antecedentes de enfermedad hepática. Se recomienda el monitoreo de la función hepática en los pacientes que toman OMNIPIL®. Pacientes de edad avanzada: En pacientes de edad avanzada y frágiles, el tratamiento debe iniciarse con precaución por un mayor riesgo de efectos secundarios. Forma de administración: OMNIPIL® cápsulas se administra por vía oral. OMNIPIL® debe administrarse por vía oral como cápsula única diaria, preferiblemente por la noche, después de la cena. OMNIPIL® debe tragarse con líquido. No debe abrirse, masticarse ni aplastarse. Evitar consumir jugo de pomelo mientras se tome OMNIPIL®.
Contraindicaciones.
Hipersensibilidad a los principios activos o a alguno de los excipientes, a otros salicilatos, a los antiinflamatorios no esteroideos (AINE), a cualquier otro inhibidor de la ECA (enzima convertidora de angiotensina) o a la tartrazina. Hipersensibilidad a la soja o a los cacahuetes. En caso de antecedentes de crisis asmática u otra reacción alérgica al ácido salicílico y otros analgésicos/antiinflamatorios no esteroideos. Úlcera péptica recurrente activa o antecedentes y/o hemorragia gástrica/intestinal, u otras clases de hemorragia como hemorragias cerebrovasculares. Hemofilia y otros trastornos de la coagulación. Insuficiencia hepática y renal graves. Pacientes en hemodiálisis Insuficiencia cardíaca grave. Tratamiento concomitante con metotrexato en dosis semanales iguales o superiores a 15 mg. Uso concomitante de OMNIPIL® con medicamentos con aliskirén está contraindicado en pacientes con diabetes mellitus o insuficiencia renal (TFG < 60 ml/min/1,73 m²). Pacientes con pólipos nasales asociados al asma inducido o exacerbado por el ácido acetilsalicílico. Hepatopatía activa o elevaciones persistentes no explicadas de las transaminasas séricas que excedan en 3 veces el límite superior normal. Durante el embarazo y la lactancia y en mujeres en edad fértil que no utilicen métodos anticonceptivos fiables. Debido al riesgo de rabdomiólisis, tratamiento concomitante con tipranavir o ritonavir. Debido al riesgo de rabdomiólisis, tratamiento concomitante con ciclosporina. Antecedentes de angioedema (hereditario, idiopático o por angioedema previo con inhibidores de la ECA o antagonistas de los receptores de la angiotensina II [ARAII]). Tratamientos extracorpóreos que impliquen el contacto de la sangre con superficies de carga negativa. Estenosis bilateral significativa de la arteria renal o estenosis de la arteria renal en un solo riñón funcionante. No debe administrarse ramipril a pacientes hipotensos o hemodinámicamente inestables. Niños y adolescentes menores de 18 años. En niños menores de 16 años con fiebre, gripe o varicela, existe riesgo de síndrome de Reye. Pacientes tratados con los antivirales contra la hepatitis C glecaprevir/ pibrentasvir.
Reacciones adversas.
Resumen del perfil de seguridad OMNIPIL®: La reacción adversa más común asociada al tratamiento con ácido acetilsalicílico, son las molestias gastrointestinales. Las úlceras y las hemorragias son poco frecuentes (menos de 1 caso por 100). Las perforaciones del tracto gastrointestinal son muy raras (menos de 1 caso por 10.000). Las reacciones adversas conocidas con la terapia con ramipril incluyen tos seca persistente y reacciones debido a la hipertensión. Las reacciones adversas asociadas a la terapia con ramipril poco frecuentes (menos de 1 caso por 100) incluyen angioedema, insuficiencia renal y hepática. Se ha comunicado neutropenia y agranulocitosis raramente (menos de 1 caso por 1.000). La mialgia (dolor muscular, espasmos musculares, hinchazón de las articulaciones) es una reacción adversa común con el tratamiento con estatinas. La miopatía y la rabdomiólisis son raras (menos de 1 caso por 1.000). La monitorización de CK debe ser considerada como parte de la evaluación de los pacientes con niveles de CK significativamente elevados al momento basal ( > 5 x LNS). En la base de datos de ensayos clínicos controlados con placebo sobre atorvastatina, de los 16.066 pacientes tratados (8.755 con atorvastatina frente a 7.311 con placebo) el 5,2 % de los que recibieron atorvastatina abandonaron el tratamiento debido a reacciones adversas, en comparación con el 4,0 % de los tratados con placebo. Al igual que con otros inhibidores de la HMG-CoA reductasa, se han comunicado elevaciones de las transaminasas séricas en pacientes que reciben atorvastatina. Dichos cambios suelen ser transitorios y de carácter leve, y no requieren la interrupción del tratamiento. En el 0,8 % de los pacientes que recibían atorvastatina se produjeron elevaciones clínicamente importantes ( > 3 veces el LSN) de las transaminasas séricas. Tales elevaciones estaban relacionadas con la dosis y fueron reversibles en todos los pacientes. En el 2,5 % de los pacientes que recibían atorvastatina se produjo un incremento de la creatina cinasa (CK) superiores a 3 veces el LSN, similar a lo observado con otros inhibidores de la HMG-CoA reductasa en los ensayos clínicos. El 0,4 % de los pacientes tratados con atorvastatina sufrieron un incremento superior a 10 veces el LSN. Con algunas estatinas se han comunicado los siguientes acontecimientos adversos: Disfunción sexual. Depresión. Se han comunicado casos excepcionales de neumopatía intersticial, especialmente con el tratamiento a largo plazo. Diabetes mellitus: La frecuencia dependerá de la presencia o ausencia de factores de riesgo. Resumen de reacciones adversas. Muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 < 1/10); poco frecuentes (≥ 1/1000 < 1/100); raras (≥ 1/10.000 < 1/1000); muy raras (≥ 1/10.000) frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Clasificación por órganos y sistemas de MedDRA. Trastornos de la sangre y del sistema linfático: Eosinofilia. Ramipril: Poco frecuente. Disminución del recuento de leucocitos incluidas neutropenia o agranulocitosis), disminución del recuento de eritrocitos, disminución de la hemoglobina, disminución del recuento plaquetario (trombocitopenia). Ramipril: Rara. Se han notificado hemorragias graves que en algunos casos pueden ser potencialmente mortales, por ejemplo hemorragia cerebral, especialmente en pacientes con hipertensión arterial no controlada y/o tratamiento concomitante con anticoagulantes. Ácido acetilsalicílico: Rara: Se observan hemorragias como epistaxis, hemorragia gingival, hemorragia cutánea o hemorragia genitourinaria, con una posible prolongación del tiempo de coagulación. Este efecto puede durar de 4 a 8 días tras la ingestión. Ácido acetilsalicílico: Rara. Trombocitopenia. Atorvastatina: Rara. Mielosupresión, pancitopenia, anemia hemolítica. Ramirpil: No conocida. Trastornos gastrointestinales: Molestias gastrointestinales como pirosis, náuseas, vómitos, gastralgia y diarrea. Ácido acetilsalicílico: Muy frecuentes. Hemorragia gastrointestinal menor (microhemorragia). Ácido acetilsalicílico: Muy frecuentes. Dispepsia, náuseas, diarrea. Ramipril: Frecuente. Atorvastatina: Frecuente. Trastornos digestivos, malestar abdominal. Ramipril: Frecuente. Inflamación gastrointestinal. Ramipril: Frecuente. Ácido acetilsalicílico: Poco frecuente. Estreñimiento. Ramipril: Poco frecuente. Atorvastatina: Frecuente. Flatulencia. Atorvastatina: Frecuente. Úlceras gastrointestinales. Ácido acetilsalicílico: Poco frecuente. Hemorragia gastrointestinal. Ácido acetilsalicílico: Poco frecuente. Anemia ferropénica por hemorragias gastrointestinales ocultas tras un uso a largo plazo. Ácido acetilsalicílico: Poco frecuente Dolor abdominal superior e inferior, eructos, pancreatitis. Atorvastatina: Poco frecuente. Pancreatitis (se han descrito casos muy excepcionales de muerte con inhibidores de la ECA), aumento de las enzimas pancreáticas, angioedema del intestino delgado, dolor abdominal superior (incluida gastritis), sequedad de boca. Ramipril: Poco frecuente. Glositis. Ramipril: Rara. Perforación de una úlcera gastrointestinal. Informe inmediatamente a su médico si observa heces negras o sangre en vómitos (signos de hemorragia gástrica grave). Ácido acetilsalicílico: Muy rara. Estomatitis aftosa. Ramipril: No conocida. Trastornos respiratorios torácicos y mediastínicos: Broncoespasmo paroxístico, disnea grave, rinitis, congestión nasal. Ácido acetilsalicílico: Frecuente. Dolor faringolaríngeo, epistaxis. Atorvastatina: Frecuente. Tos irritativa no productiva. Ramipril: Frecuente. Bronquitis, sinusitis, disnea, broncoespasmo (incluido empeoramiento del asma), congestión nasal. Ramipril: Poco frecuente. Infecciones e infestaciones: Nasofaringitis. Atorvastatina: Frecuente. Trastornos del sistema nervioso. Cefalea. Ramipril: Frecuente. Atorvastatina: Frecuente. Mareos. Ramipril: Frecuente. Atorvastatina: Poco frecuente. Vértigo, ageusia. Ramipril: Poco frecuente. Parestesia, disgeusia. Ramipril: Poco frecuente. Atorvastatina: Poco frecuente. Hipoestesia, amnesia. Atorvastatina: Poco frecuente. Neuropatía periférica. Atorvastatina: Rara. Temblor, trastorno del equilibrio. Ramipril: Rara. Isquemia cerebral (incluidos accidente cerebrovascular isquémico y accidente isquémico transitorio), deterioro de las habilidades psicomotoras, sensación de quemazón, parosmia. Ramipril: No conocida. Trastornos de la piel y del tejido subcutáneo: Exantema, en especial maculopapular. Ramipril: Frecuente. Reacciones cutáneas. Ácido acetilsalicílico: Poco frecuente. Urticaria, prurito, alopecia. Atorvastatina: Poco frecuente. Angioedema; en muy raras ocasiones, la obstrucción de las vías respiratorias por angioedema puede producir la muerte; prurito, hiperhidrosis. Ramipril: Poco frecuente. Edema angioneurótico, dermatitis ampollosa incluido eritema multiforme, síndrome de Stevens-Johnson y necrólisis epidérmica tóxica. Atorvastatina: Rara. Dermatitis exfoliativa, urticaria, onicólisis. Ramipril: Rara. Reacción de fotosensibilidad. Ramipril: Muy rara. Eritema multiforme. Ramipril: No conocida. Ácido acetilsalicílico: Muy rara. Pénfigo, empeoramiento de la psoriasis, dermatitis psoriasiforme, exanterna o enantema penfigoide o liquenoide, alopecia. Ramipril: No conocida. Trastornos del sistema inmunológico: Reacciones alérgicas. Atorvastatina: Frecuente. Reacciones alérgicas de la piel, las vías respiratorias, el tubo digestivo y el sistema cardiovascular, sobre todo en pacientes asmáticos (con los siguientes posibles síntomas: reducción de la presión arterial, disnea, rinitis, congestión nasal, choque anafiláctico, edema de Quincke). Ácido acetilsalicílico: Rara. Anafilaxia. Atorvastatina: Muy rara. Reacciones anafilácticas o anafilactoides, aumento de anticuerpos antinucleares. Ramipril: No conocida. Trastornos hepatobiliares: Hepatitis. Atorvastatina: Poco frecuente. Elevación de las enzimas hepáticas y/o de la bilirrubina conjugada. Ramipril: Poco frecuente. Colestasis. Atorvastatina: Rara. Ictericia colestásica, daño hepatocelular. Ramipril: Rara. Insuficiencia hepática. Atorvastatina: Muy rara. Elevación de los valores en las pruebas de función hepática. Ácido acetilsalicílico: Muy rara. Insuficiencia hepática aguda, hepatitis citolítica o colestásica (en muy raras ocasiones con desenlace mortal). Ramipril: No conocida Trastornos renales y urinarios: Insuficiencia renal (incluida insuficiencia renal aguda), aumento de la diuresis, empeoramiento de una proteinuria previa, aumento de la urea sanguínea, aumento de la creatinina sanguínea. Ramipril: Poco frecuente. Insuficiencia renal. Ácido acetilsalicílico: Muy rara. Trastornos del metabolismo y la nutrición: Hiperglucemia. Atorvastatina: Frecuente. Hiperpotasemia. Ramipril: Frecuente. Hipoglucemia. Atorvastatina: Poco frecuente. Ácido acetilsalicílico: Muy rara. Incremento ponderal. Atorvastatina: Poco frecuente. Anorexia. Ramipril: Poco frecuente. Atorvastatina: Poco frecuente. Hiporexia. Ramipril: Poco frecuente. En dosis bajas, el ácido acetilsalicílico reduce la excreción de ácido úrico. En los pacientes predispuestos puede provocar crisis de gota. Ácido acetilsalicílico: Muy rara. Hiponatremia. Ramipril: No conocida. Trastornos psiquiátricos: Pesadillas, insomnio. Atorvastatina: Poco frecuente. Depresión, ansiedad, nerviosismo, inquietud, trastornos del sueño, incluida la somnolencia. Ramipril: Poco frecuente. Estado confusional. Ramipril: Rara. Trastornos de la atención. Ramipril: No conocida. Trastornos oculares: Visión borrosa. Ramipril: Poco frecuente. Atorvastatina: Poco frecuente. Trastornos visuales. Ramipril: Poco frecuente. Atorvastatina: Rara. Conjuntivitis. Ramipril: Rara. Trastornos del oído y del laberinto: Acúfenos. Ramipril: Rara. Atorvastatina: Poco frecuente. Hipoacusia. Ramipril: Rara. Pérdida de audición. Atorvastatina: Muy rara. Trastornos musculoesqueléticos y del tejido conjuntivo Mialgia, espasmos musculares. Ramipril: Frecuente. Atorvastatina: Frecuente. Dolores en las extremidades, inflamación articular, dolor de espalda. Atorvastatina: Frecuente. Artralgia. Ramipril: Poco frecuente. Atorvastatina: Frecuente. Dolor cervical, fatiga muscular. Atorvastatina: Poco frecuente. Miopatía, miosítis, rabdomiólisis, tendinopatía a veces complicada por rotura. Atorvastatina: Rara. Miopatía necrotizante inmunomediada. Atorvastatina: No conocida Trastornos del aparato reproductor y de la mama: Disfunción eréctil transitoria, disminución de la libido. Ramipril: Poco Frecuente. Ginecomastia. Ramipril: No conocida. Atorvastatina: Muy rara. Trastornos generales y alteraciones en el lugar de administración: Dolor torácico, fatiga. Ramipril: Frecuente. Atorvastatina: Poco frecuente -Pirexia. Ramipril: Poco frecuente. Atorvastatina: Poco frecuente. Malestar, edema periférico. Atorvastatina: Poco frecuente. Astenia. Ramipril: Rara. Atorvastatina: Poco frecuente. Exploraciones complementarias: Anomalías en las pruebas de función hepática, aumento de la creatina cinasa en sangre. Atorvastatina: Frecuente. Presencia de leucocitos en la orina. Atorvastatina: Poco frecuente Trastornos Cardiacos: Isquemia miocárdica (incluidos angina de pecho o infarto de miocardio), taquicardia, arritmia, palpitaciones y edema periférico. Ramipril: Poco frecuente. Hipotensión ortostática, síncope. Ramipril: Frecuente. Rubefacción. Ramipril: Poco frecuente. Estenosis vascular, hipoperfusión, vasculitis. Ramipril: Rara. Fenómeno de Raynaud. Ramipril: No conocida. Notificación de Sospecha de Reacciones Adversas: Es importante notificar la sospecha de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia al siguiente link: http://sistemas. anmat.gov.ar/aplicaciones_net/fvg_eventos_adversos_nuevo/ index.htlm y/o al Departamento de Farmacovigilancia de GADOR S.A. vía email a farmacovigilancia@gador.com o telefónicamente al 0800- 220-2273.
Advertencias.
OMNIPIL® debe utilizarse solo como tratamiento de sustitución en pacientes controlados de forma adecuada con los monocomponentes administrados concomitantemente en dosis terapéuticas equivalentes. Advertencias para poblaciones especiales: Se requiere una vigilancia médica especialmente estrecha en caso de: Hipersensibilidad a otros analgésicos/antiinflamatorios/antipiréticos/ antirreumáticos u otros alérgenos. Otras alergias conocidas (p. ej., reacciones cutáneas, prurito, urticaria), asma bronquial, rinitis alérgica, inflamación de las mucosas nasales (hiperplasia adenoidea) y otras enfermedades respiratorias crónica