Ocrevus®
ROCHE
Inmunosupresor selectivo. Anticuerpo monoclonal humanizado recombinante anti-CD20.
Composición.
Cada vial contiene 300 mg de ocrelizumab en 10 ml a una concentración de 30 mg/ml, en un excipiente compuesto por acetato de sodio trihidratado 21,4 mg, ácido acético glacial 2,5 mg, a,a-trehalosa dihidratada 400,0 mg, polisorbato 20: 2,0 mg y agua para inyectables c.s.p. 10,0 ml.
Farmacología.
Código ATC: L04AA36. Grupo farmacoterapéutico: Grupo de inmunosupresores selectivos. Propiedades farmacodinámicas: Mecanismo de acción: Ocrelizumab es un anticuerpo monoclonal humanizado recombinante que actúa de forma selectiva contra linfocitos B que expresan CD20. CD20 es un antígeno de superficie celular que se encuentra en linfocitos pre-B, linfocitos B maduros y linfocitos B de memoria, pero que no se expresan en las células madre linfoides ni en las células plasmáticas. Los mecanismos exactos a través de los cuales ocrelizumab ejerce sus efectos clínicos terapéuticos en la EM no se han esclarecido completamente, pero se cree que está implicado en la inmunomodulación a través de la reducción del número y de la función de los linfocitos B que expresan CD20. Tras la unión a la superficie celular, ocrelizumab reduce de forma selectiva los linfocitos B que expresan CD20 a través de fagocitosis celular dependiente de anticuerpos (ADCP), citotoxicidad celular dependiente de anticuerpos (ADCC), citotoxicidad dependiente del complemento (CDC) y apoptosis. Se conservan la capacidad de reconstitución de los linfocitos B y la inmunidad humoral preexistente. Además, la inmunidad innata y el número total de linfocitos T no se ven afectados. Efectos farmacodinámicos: El tratamiento con Ocrevus produce la rápida depleción de linfocitos B CD19+ en la sangre al cabo de 14 días después del tratamiento (primer punto temporal de la evaluación), siendo este un efecto farmacológico esperado. Esto se mantuvo a lo largo del período de tratamiento. Para los recuentos de linfocitos B se emplea CD19, ya que la presencia de Ocrevus interfiere con el reconocimiento de CD20 por parte del ensayo. En los estudios de fase III, entre cada dosis de Ocrevus, hasta el 5% de los pacientes mostraron repleción de linfocitos B ( > límite inferior de la normalidad [LIN] o el valor inicial) al menos en un momento determinado. La magnitud y la duración de la depleción de linfocitos B fueron consistentes en los ensayos en la EMPP y la EMR. El tiempo de seguimiento más prolongado después de la última infusión de Ocrevus (estudio en fase II WA21493, n = 51) indica que la mediana del tiempo hasta la repleción de linfocitos B (retorno al valor inicial/LIN, lo que se produzca antes) fue de 72 semanas (intervalo de 27 - 175 semanas). El 90% de todos los pacientes presentaron repleción de linfocitos B hasta el LIN o hasta el valor inicial al cabo de aproximadamente dos años y medio después de la última infusión. Eficacia clínica y seguridad: Formas recurrentes de EM: La eficacia y la seguridad de Ocrevus se evaluaron en dos ensayos clínicos aleatorizados, doble ciego, con doble simulación y controlados con comparador activo (WA21092 y WA21093), con diseño idéntico, en pacientes con formas recurrentes de EM (de acuerdo con los criterios de McDonald de 2010) y con evidencia de enfermedad activa (definida por características clínicas o de imagen) en los dos años anteriores. El diseño del estudio y las características iniciales de la población del estudio se resumen en la Tabla 1. Las características demográficas e iniciales estaban bien equilibrada en los dos grupos de tratamiento. Los pacientes que recibían Ocrevus (grupo A) recibieron 600 mg cada 6 meses (dosis 1 consta de dos infusiones intravenosas de 300 mg, administradas con un intervalo de separación de 2 semanas, y las dosis posteriores se administraron como una única infusión intravenosa de 600 mg). Los pacientes del grupo B recibieron interferón beta-1a (Rebif) 44 g mediante inyección por vía subcutánea 3 veces por semana.
Los resultados principales de eficacia clínicos y de RM se presentan en la Tabla 2 y la Figura 1. Los resultados de estos estudios revelan que Ocrevus suprimió de forma significativa las recaídas, la actividad de la enfermedad subclínica medida por RM y la progresión de la enfermedad en comparación con la administración subcutánea de 44 mg de interferón beta-1a.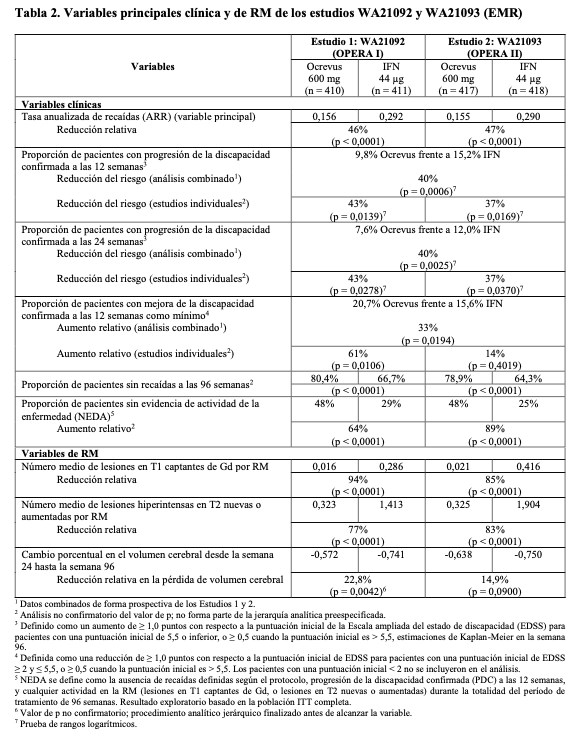
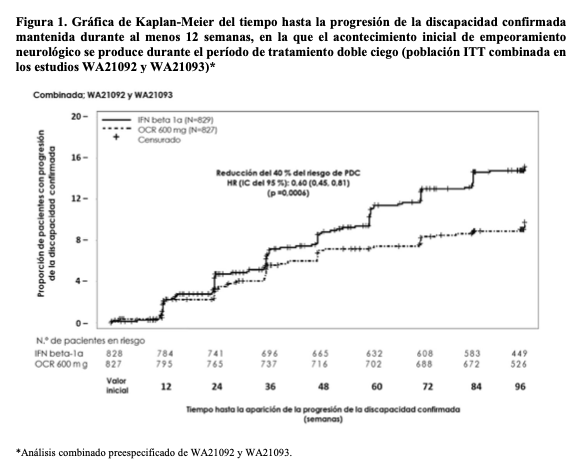
Los resultados de los análisis combinados preespecificados del tiempo hasta la PDC mantenida durante al menos 12 semanas (reducción del 40% del riesgo para Ocrevus en comparación con interferón beta-1a, p = 0,0006) coincidieron en gran medida con los resultados mantenidos durante al menos 24 semanas (reducción del 40% del riesgo para Ocrevus en comparación con interferón beta-1a, p = 0,0025). Los estudios incluyeron pacientes con enfermedad activa. Estos incluyeron tanto pacientes activos sin tratamiento previo como pacientes con respuesta subóptima a tratamientos previos, definido por características clínicas o de imagen. El análisis de las poblaciones de pacientes con diferentes niveles iniciales de actividad de la enfermedad, incluida la enfermedad activa y muy activa, mostró que la eficacia de Ocrevus sobre la ARR (tasa anualizada de recaídas) y la PDC de 12 semanas coincidía con la de la población general. EM primaria progresiva: La eficacia y la seguridad de Ocrevus también se evaluaron en un ensayo clínico aleatorizado, doble ciego y controlado con placebo en pacientes con EM primaria progresiva (estudio WA25046) que se encontraban en una fase temprana de la enfermedad según los principales criterios de inclusión, por ejemplo, entre los 18 y los 55 años, incluidos; EDSS en el cribado de 3,0 a 6,5 puntos; duración de la enfermedad menor de 10 años desde el inicio de los síntomas de la EM en los pacientes con EDSS en el cribado ≤ 5,0 o menor de 15 años en los pacientes con un EDSS en el cribado > 5,0. En relación con la actividad de la enfermedad, las características propias de actividad inflamatoria, incluso en EM progresiva, pueden estar relacionadas con las imágenes, (por ejemplo, las lesiones en T1 captantes de Gd y/o las lesiones T2 activas [nuevas o aumentadas]). La evidencia de las imágenes de RM es importante para confirmar la actividad inflamatoria en todos los pacientes. No se estudiaron pacientes mayores de 55 años de edad. El diseño del estudio y las características iniciales de la población del estudio se presentan en la Tabla 3. Las características demográficas e iniciales estaban bien equilibradas en los dos grupos de tratamiento. En las imágenes de la RM cerebral se observó actividad inflamatoria por la presencia de lesiones captantes de Gd en T1 o lesiones en T2. Durante el estudio en fase III de la EMPP, los pacientes recibieron 600 mg de Ocrevus cada 6 meses como dos infusiones de 300 mg, administradas con un intervalo de dos semanas entre sí, a lo largo del período de tratamiento. Las infusiones de 600 mg en la EMR y las 2 infusiones de 300 mg en la EMPP demostraron perfiles de FC/FD coincidentes. Los perfiles de RRI por infusión fueron también similares, independientemente de si la dosis de 600 mg se administró como infusión única de 600 mg o como dos infusiones de 300 mg separadas por un intervalo de dos semanas (véanse Reacciones adversas y Características farmacológicas - Propiedades, Propiedades farmacocinéticas), pero debido a que se administraron más infusiones con la pauta de 2 infusiones de 300 mg, el número total de RRI fue superior. Por tanto, después de la dosis 1 se recomienda administrar Ocrevus como infusión única de 600 mg (véase Posología y formas de administración) para reducir el número total de infusiones (con exposición simultánea a metilprednisolona profiláctica y un antihistamínico) y las reacciones relacionadas con la infusión.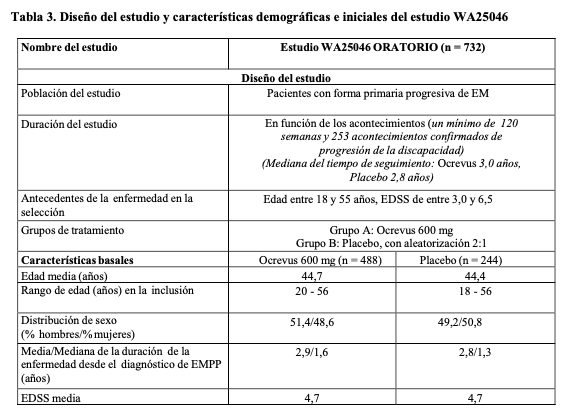
Los resultados principales de eficacia clínicos y de RM se presentan en la Tabla 4 y la Figura 2. Los resultados de este estudio revelan que Ocrevus retrasa de forma significativa la progresión de la enfermedad y reduce el deterioro en la velocidad de la marcha en comparación con placebo.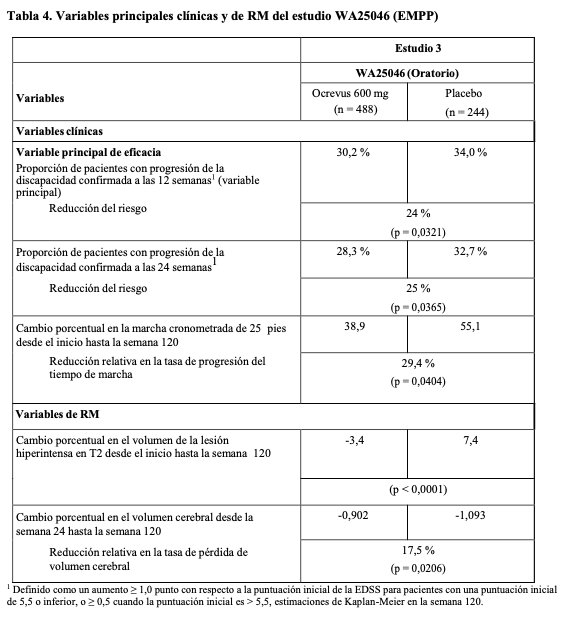
Un análisis de subgrupos preespecificado de la primera variable, pero sin estimación de su potencia, sugiere que los pacientes más jóvenes o aquellos con lesiones en T1 captantes de Gd al inicio obtuvieron mayor beneficio del tratamiento que los pacientes mayores o sin lesiones en T1 captantes de Gd (≤ 45 años: HR 0,64 [0,45-0,92], > 45 años: HR 0,88 [0,62-1,26]; con lesiones en T1 captantes de Gd al inicio: HR 0,65 [0,40-1,06], sin lesiones en T1 captantes de Gd al inicio: HR 0,84 [0,62-1,13]). Además, análisis post-hoc sugirieron que los pacientes más jóvenes con lesiones en T1 captantes de Gd al inicio consiguen el mejor efecto del tratamiento (≤ 45 años: HR 0,52 [0,27-1,00]; ≤ 46 años [mediana de edad del estudio WA25046]: HR 0,48 [0,25-0,92]; < 51 años: HR 0,53 [0,31-0,89]. Los análisis post-hoc se realizaron en el Período Controlado Extendido (PCE), que incluye tratamiento doble ciego y aproximadamente 9 meses adicionales de seguimiento controlado antes de continuar con la fase de extensión abierta (OLE) o hasta la retirada del tratamiento del estudio. En la semana 144, la proporción de pacientes con Progresión de la Discapacidad Confirmada a las 24 semanas con EDSS ≥ 7,0 (PDC 24 semanas con EDSS ≥ 7,0, tiempo hasta la necesidad de utilizar silla de ruedas) fue del 9,1% en el grupo placebo en comparación con el 4,8% en el grupo Ocrevus, lo que significó una reducción del riesgo de tiempo hasta la necesidad de utilizar silla de ruedas del 47% (HR 0,53, [0,31}, 0,92]) durante el PCE. Estos resultados deben interpretarse con cautela debido a que fueron de naturaleza exploratoria e incluyeron datos después del desenmascaramiento. Subestudio de infusión más corta: La seguridad de la infusión más corta de Ocrevus (2 horas) se evaluó en un subestudio prospectivo, multicéntrico, aleatorizado, doble ciego, controlado, de grupos paralelos, dentro del estudio MA30143 (Ensemble) en pacientes con esclerosis múltiple remitente recurrente que eran naive a otros tratamientos modificadores de la enfermedad. La primera dosis de Ocrevus se administró en dos infusiones de 300 mg (600 mg en total) separadas por 14 días. Los pacientes fueron aleatorizados a partir de la segunda dosis y en adelante (a partir de la 2a hasta la 6a dosis) en una ratio 1:1 bien al grupo que recibía la infusión convencional con Ocrevus, infundida durante aproximadamente 3,5 horas cada 24 semanas, o bien al grupo que recibía la infusión más corta con Ocrevus, infundida durante aproximadamente 2 horas cada 24 semanas. La aleatorización se estratificó por regiones y según la dosis en la que los pacientes fueron asignados de manera aleatorizada por primera vez. La variable primaria fue la proporción de pacientes que tuvieron RRI durante o dentro de las 24 horas siguientes a la primera infusión de Ocrevus aleatorizada. El análisis primario se realizó cuando se aleatorizaron a 580 pacientes. La proporción de pacientes con RRI que ocurrieron durante la infusión o dentro de las 24 horas siguientes fue de 24,6% en el grupo de la infusión más corta comparado con el 23,1% en el grupo de la infusión convencional. La diferencia entre los grupos estratificados fue similar. En general, en todas las dosis aleatorizadas, la mayoría de las RRI fueron de leves a moderadas y solo dos RRI fueron de intensidad severa, una RRI en cada grupo. No hubo RRI con riesgo para la vida, mortales o graves. Inmunogenicidad: Los pacientes de los ensayos de EM (WA21092, WA21093 y WA25046) fueron analizados en múltiples puntos temporales (al inicio y cada 6 meses después del tratamiento durante todo el ensayo) para detectar la presencia de anticuerpos antifármaco (AAF). De los 1.311 pacientes tratados con Ocrevus, 12 (~1%) dieron positivo para AAF producidos durante el tratamiento, de los cuales 2 pacientes dieron positivo para anticuerpos neutralizantes. El impacto de los AAF surgidos durante el tratamiento sobre la seguridad y la eficacia no puede evaluarse debido a la baja incidencia de AAF asociados con Ocrevus. Inmunización: En un estudio aleatorizado, abierto, en pacientes con EMR (n = 102), el porcentaje de pacientes con una respuesta positiva a la vacuna del tétanos transcurridas 8 semanas desde la vacunación fue del 23,9% en el grupo con ocrelizumab en comparación con el 54,5% en el grupo control (tratamiento no modificador de la enfermedad excepto interferón beta). La media geométrica de los títulos de anticuerpos específicos frente al toxoide tetánico tras 8 semanas fue 3,74 y 9,81 UI/ml, respectivamente. La respuesta positiva a ≥ 5 serotipos en 23-PPV a las 4 semanas de la vacunación fue del 71,6% en el grupo con ocrelizumab y del 100% en el grupo control. La administración de una vacuna de refuerzo (3-PCV) cuatro semanas después de la 23-PPV en pacientes tratados con ocrelizumab no mejoró de forma relevante la respuesta frente a 12 serotipos en común con 23-PPV. Antes de la vacunación, el porcentaje de pacientes con títulos seroprotectores frente a cinco cepas de gripe osciló entre el 20,0 - 60,0% y entre el 16,7 - 43,8%, y 4 semanas después de la vacunación osciló entre el 55,6 - 80,0% y entre el 75,0 - 97,0%, en pacientes tratados con ocrelizumab y en el grupo control, respectivamente (véanse Precauciones y advertencias e Interacciones). Población pediátrica: La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Ocrevus en uno o más grupos de la población pediátrica en el tratamiento de la esclerosis múltiple. Véase Posología y formas de administración para consultar la información sobre el uso en la población pediátrica. Propiedades farmacocinéticas: La farmacocinética de ocrelizumab en los estudios de EM se describió mediante un modelo de dos compartimentos con aclaramiento dependiente del tiempo y con parámetros de FC típicos para un anticuerpo monoclonal IgG1. La exposición total (ABC a lo largo del intervalo de administración de 24 semanas) fue idéntica en la pauta de 2 infusiones de 300 mg de los estudios de EMPP y en la pauta de 1 infusión de 600 mg de los estudios de EMR, tal como se esperaba ya que se administró una dosis idéntica. El área bajo la curva (ABC) después de la 4a dosis de 600 mg de ocrelizumab fue de 3.510 g/mldía, y la concentración máxima (Cmáx) media fue de 212 g/ml en la EMR (infusión de 600 mg) y de 141 g/ml en la EMPP (infusiones de 300 mg). Absorción: Ocrevus se administró como infusión intravenosa. No se han realizado estudios con otras vías de administración. Distribución: La estimación de la farmacocinética poblacional del volumen de distribución central fue de 2,78 litros. Las estimaciones del volumen periférico y del aclaramiento intercompartimental fueron de 2,68 litros y 0,294 litros/día. Biotransformación: No se ha estudiado directamente el metabolismo de Ocrevus, ya que los anticuerpos se eliminan principalmente por catabolismo (por ejemplo, rotura en péptidos y aminoácidos). Eliminación: El aclaramiento constante se estimó en 0,17 litro/día y el aclaramiento inicial dependiente del tiempo en 0,0489 litro/día, que se redujo con una vida media de 33 semanas. La vida media de eliminación terminal de ocrelizumab fue de 26 días. Farmacocinética en poblaciones especiales: Población pediátrica: No se han realizado estudios para investigar la farmacocinética de ocrelizumab en niños y adolescentes < 18 años de edad. Pacientes de edad avanzada: No se han realizado estudios de FC de ocrelizumab en pacientes ≥ 55 años debido a la limitada experiencia clínica (véase Posología y formas de administración). Pacientes con insuficiencia renal No se ha realizado ningún estudio farmacocinético formal. Se incluyó en los ensayos clínicos a pacientes con insuficiencia renal leve y no se observó ningún cambio en la farmacocinética de Ocrevus en estos pacientes. No se dispone de información de la FC en pacientes con insuficiencia renal moderada o grave. Pacientes con insuficiencia hepática: No se ha realizado ningún estudio farmacocinético formal. Se incluyó en los ensayos clínicos a pacientes con insuficiencia hepática leve y no se observó ningún cambio en la farmacocinética en estos pacientes. No se dispone de información de la FC en pacientes con insuficiencia hepática moderada o grave. Datos preclínicos sobre seguridad: Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas y desarrollo embriofetal. No se han realizado estudios de carcinogenicidad ni mutagenicidad con ocrelizumab. En dos estudios de desarrollo pre y posnatal en monos cynomolgus, la administración de ocrelizumab desde el día gestacional 20 hasta al menos la fase de inducción al parto se asoció con glomerulopatía, formación de folículos linfoides en la médula ósea, inflamación renal linfoplasmacítica y reducción del peso testicular en la descendencia. Las dosis maternas administradas en estos estudios produjeron concentraciones séricas máximas (Cmáx) medias entre 4,5 hasta 21 veces más elevadas que las previstas en el entorno clínico. Hubo cinco casos de neonatos moribundos, uno atribuido a debilidad debida a nacimiento prematuro acompañado de infección oportunista bacteriana, otro debido a una meningoencefalitis infecciosa que afectaba al cerebelo del neonato cuya madre presentaba una infección bacteriana activa (mastitis) y tres con evidencia de ictericia y daño hepático, con sospecha de etiología viral, posiblemente un poliomavirus. La evolución de estas cinco infecciones confirmadas o sospechosas podría haberse visto afectada por la depleción de linfocitos B. Se observó que las crías de madres expuestas a ocrelizumab presentaban poblaciones reducidas de linfocitos B durante la fase posnatal. Se detectaron niveles medibles de ocrelizumab en la leche materna (aproximadamente 0,2% de los niveles séricos mínimos en situación de equilibrio) durante el período de lactancia.
Indicaciones.
Ocrevus está indicado para el tratamiento de pacientes adultos con formas recurrentes de esclerosis múltiple (EMR) con enfermedad activa definida por características clínicas o de imagen (véase Características farmacológicas - Propiedades, Propiedades farmacodinámicas). Ocrevus está indicado para el tratamiento de pacientes adultos con esclerosis múltiple primaria progresiva (EMPP) temprana, en referencia a la duración de la enfermedad y al nivel de discapacidad, y presenten actividad inflamatoria en las pruebas de imagen (véase Características farmacológicas - Propiedades, Propiedades farmacodinámicas).
Dosificación.
La sustitución por cualquier otro medicamento biológico requiere el consentimiento del médico prescriptor. El tratamiento con Ocrevus debe iniciarlo y supervisarlo un médico especialista con experiencia en el diagnóstico y tratamiento de enfermedades neurológicas y con acceso a un apoyo médico adecuado para el manejo de reacciones graves, como las reacciones graves relacionadas con la infusión (RRI). Premedicación para reacciones relacionadas con la infusión (RRI): Se deben administrar las siguientes dos premedicaciones antes de cada infusión de Ocrevus para reducir la frecuencia y la gravedad de las RRI (para consultar la información acerca de los pasos adicionales para reducir las RRI véase Reacciones relacionadas con la infusión en Precauciones y advertencias): 100 mg de metilprednisolona intravenosa (o equivalente) aproximadamente 30 minutos antes de cada infusión de Ocrevus; antihistamínico aproximadamente 30 - 60 minutos antes de cada infusión de Ocrevus. Adicionalmente, se puede considerar utilizar premedicación con un antipirético (por ejemplo, paracetamol) aproximadamente 30 - 60 minutos antes de cada infusión de Ocrevus. Posología: Dosis inicial: La dosis inicial de 600 mg se administra en dos infusiones intravenosas separadas; primero una infusión de 300 mg seguida de una segunda infusión de 300 mg dos semanas más tarde (Tabla 5). Dosis posteriores: A partir de entonces, las siguientes dosis de Ocrevus se administran en dosis únicas de 600 mg en infusión intravenosa, cada 6 meses (Tabla 5). La dosis subsiguiente de 600 mg debe administrarse 6 meses después de la primera infusión de la dosis inicial. Se debe mantener un intervalo mínimo de 5 meses entre cada dosis de Ocrevus. Si los pacientes no experimentan reacciones graves relacionadas con la infusión (RRI), con ninguna infusión previa de Ocrevus, se puede administrar una infusión más corta (2 horas) en dosis posteriores (Tabla 5, Opción 2). Ajustes de la infusión en caso de RRI: En caso de RRI durante cualquier infusión, considere los siguientes ajustes. Puede encontrar información adicional sobre RRI en Precauciones y advertencias. RRI potencialmente mortales: Si durante la infusión hay signos de una RRI potencialmente mortal o incapacitante, como por ejemplo hipersensibilidad aguda o síndrome de insuficiencia respiratoria aguda, se debe finalizar la infusión inmediatamente y administrar al paciente el tratamiento adecuado. Se debe suspender Ocrevus permanentemente en esos pacientes (véase Contraindicaciones). RRI graves: Si un paciente experimenta una RRI grave (como por ejemplo disnea) o una combinación de síntomas como enrojecimiento, fiebre o dolor de garganta, se debe finalizar la infusión inmediatamente y administrar al paciente el tratamiento sintomático. Solo se debe reiniciar la infusión una vez que se hayan resuelto todos los síntomas. La infusión se debe reiniciar a la mitad de la velocidad de infusión en el momento en el que se produjo la reacción. No es necesario realizar un ajuste de la infusión en las infusiones posteriores, a menos que el paciente experimente una RRI. RRI de leves a moderadas: Si un paciente experimenta una RRI de leve a moderada (por ejemplo, dolor de cabeza), la velocidad de la infusión se debe reducir a la mitad de la velocidad en el momento en el que se produjo el acontecimiento. Esta velocidad reducida se debe mantener durante al menos 30 minutos. Si se tolera, la velocidad de la infusión se puede aumentar conforme a la velocidad inicial de infusión del paciente. No es necesario realizar un ajuste de la infusión en las infusiones posteriores, a menos que el paciente experimente una RRI. Modificaciones de la dosis durante el tratamiento: Los ejemplos anteriores de interrupción y ralentización de la dosis (para las RRI leves/moderadas y graves) ocasionarán un cambio en la velocidad de infusión y aumentarán la duración total de la misma, sin modificar la dosis total. No se recomienda reducir la dosis de Ocrevus. Retraso u olvido de dosis: Si se olvida administrar una infusión de Ocrevus, se debe administrar tan pronto como sea posible; no se debe esperar a la siguiente dosis que esté planificada. Se debe mantener el intervalo de tratamiento de 6 meses (con un mínimo de 5 meses) entre cada dosis de Ocrevus (Tabla 5). Poblaciones especiales: Población pediátrica: No se ha establecido todavía la seguridad y eficacia de Ocrevus en niños y adolescentes de 0 a 18 años. No se dispone de datos. Adultos mayores de 55 años y pacientes de edad avanzada No es necesario un ajuste de dosis en pacientes mayores de 55 años según se deduce de los datos limitados de los que se dispone (véanse Características farmacológicas - Propiedades, Propiedades farmacodinámicas y Propiedades farmacocinéticas). Los pacientes incluidos en los ensayos clínicos en marcha siguen en tratamiento con dosis de 600 mg de ocrelizumab cada seis meses después de cumplir los 55 años de edad. Pacientes con insuficiencia renal: La seguridad y la eficacia de Ocrevus en pacientes con insuficiencia renal no se han estudiado formalmente. En los ensayos clínicos se incluyeron pacientes con insuficiencia renal leve. No hay experiencia en pacientes con insuficiencia renal moderada y grave. Ocrevus es un anticuerpo monoclonal que se elimina por catabolismo (por ejemplo, rotura de péptidos y aminoácidos), por lo que no se prevé que sea necesario realizar un cambio de la dosis en pacientes con insuficiencia renal (véase Características farmacológicas - Propiedades, Propiedades farmacocinéticas). Pacientes con insuficiencia hepática: La seguridad y la eficacia de Ocrevus en pacientes con insuficiencia hepática no se han estudiado formalmente. En los ensayos clínicos se incluyeron pacientes con insuficiencia hepática leve. No hay experiencia en pacientes con insuficiencia hepática moderada y grave. Ocrevus es un anticuerpo monoclonal que se elimina por catabolismo (en lugar del metabolismo hepático), por lo que no se prevé que sea necesario realizar un cambio de dosis en pacientes con insuficiencia hepática (véase Características farmacológicas - Propiedades, Propiedades farmacocinéticas). Formas de administración: Ocrevus se administra por infusión intravenosa a través de una línea exclusiva, después de su dilución. Las infusiones de Ocrevus no se deben administrar en inyección intravenosa rápida o bolo.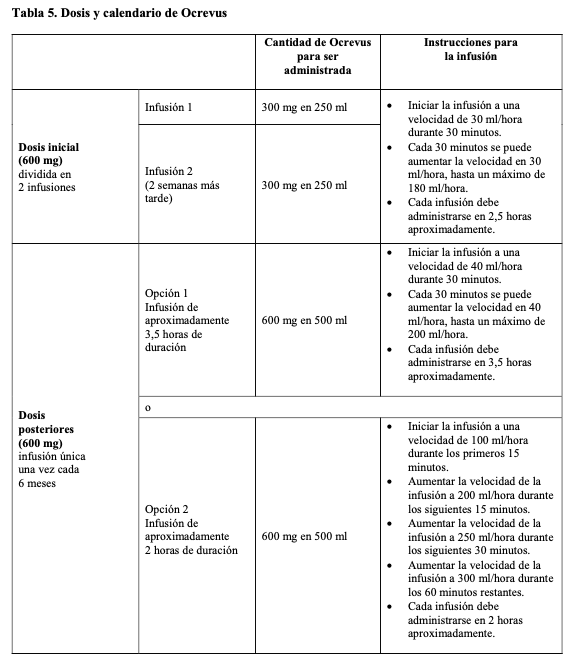
Las soluciones de Ocrevus para infusión intravenosa se preparan mediante dilución del medicamento en una bolsa de infusión que contiene cloruro sódico al 0,9%, hasta alcanzar una concentración final de aproximadamente 1,2 mg/ml. Para consultar las instrucciones de dilución del medicamento antes de la administración, véase Observaciones particulares. Se debe vigilar a los pacientes durante la infusión y durante al menos una hora tras finalizar la misma (véase Precauciones y advertencias).
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes. Infección activa presente (véase Precauciones y advertencias). Pacientes en un estado inmunocomprometido grave (véase Precauciones y advertencias). Neoplasias malignas activas conocidas (véase Precauciones y advertencias).
Reacciones adversas.
Resumen del perfil de seguridad: Las reacciones adversas (RAMs) más importantes y notificadas con mayor frecuencia fueron reacciones relacionadas con la infusión (RRI) e infecciones. Para más información véanse Precauciones y advertencias y Reacciones adversas, en Descripción de reacciones adversas seleccionadas. Tabla de reacciones adversas: El perfil se seguridad global de Ocrevus en Esclerosis Múltiple se basa en los datos de pacientes de ensayos clínicos pivotales en EM (EMR y EMPP). La Tabla 6 resume las RAMs notificadas en relación con el uso de Ocrevus en 1.311 pacientes (3.054 años- paciente) durante los períodos de tratamiento controlado de los ensayos clínicos en EM. Las frecuencias se definen como muy frecuente (≥ 1/10), frecuente (≥ 1/100 a < 1/10), poco frecuente (≥ 1/1.000 a < 1/100), rara (≥ 1/10.000 a < 1/1.000), muy rara ( < 1/10.000) y desconocida (no se puede estimar a partir de los datos disponibles). Dentro del Sistema de Clasificación de Órganos, las reacciones adversas se presentan en orden decreciente de frecuencia.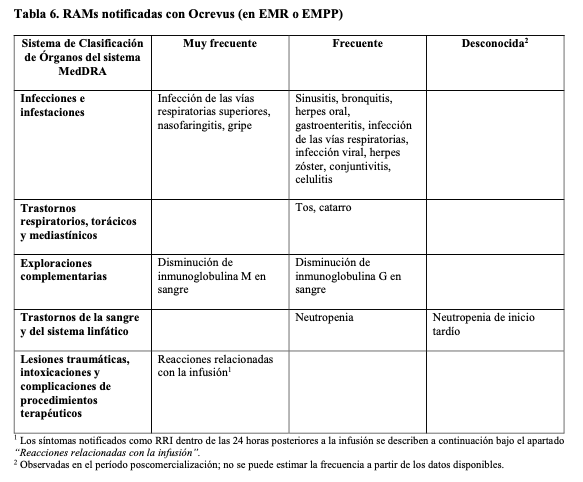
Descripción de reacciones adversas seleccionadas: Reacciones relacionadas con la infusión: En los ensayos de EMR y EMPP, los síntomas asociados con las RRI incluyeron, entre otros: prurito, erupción cutánea, urticaria, eritema, rubor, hipotensión, fiebre, fatiga, cefalea, mareo, irritación de garganta, dolor orofaríngeo, disnea, edema faríngeo o laríngeo, náuseas, taquicardia. En los ensayos controlados no hubo RRI mortales. Además, los síntomas de RRI en el entorno poscomercialización incluyeron anafilaxia. En los ensayos clínicos con control activo (EMR), la RRI fue el acontecimiento adverso más frecuente en pacientes tratados con Ocrevus, con una incidencia global del 34,3% en comparación con una incidencia del 9,9% en el grupo de tratamiento con interferón beta-1a (infusión de placebo). La incidencia de RRI fue más elevada durante la infusión 1 de la dosis 1 (27,5%) y se redujo con el paso del tiempo hasta < 10% en la dosis 4. La mayoría de las RRI en ambos grupos de tratamiento fueron de leves a moderadas. El 21,7% y el 10,1% de los pacientes tratados con Ocrevus experimentaron RRI leves y moderadas, respectivamente, el 2,4% experimentó RRI graves y el 0,1% experimentó RRI potencialmente mortales (véase Precauciones y advertencias). En el ensayo clínico controlado con placebo (EMPP), la RRI fue el acontecimiento adverso más frecuente en pacientes tratados con Ocrevus, con una incidencia global del 40,1% en comparación con una incidencia del 25,5% en el grupo de placebo. La incidencia de RRI fue más elevada durante la primera infusión de la dosis 1 (27,4%) y se redujo con dosis posteriores hasta < 10% con la dosis 4. Una mayor proporción de pacientes de cada grupo experimentó RRI con la primera infusión de cada dosis en comparación con la segunda infusión de la misma dosis. La mayoría de las RRI fueron de leves a moderadas. El 26,7% y el 11,9% de los pacientes tratados con Ocrevus experimentaron RRI leves y moderadas, respectivamente, y el 1,4% experimentó RRI graves. No hubo RRI potencialmente mortales (véase Precauciones y advertencias). Alternativa de infusión de menor duración en dosis posteriores: En un estudio (MA30143 Subestudio de infusión más corta) diseñado para caracterizar el perfil de seguridad de las infusiones de Ocrevus más corta (2 horas) en pacientes con esclerosis múltiple remitente recurrente, la incidencia, la intensidad y los tipos de síntomas de RRI fueron equivalentes a los que se observan con infusiones administradas en 3,5 horas (véase Características farmacológicas - Propiedades, Propiedades farmacodinámicas). El número total de intervenciones que fueron necesarias fue bajo en ambos grupos de infusión, sin embargo, se necesitaron más intervenciones (reducción de la velocidad de infusión o interrupciones temporales) para manejar las RRI en el grupo de infusión más corta (2 horas) en comparación con el grupo de infusión en 3,5 horas (8,7% versus 4,8%, respectivamente). Infección: En los estudios con control activo en la EMR, se produjeron infecciones en el 58,5% de los pacientes que recibían Ocrevus frente al 52,5% que recibían interferón beta-1a. Se produjeron infecciones graves en el 1,3% de los pacientes que recibían Ocrevus frente al 2,9% de los pacientes que recibían interferón beta-1a. En el estudio controlado con placebo de la EMPP, se produjeron infecciones en el 72,2% de los pacientes que recibían Ocrevus frente al 69,9% de los pacientes que recibían placebo. Se produjeron infecciones graves en el 6,2% de los pacientes que recibían Ocrevus frente al 6,7% de los pacientes que recibían placebo. Entre los años 2 y 3 de tratamiento se observó un aumento en la tasa de infecciones graves en los pacientes con EMR que no se observó en los años siguientes. No se observó un aumento en EMPP. Infección de las vías respiratorias: La proporción de infecciones de las vías respiratorias fue mayor en los pacientes tratados con Ocrevus en comparación con interferón beta-1a y placebo. En los ensayos clínicos de EMR, el 39,9% de los pacientes tratados con Ocrevus y el 33,2% de los pacientes tratados con interferón beta-1a experimentaron infección de las vías respiratorias superiores y el 7,5% de los pacientes tratados con Ocrevus y el 5,2% de los pacientes tratados con interferón beta-1a experimentaron infección de las vías respiratorias inferiores. En el ensay clínico de EMPP, el 48,8% de los pacientes tratados con Ocrevus y el 42,7% de los pacientes que recibieron placebo experimentaron infección de las vías respiratorias superiores y el 9,9% de los pacientes tratados con Ocrevus y el 9,2% de los pacientes que recibieron placebo experimentaron infección de las vías respiratorias inferiores. Las infecciones de las vías respiratorias notificadas en los pacientes tratados con Ocrevus fueron predominantemente de leves a moderadas (80 - 90%). Herpes: En los ensayos clínicos con control activo (EMR), las infecciones por herpes se notificaron con mayor frecuencia en los pacientes tratados con Ocrevus que en los pacientes tratados con interferón beta-1a, incluyendo la infección por herpes zóster (2,1% frente al 1,0%), herpes simplex (0,7% frente al 0,1%), herpes oral (3,0% frente al 2,2%), herpes genital (0,1% frente al 0%) y herpes virus (0,1% frente al 0%). Las infecciones fueron predominantemente de leves a moderadas en intensidad y los pacientes se recuperaron con tratamientos estándar. En el ensayo controlado con placebo (EMPP), se observó en el grupo de tratamiento con Ocrevus una mayor proporción de pacientes con herpes oral (2,7% frente al 0,8%). Anomalías analíticas: Inmunoglobulinas: El tratamiento con Ocrevus produjo una disminución de las imnunoglobulinas totales durante el período controlado de los estudios, motivada principalmente por la reducción de IgM. Los datos obtenidos de los ensayos clínicos han mostrado una asociación entre una disminución de los niveles de IgG (e igualmente de IgM o IgA, aunque inferior) y las infecciones graves. Linfocitos: En EMR, se observó una disminución en el valor de los linfocitos < LIN en el 20,7% de los pacientes con Ocrevus en comparación con el 32,6% de los pacientes tratados con interferón beta-1a. En EMPP, se observó una disminución de los linfocitos < LIN en el 26,3% de los pacientes tratados con Ocrevus frente al 11,7% de los pacientes tratados con placebo. En pacientes tratados con Ocrevus la mayoría de estas disminuciones notificadas fueron de gravedad de grado 1 ( < LIN - 800 células/mm3) y de grado 2 (entre 500 y 800 células/mm3). Aproximadamente el 1% de los pacientes del grupo de Ocrevus tenían linfopenia de grado 3 (entre 200 y 500 células/mm3). En ninguno de los pacientes se notificó linfopenia de grado 4 ( < 200 células/mm3). Se observó un incremento en la tasa de infecciones graves durante episodios con un descenso confirmado en el recuento de linfocitos totales en pacientes tratados con ocrelizumab. No se pudieron extraer conclusiones definitivas debido a que el número de infecciones graves fue demasiado bajo. Neutrófilos: En el período de tratamiento con control activo (EMR), se observó un descenso de los neutrófilos < LIN en el 14,7% de los pacientes tratados con Ocrevus en comparación con el 40,9% de los pacientes tratados con interferón beta-1a. En el ensayo clínico controlado con placebo (EMPP), la proporción de pacientes tratados con Ocrevus que presentaron un descenso de los neutrófilos fue superior (12,9%) que los pacientes tratados con placebo (10,0%); entre ellos, un porcentaje mayor de pacientes (4,3%) en el grupo de Ocrevus presentaron neutropenia de grado 2 o superior en comparación con el 1,3% de los pacientes en el grupo de placebo; aproximadamente el 1% de los pacientes en el grupo de Ocrevus presentaron neutropenia de grado 4 en comparación con el 0% de los pacientes en el grupo de placebo. La mayoría de los descensos de los neutrófilos fue de carácter transitorio (solamente se observó una vez en un paciente tratado con Ocrevus) y de gravedad de grado 1 (entre < LIN y 1.500 células/mm3) y de grado 2 (entre 1.000 y 1.500 células/mm3). Un paciente con neutropenia de grado 3 (entre 500 y 1.000 células/mm3) y un paciente con neutropenia de grado 4 ( < 500 células/mm3) requirieron tratamiento específico con factor estimulador de colonias de granulocitos, y permanecieron en el grupo de ocrelizumab después del episodio. Se puede producir neutropenia varios meses después de la administración de Ocrevus (véase Precauciones y advertencias). Otras: Un paciente, que recibió 2.000 mg de Ocrevus, murió de síndrome de respuesta inflamatoria sistémica (SRIS) de etiología desconocida, tras un examen por resonancia magnética (RM) 12 semanas después de la última infusión; una reacción anafiláctica al agente de contraste con gadolinio (Gd) para RM podría haber contribuido al SRIS. Comunicación de reportes de reacciones adversas Es importante comunicar las presuntas reacciones adversas después de la autorización del medicamento. Esto permite la monitorización continua de la relación riesgo/beneficio. Se solicita a los profesionales de la salud informar sobre cualquier sospecha de eventos adversos asociados con el uso de Ocrevus® al Área de Farmacovigilancia de Roche al siguiente teléfono 0800-77-ROCHE (76243) o escribiendo a argentina.safety@roche.com. En forma alternativa, esta información puede ser reportada ante ANMAT. Ante cualquier inconveniente con el producto, el paciente puede llenar la ficha que está en la Página Web de la ANMAT: https://www.argentina.gob.ar/anmat/farmacovigilancia/notificanos o llamar a ANMAT responde al 0800-333-1234"
Precauciones.
Trazabilidad: Con el fin de mejorar la trazabilidad de los medicamentos biológicos, el nombre comercial y el número de lote del producto administrado deben estar claramente registrados. Reacciones relacionadas con la infusión (RRI): Se asocia Ocrevus con RRI, las cuales pueden estar relacionadas con la liberación de citoquinas y/u otros mediadores químicos. Los síntomas de RRI pueden ocurrir durante cualquier infusión, pero se han notificado con mayor frecuencia durante la primera infusión. Las RRI pueden ocurrir dentro de las 24 horas posteriores a la infusión (véase Reacciones adversas). Estas reacciones pueden presentarse como prurito, erupción cutánea, urticaria, eritema, irritación de garganta, dolor orofaríngeo, disnea, edema faríngeo o laríngeo, enrojecimiento, hipotensión, fiebre, cansancio, dolor de cabeza, mareos, náuseas, taquicardia y anafilaxia. Antes de la infusión: Manejo de reacciones graves: Se debe disponer de los recursos necesarios para el manejo de reacciones graves como las RRI graves, reacciones de hipersensibilidad y/o reacciones anafilácticas. Hipotensión: Durante las infusiones de Ocrevus, se puede presentar hipotensión, como síntoma de RRI. Por lo tanto, se debe considerar la suspensión de los tratamientos antihipertensivos en las 12 horas previas, y durante cada infusión con Ocrevus. No se ha estudiado en pacientes con antecedentes de insuficiencia cardíaca congestiva (clases III y IV de la New York Heart Association). Premedicación: Los pacientes deben recibir premedicación para reducir la frecuencia y la gravedad de las RRI (véase Posología y formas de administración). Durante la infusión: Se deben tomar las siguientes medidas en aquellos pacientes que experimenten síntomas respiratorios graves, tales como broncoespasmo o exacerbación del asma: se debe interrumpir la infusión de forma inmediata y permanente; se debe administrar tratamiento sintomático; se debe monitorizar al paciente hasta que los síntomas respiratorios se hayan resuelto, ya que la mejora inicial de los síntomas clínicos podría estar seguida de un deterioro. Puede haber dificultad para distinguir la hipersensibilidad de una RRI de acuerdo a los síntomas. Si se sospecha que durante la infusión se produce una reacción de hipersensibilidad, la infusión debe interrumpirse de forma inmediata y permanente (véase a continuación Reacciones de hipersensibilidad). Después de la infusión: Se debe vigilar a los pacientes tratados con Ocrevus durante al menos una hora tras finalizar la infusión, por si apareciera cualquier síntoma de RRI. Los médicos deben alertar a los pacientes de que las RRI pueden ocurrir dentro de las 24 horas posteriores a la infusión. Para más información