NULOJIX®
BRISTOL-M.S.
Agente inmunosupresor selectivo.
Advertencia: Trastorno linfoproliferativo postrasplante, otras malignidades e infecciones serias: Mayor riesgo de desarrollar trastorno linfoproliferativo postrasplante (PTLD), que afecta principalmente el sistema nervioso central (SNC). En particular, los receptores sin inmunidad al virus de Epstein-Barr (EBV) corren mayor riesgo; por lo tanto, usar únicamente en pacientes EBV seropositivos. No usar NULOJIX en receptores de trasplante EBV seronegativos o cuya serología se desconozca [véase Contraindicaciones y Advertencias]. Sólo deben recetar NULOJIX médicos con experiencia en el tratamiento inmunosupresivo y el manejo de pacientes con trasplante renal. Los pacientes que reciban el fármaco deben recibir atención en instalaciones equipadas y con el personal necesario, y con los recursos de laboratorio y el respaldo médico adecuados. El médico responsable del tratamiento de mantenimiento debe contar con toda la información necesaria acerca del paciente para el seguimiento [véase Advertencias]. La inmunosupresión puede causar mayor susceptibilidad a las infecciones y el posible desarrollo de neoplasias malignas [véase Advertencias]. No se recomienda el uso en pacientes con trasplante hepático debido a un mayor riesgo de pérdida del injerto y muerte [véase Advertencias].
Descripción.
NULOJIX® (belatacept), un bloqueante selectivo de la coestimulación de células T, es una proteína de fusión soluble que consiste en el dominio extracelular modificado del CTLA-4 unido a una porción (dominios de bisagra-CH2-CH3) del dominio Fc de un anticuerpo inmunoglobulina G1 humano. Belatacept se produce por medio de tecnología de ADN recombinante en un sistema de expresión celular de mamífero. Se realizaron dos sustituciones de aminoácidos (L104 a E; A29 a Y) en la región de unión al ligando del CTLA-4. Como resultado de estas modificaciones, belatacept se une a CD80 y CD86 con más avidez que abatacept, la molécula original de CTLA4-inmunoglobulina (CTLA4-Ig) de la cual se deriva. El peso molecular de belatacept es aproximadamente 90 kilodaltones. NULOJIX se presenta como polvo liofilizado estéril, entre blanco y blanquecino, para administración intravenosa. Antes de usar, el liófilo se reconstituye con un líquido adecuado para obtener una solución de transparente a levemente opalescente, entre incolora y de color amarillo pálido, con un pH en el rango de 7,2 a 7,8. Los líquidos adecuados para la reconstitución del liófilo incluyen agua estéril para uso inyectable (SWFI), cloruro de sodio al 0,9% (NS) o dextrosa en agua al 5% (D5W) [véase Dosificación]. Cada vial para uso único de 250 mg de NULOJIX contiene además fosfato de sodio monobásico (34,5 mg), cloruro de sodio (5,8 mg) y sacarosa (500 mg).
Composición.
Cada vial de dosis única de NULOJIX contiene: Belatacept 250 mg. Excipientes: sacarosa 500 mg, fosfato sódico monobásico 34,5 mg, cloruro de sodio 5,8 mg. Hidróxido de sodio 1N / Acido clorhídrico 1N c.s.p. pH 7,5 para el ajuste del pH.
Estudios clínicos.
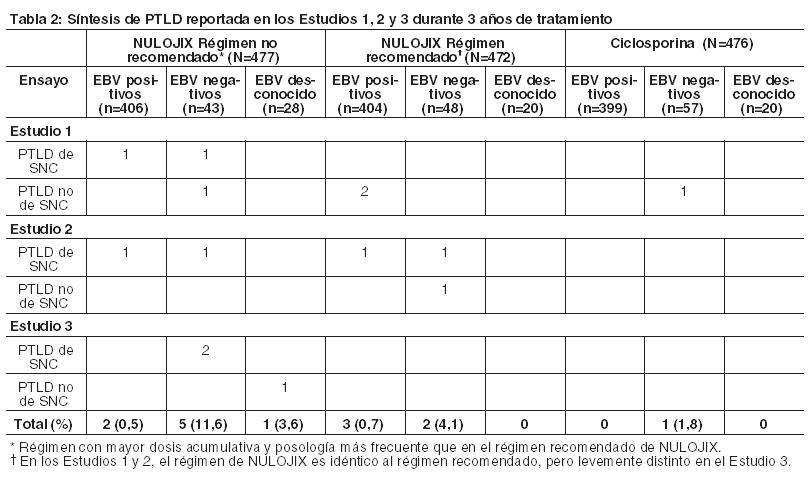
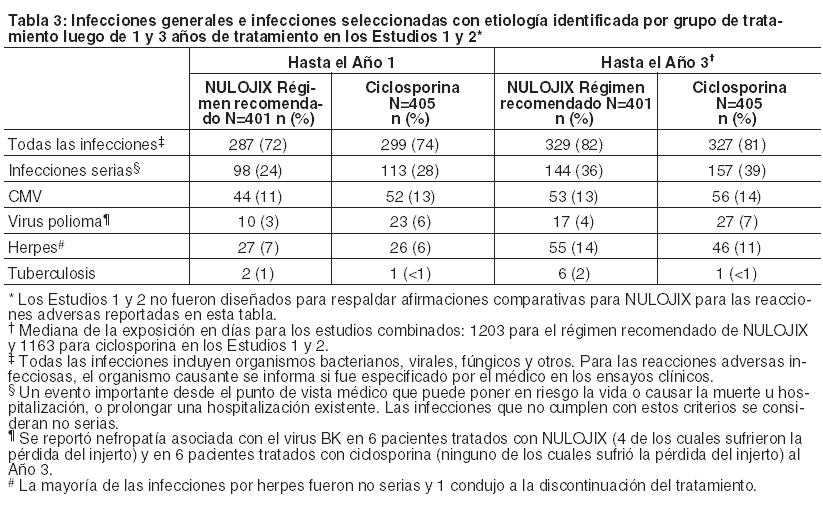
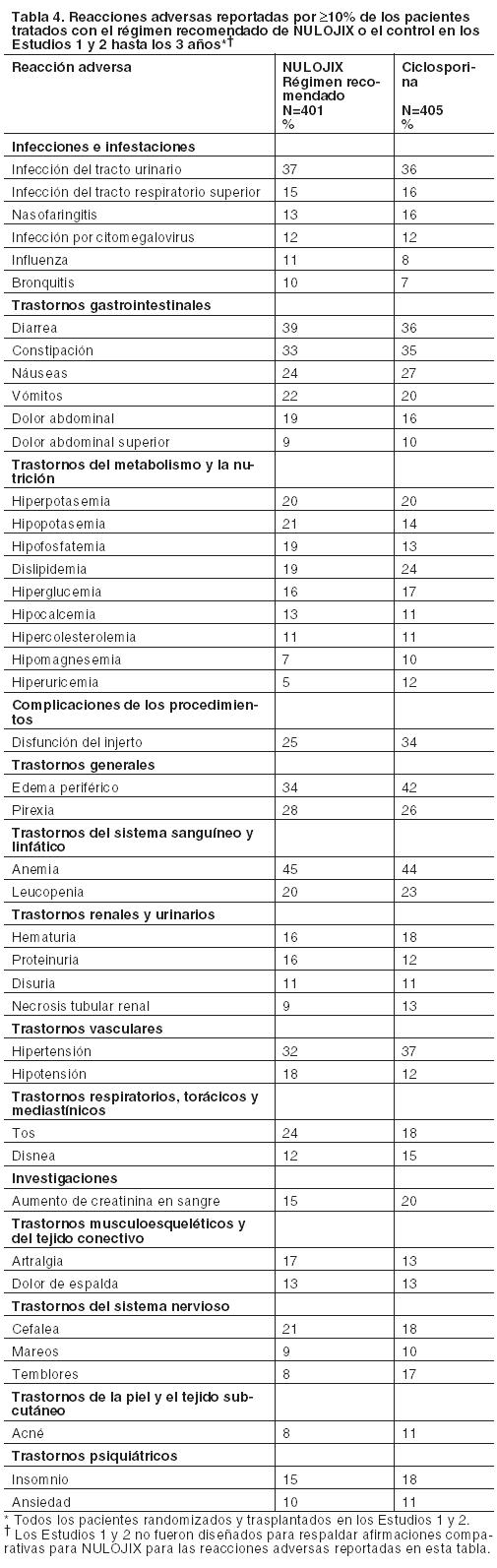
Prevención del rechazo de órgano en receptores de trasplante renal: La eficacia y la seguridad de NULOJIX en el trasplante renal de novo se evaluaron en dos estudios abiertos, randomizados, multicéntricos, con control activo (Estudio 1 y Estudio 2). Estos estudios evaluaron dos regímenes posológicos de NULOJIX: el régimen posológico recomendado [véase Dosificación] y un régimen con mayores dosis acumulativas y posología más frecuente que las recomendadas, en comparación con un régimen de control con ciclosporina. Todos los grupos de tratamiento también recibieron inducción con basiliximab, micofenolato mofetilo (MMF) y corticosteroides. Régimen de tratamiento: El régimen recomendado de NULOJIX consistió en una dosis de 10 mg por kilogramo administrada el Día 1 (el día del trasplante, antes del implante), el Día 5 (aproximadamente 96 horas después de la dosis del Día 1), al final de las Semanas 2 y 4; luego cada 4 semanas hasta la Semana 12 después del trasplante. Comenzando en la Semana 16 luego del trasplante, se administró NULOJIX en la dosis de mantenimiento de 5 mg por kg cada 4 semanas (más/menos 3 días). NULOJIX se administró en forma de infusión intravenosa durante 30 minutos [véase Dosificación]. Basiliximab 20 mg se administró por vía intravenosa el día del trasplante y 4 días después. La dosis inicial de MMF fue de 1 gramo dos veces por día y se ajustó, según fuera necesario, en función de los signos clínicos de eventos adversos o falla de eficacia. La dosificación de corticosteroides especificada en el protocolo en los Estudios 1 y 2 el Día 1 fue metilprednisolona (como succinato de sodio) 500 mg IV al ingresar en el quirófano, el Día 2, metilprednisolona 250 mg IV, y el Día 3, prednisona 100 mg por vía oral. Las medianas reales de las dosis de corticosteroides usadas con el régimen recomendado de NULOJIX desde la Semana 1 hasta el Mes 6 se sintetizan en la tabla a continuación (Tabla 6).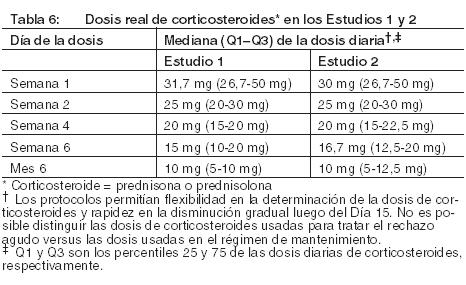
El Estudio 1 enroló receptores de órganos de donantes vivos y de donantes cadavéricos según los criterios estándar, y el Estudio 2 enroló receptores de órganos de donantes según criterios expandidos. Los órganos de donantes según criterios estándar se definieron como órganos de un donante cadavérico con isquemia fría anticipada durante < 24 horas y que no cumplían con la definición de órganos de donante según criterios expandidos. Los donantes según criterios expandidos se definieron como donantes cadavéricos con al menos uno de los siguientes factores: (1) donante de ≥ 60 años de edad; (2) donante de ≥ 50 años de edad y otras comorbilidades (≥ 2 de las siguientes: accidente cerebrovascular, hipertensión, creatinina sérica > 1,5 mg/dl); (3) donación de órgano después de la muerte cardíaca; o isquemia fría anticipada del órgano ≥ 24 horas. El Estudio 1 excluyó a los receptores con un primer trasplante cuyo valor actual de Panel de Anticuerpos Reactivos (PRA) era ≥50% y a los receptores de un nuevo trasplante cuyo valor actual de PRA era ≥30%; el Estudio 2 excluyó a receptores con un valor de PRA actual ≥ 30%. Ambos estudios excluyeron a receptores con VIH, hepatitis C o evidencia de infección actual por hepatitis B; receptores con tuberculosis activa; y receptores en quienes era difícil lograr acceso intravenoso. Se presentan datos de eficacia para el régimen recomendado de NULOJIX y el régimen de ciclosporina en los Estudios 1 y 2. El régimen de NULOJIX con mayores dosis acumulativas y dosificación más frecuente de belatacept se asoció con más fracasos en materia de eficacia. No se recomiendan mayores dosis ni una dosificación más frecuente de NULOJIX [véase Dosificación, Advertencias, y Reacciones Adversas]. Estudio 1: Receptores de riñones de donantes vivos y cadavéricos según criterios estándar: En el Estudio 1, 666 pacientes fueron enrolados, randomizados y trasplantados: 226 al régimen recomendado de NULOJIX, 219 al régimen de NULOJIX con mayores dosis acumulativas y dosificación más frecuente que las recomendadas, y 221 al régimen de control con ciclosporina. La mediana de la edad fue de 45 años; el 58% de los órganos provinieron de donantes vivos; el 3% fueron retrasplantados; el 69% de la población del estudio era de sexo masculino; el 61% de los pacientes eran caucásicos, el 8% eran negros/afroamericanos, y el 31% se categorizaron como pertenecientes a otras razas; el 16% tenía un valor de PRA ≥ 10%; el 41% tenía de 4 a 6 desajustes de HLA; el 27% tenía diabetes antes del trasplante. La incidencia de retraso en el funcionamiento del injerto fue similar en todas las ramas de tratamiento (14%-18%). Se produjo la discontinuación prematura del tratamiento al final del primer año en el 19% de los pacientes que recibieron el régimen recomendado de NULOJIX y en el 19% de los pacientes que recibieron el régimen de ciclosporina. Entre los pacientes que recibieron el régimen recomendado de NULOJIX, el 10% discontinuó el tratamiento debido a falta de eficacia, el 5% debido a eventos adversos y el 4% por otros motivos. Entre los pacientes que recibieron el régimen de ciclosporina, el 9% discontinuó el tratamiento debido a eventos adversos, el 5% debido a falta de eficacia y el 5% por otros motivos. Al final de los tres años, el 25% de los pacientes que recibieron el régimen recomendado de NULOJIX y el 34% de los pacientes que recibieron el régimen de ciclosporina habían discontinuado el tratamiento. Entre los pacientes que recibieron el régimen recomendado de NULOJIX, el 12% discontinuó debido a falta de eficacia, el 7% debido a eventos adversos y el 6% por otros motivos. Entre los pacientes que recibieron el régimen de ciclosporina, el 15% discontinuó debido a eventos adversos, el 8% debido a falta de eficacia y el 11% por otros motivos. Evaluación de eficacia: La Tabla 7 sintetiza los resultados del Estudio 1 luego de 1 y 3 años de tratamiento con el régimen posológico recomendado de NULOJIX y el régimen de control con ciclosporina. El fracaso de la eficacia al año se definió como la ocurrencia de rechazo agudo probado por biopsia (BPAR), pérdida del injerto, muerte o pérdida durante el seguimiento. El BPAR se definió como un rechazo agudo confirmado histológicamente por un patólogo central en una biopsia realizada por cualquier razón, esté o no acompañada por signos clínicos de rechazo. La sobrevida del paciente y del injerto también se evaluaron por separado.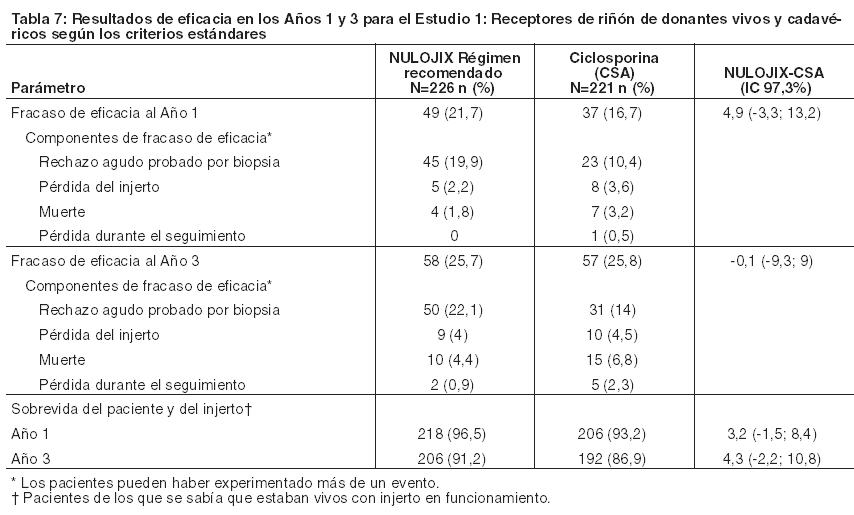
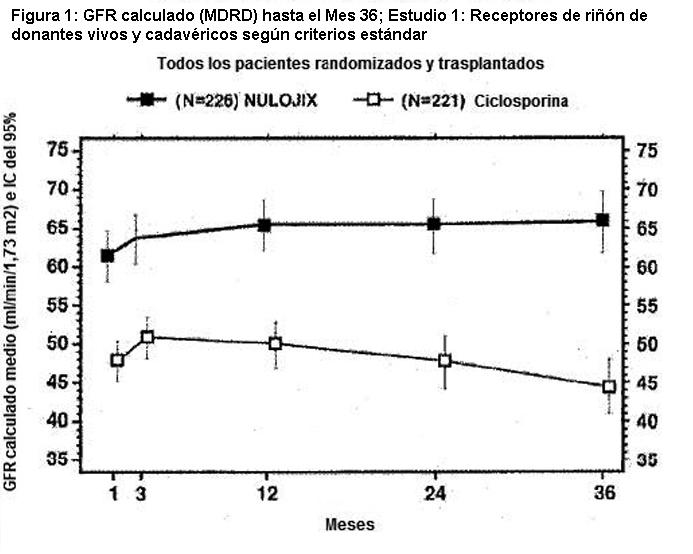
En el Estudio 1, el índice de BPAR al año y a los 3 años fue mayor en pacientes tratados con el régimen recomendado de NULOJIX que en aquellos tratados con el régimen de ciclosporina. De los pacientes que experimentaron BPAR con NULOJIX, el 70% lo experimentaron al Mes 3, y el 84% lo experimentaron al Mes 6. A los 3 años, se produjo BPAR recurrente con una frecuencia similar entre los grupos de tratamiento ( < 3%). El componente de BPAR determinado por biopsia solamente (rechazo agudo subclínico definido por el protocolo) fue del 5% en ambos grupos de tratamiento. Los pacientes tratados con el régimen recomendado de NULOJIX experimentaron episodios de BPAR clasificados como Banff grado lIb o mayor (6% [14/226] tras 1 año y 7% [15/226] tras 3 años) más frecuentemente que los pacientes tratados con el régimen de ciclosporina (2% [4/221] tras 1 año y 2% [5/221] tras 3 años). Además, la terapia de depleción de células T se usó con mayor frecuencia para tratar episodios de BPAR en los pacientes tratados con NULOJIX (10%; 23/226) en comparación con los pacientes tratados con ciclosporina (2%; 5/221). En el Mes 12, la diferencia en el índice de filtración glomerular (GFR) calculado medio entre pacientes con y sin antecedentes de BPAR fue 19 mL/min/l,73 m2 entre pacientes tratados con NULOJIX en comparación con 7 mL/min/1,73 m2 entre pacientes tratados con ciclosporina. A los 3 años, el 22% (11/50) de los pacientes tratados con NULOJIX que tenían antecedentes de BPAR experimentaron pérdida del injerto y/o muerte en comparación con el 10% (3/31) de los pacientes tratados con ciclosporina que presentaban antecedentes de BPAR; en este punto temporal, el 10% (5/50) de los pacientes tratados con NULOJIX experimentaron pérdida del injerto y el 12% (6/50) de los pacientes tratados con NULOJIX habían muerto luego de un episodio de BPAR, mientras que el 7% (2/31) de los pacientes tratados con ciclosporina experimentaron pérdida del injerto y el 7% (2/31) de los pacientes tratados con ciclosporina habían muerto luego de un episodio de BPAR. La prevalencia general de anticuerpos específicos del donante fue del 5% y el 11% para el régimen recomendado de NULOJIX y para ciclosporina, respectivamente, hasta 36 meses luego del trasplante. Aunque la diferencia en el GFR en los pacientes con BPAR versus aquellos sin BPAR fue mayor en los pacientes tratados con NULOJIX que en aquellos tratados con ciclosporina, el GFR medio luego de BPAR fue similar entre los individuos tratados con NULOJIX (49 mL/min/1,73 m2) y aquellos tratados con ciclosporina (43 mL/min/1,73 m2) al cabo de 1 año. La relación entre BPAR, GFR, y la sobrevida de paciente e injerto es incierta debido a la limitada cantidad de pacientes que experimentaron BPAR, a las diferencias en la hemodinamia renal (y, en consecuencia, en el GFR) entre los regímenes inmunosupresores de mantenimiento, y el alto índice de regímenes terapéuticos de cambio luego de BPAR. Evaluación de la eficacia en la subpoblación EBV seropositive: NULOJIX se recomienda para usar únicamente en pacientes EBV seropositivos [véase Indicaciones y Uso]. En el Estudio 1, aproximadamente el 87% de los pacientes eran EBV seropositivos antes del trasplante. Los resultados de eficacia en la subpoblación EBV seropositiva fueron consistentes con aquellos en la población total estudiada. Al cabo de 1 año, el índice de fracaso en cuanto a eficacia en la población EBV seropositiva era del 21% (42/202) en pacientes tratados con el régimen recomendado de NULOJIX y del 17% (31/184) en pacientes tratados con ciclosporina (diferencia=4%, IC del 97,3% [-4,8; 12,8]). La sobrevida del paciente y del injerto fue del 98% (198/202) en los pacientes tratados con NULOJIX y del 92% (170/184) en los pacientes tratados con ciclosporina (diferencia=5,6%, IC del 97,3% [0,8; 10,4]). A los 3 años, el fracaso en materia de eficacia fue del 25% en ambos grupos de tratamiento, y la sobrevida del paciente y del injerto fue del 94% (187/202) en los pacientes tratados con NULOJIX en comparación con el 88% (162/184) en los pacientes tratados con ciclosporina (diferencia=4,6%, IC del 97,3% [-2,1; 11,3]). Evaluación del índice de filtración glomerular (GFR): El índice de filtración glomerular (GFR) se midió tras 1 y 2 años, y se calculó usando la fórmula de la Modificación de la Dieta en la Enfermedad Renal (MDRD) tras 1, 2 y 3 años luego del trasplante. Como se muestra en la Tabla 8, tanto el GFR medido como el calculado fue mayor en los pacientes tratados con el régimen recomendado de NULOJIX en comparación con los pacientes tratados con el régimen de control con ciclosporina en todos los puntos temporales. Como se muestra en la Figura 1, las diferencias en el GFR fueron evidentes en el primer mes luego del trasplante y se mantuvieron hasta 3 años (36 meses). Un análisis del cambio en el GFR calculado medio entre los 3 y los 36 meses demostró un incremento de 0,8 mL/min/año (IC del 95% [-0,2; 1,8]) para los pacientes tratados con NULOJIX y una disminución de 2,2 mL/min/año (IC del 95% [-3,2; -1,2]) para los pacientes tratados con ciclosporina.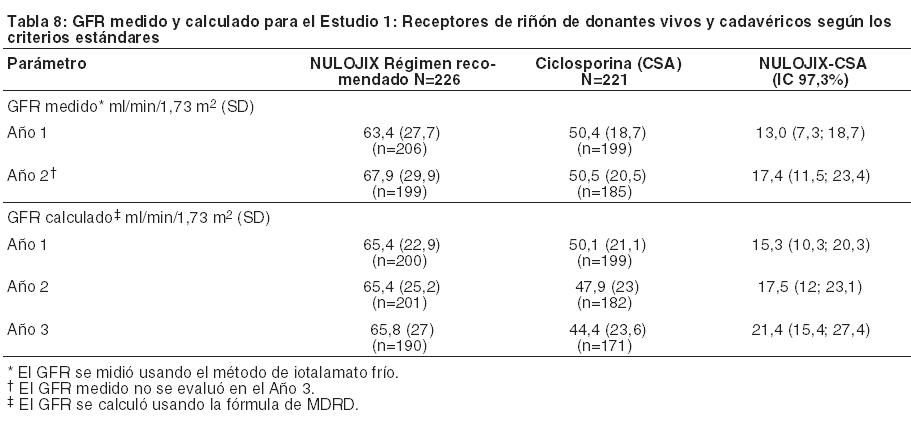
Evaluación de nefropatía crónica del aloinjerto (CAN): La prevalencia de nefropatía crónica del aloinjerto (CAN) al cabo de 1 año, según se define por el sistema de clasificación Banff '97, fue del 24% (54/226) en pacientes tratados con el régimen recomendado de NULOJIX y del 32% (71/219) en pacientes tratados con el régimen de control con ciclosporina. No se evaluó CAN luego del primer año tras el trasplante. Se desconoce la significación clínica de este hallazgo. Estudio 2: Receptores de riñón de donantes según criterios expandidos: En el Estudio 2, 543 pacientes fueron enrolados, randomizados y trasplantados: 175 al régimen recomendado de NULOJIX, 184 al régimen de NULOJIX con mayores dosis acumulativas y dosificación más frecuente que las recomendadas, y 184 al régimen de control con ciclosporina. La mediana de la edad fue de 58 años; el 67% de la población del estudio era de sexo masculino; el 75% de los pacientes eran blancos, el 13% eran negros/afroamericanos, el 12% fueron categorizados como de otras razas; el 3% tenía un valor de PRA ≥ 10%; el 53% tenía de 4 a 6 desajustes de HLA; y el 29% tenía diabetes antes del trasplante. La incidencia de retraso en el funcionamiento del injerto fue similar en todas las ramas de tratamiento (47% a 49%). La discontinuación prematura del tratamiento al final del primer año se produjo en el 25% de los pacientes que recibieron el régimen recomendado de NULOJIX y en el 30% de los pacientes que recibieron el régimen de control con ciclosporina. Entre los pacientes que recibieron el régimen recomendado de NULOJIX, el 14% discontinuaron el tratamiento debido a eventos adversos, el 9% debido a falta de eficacia y el 2% por otros motivos. Entre los pacientes que recibieron el régimen de ciclosporina, el 17% discontinuaron el tratamiento debido a eventos adversos, el 7% debido a falta de eficacia y el 6% por otros motivos. Al final de los 3 años, el 35% de los pacientes que recibieron el régimen recomendado de NULOJIX y el 44% de los pacientes que recibieron el régimen de ciclosporina habían discontinuado el tratamiento. Entre los pacientes que recibieron el régimen recomendado de NULOJIX, el 20% discontinuaron el tratamiento debido a eventos adversos, el 9% debido a falta de eficacia y el 6% por otros motivos. Entre los pacientes que recibieron el régimen de ciclosporina, el 25% discontinuaron el tratamiento debido a eventos adversos, el 10% debido a falta de eficacia y el 10% por otros motivos. Evaluación de eficacia: La Tabla 9 sintetiza los resultados del Estudio 2 luego de 1 y 3 años de tratamiento con el régimen posológico recomendado de NULOJIX y el régimen de control con ciclosporina. El fracaso de la eficacia al año se definió como la ocurrencia de rechazo agudo probado por biopsia (BPAR), pérdida del injerto, muerte o pérdida durante el seguimiento. El BPAR se definió como un rechazo agudo confirmado histológicamente por un patólogo central en una biopsia realizada por cualquier razón, esté o no acompañada por signos clínicos de rechazo. La sobrevida del paciente y del injerto también se evaluaron.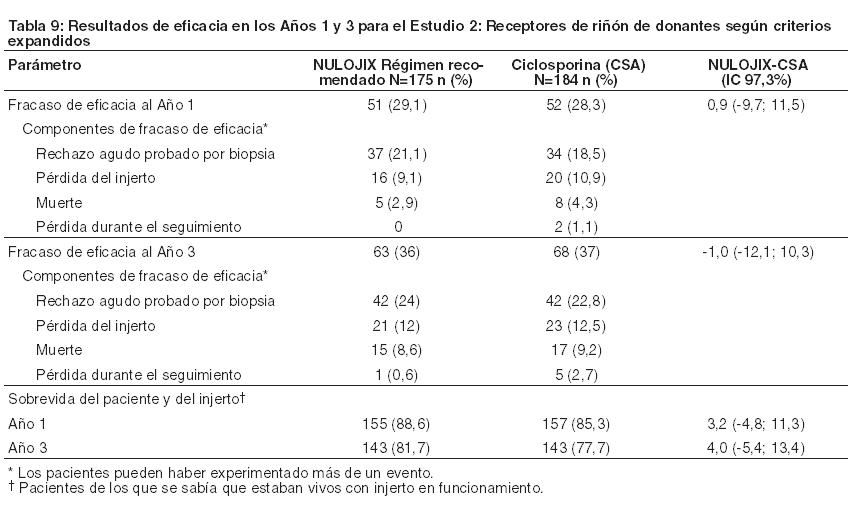
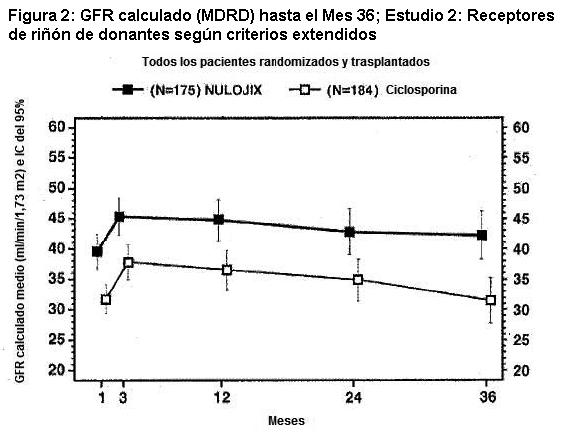
En el Estudio 2, el índice de BPAR al año y a los 3 años fue similar en pacientes tratados con NULOJIX y con ciclosporina. De los pacientes que experimentaron BPAR con NULOJIX, el 62% lo experimentaron al Mes 3, y el 76% lo experimentaron al Mes 6. A los 3 años, se produjo BPAR recurrente con una frecuencia similar entre los grupos de tratamiento ( < 3%). El componente de BPAR determinado por biopsia solamente (rechazo agudo subclínico definido por el protocolo) fue del 5% en ambos grupos de tratamiento. Una proporción similar de pacientes tratados con el régimen recomendado de NULOJIX experimentaron episodios de BPAR clasificados como Banff grado lIb o mayor (5% [9/175] tras 1 año y 6% [10/175] tras 3 años) en comparación con los pacientes tratados con el régimen de ciclosporina (4% [7/184] tras 1 año y 5% [9/184] tras 3 años). Además, la terapia de depleción de células T se usó con frecuencia similar para tratar cualquier episodio de BPAR en los pacientes tratados con NULOJIX (5%; 9/175) en comparación con los pacientes tratados con ciclosporina (4%; 7/184). En el Mes 12, la diferencia en el GFR calculado medio entre pacientes con y sin antecedentes de BPAR fue 10 mL/min/l,73 m2 entre pacientes tratados con NULOJIX en comparación con 14 mL/min/1,73 m2 entre pacientes tratados con ciclosporina. A los 3 años, el 24% (10/42) de los pacientes tratados con NULOJIX que tenían antecedentes de BPAR experimentaron pérdida del injerto y/o muerte en comparación con el 31% (13/42) de los pacientes tratados con ciclosporina que presentaban antecedentes de BPAR; en este punto temporal, el 17% (7/42) de los pacientes tratados con NULOJIX experimentaron pérdida del injerto y el 14% (6/42) de los pacientes tratados con NULOJIX habían muerto luego de un episodio de BPAR, mientras que el 19% (8/42) de los pacientes tratados con ciclosporina experimentaron pérdida del injerto y el 19% (8/42) de los pacientes tratados con ciclosporina habían muerto luego de un episodio de BPAR. La prevalencia general de anticuerpos específicos del donante fue del 6% y el 15% para el régimen recomendado de NULOJIX y para ciclosporina, respectivamente, hasta 36 meses luego del trasplante. El GFR medio luego de BPAR fue de 36 mL/min/1,73 m2 en los individuos tratados con NULOJIX y de 24 mL/min/1,73 m2 en aquellos tratados con ciclosporina al cabo de 1 año. La relación entre BPAR, GFR, y la sobrevida de paciente e injerto es incierta debido a la limitada cantidad de pacientes que experimentaron BPAR, a las diferencias en la hemodinamia renal (y, en consecuencia, en el GFR) entre los regímenes inmunosupresores de mantenimiento, y el alto índice de regímenes terapéuticos de cambio luego de BPAR. Evaluación de la eficacia en la subpoblación EBV seropositiva: NULOJIX se recomienda para usar únicamente en pacientes EBV seropositivos [véase Indicaciones]. En el Estudio 2, aproximadamente el 91% de los pacientes eran EBV seropositivos antes del trasplante. Los resultados de eficacia en la subpoblación EBV seropositiva fueron consistentes con aquellos en la población total estudiada. Al cabo de 1 año, el índice de fracaso en cuanto a eficacia en la población EBV seropositiva era del 29% (45/156) en pacientes tratados con el régimen recomendado de NULOJIX y del 28% (47/168) en pacientes tratados con ciclosporina (diferencia=0,8%, IC del 97,3% [-10,3; 11,9]). El índice de sobrevida del paciente y del injerto en la población EBV seropositiva fue del 89% (139/156) en los pacientes tratados con NULOJIX y del 86% (144/168) en los pacientes tratados con ciclosporina (diferencia=3,4%, IC del 97,3% [-4,7; 11,5]). A los 3 años, el fracaso en materia de eficacia fue del 35% (54/156) en los pacientes tratados con NULOJIX, y del 36% (61/168) en los pacientes tratados con ciclosporina. La sobrevida del paciente y del injerto fue del 83% (130/156) en los pacientes tratados con NULOJIX en comparación con el 77% (130/168) en los pacientes tratados con ciclosporina (diferencia=5,9%, IC del 97,3% [-3,8; 15,6]). Evaluación del índice de filtración glomerular (GFR): El índice de filtración glomerular (GFR) se midió tras 1 y 2 años, y se calculó usando la fórmula de la Modificación de la Dieta en la Enfermedad Renal (MDRD) tras 1, 2 y 3 años luego del trasplante. Como se muestra en la Tabla 10, tanto el GFR medido como el calculado fue mayor en los pacientes tratados con el régimen recomendado de NULOJIX en comparación con los pacientes tratados con el régimen de control con ciclosporina en todos los puntos temporales. Como se muestra en la Figura 2, las diferencias en el GFR fueron evidentes en el primer mes luego del trasplante y se mantuvieron hasta 3 años (36 meses). Un análisis del cambio en el GFR calculado medio entre los 3 y los 36 meses demostró una disminución de 0,8 mL/min/año (IC del 95% [-1,9; 0,3]) para los pacientes tratados con NULOJIX y una disminución de 2,0 mL/min/año (IC del 95% [-3,1; -0,8]) para los pacientes tratados con ciclosporina.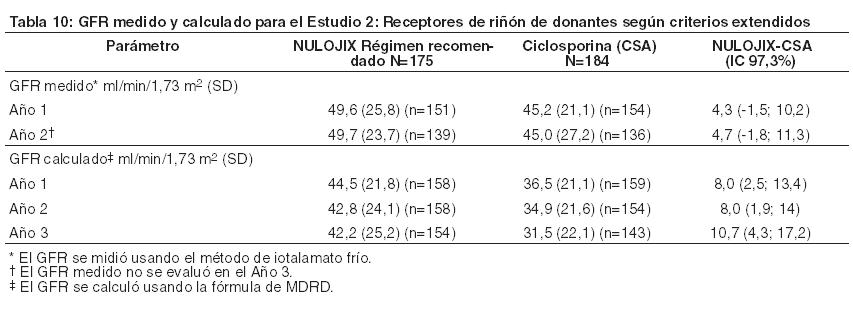
Evaluación de nefropatía crónica del aloinjerto (CAN): La prevalencia de nefropatía crónica del aloinjerto (CAN) al cabo de 1 año, según se define por el sistema de clasificación Banff '97, fue del 46% (80/174) en pacientes tratados con el régimen recomendado de NULOJIX y del 52% (95/184) en pacientes tratados con el régimen de control con ciclosporina. No se evaluó CAN luego del primer año tras el trasplante. Se desconoce la significación clínica de este hallazgo.
Farmacología.
Mecanismo de acción: Belatacept, un bloqueante selectivo de la coestimulación de células T (linfocitos), se une a CD80 y CD86 en las células presentadoras de antígenos, con lo cual bloquea la coestimulación mediada por CD28 de los linfocitos T. In vitro, belatacept inhibe la proliferación de linfocitos T y la producción de las citoquinas interleucina 2, interferón, interleucina 4 y TNF. Los linfocitos T activados son los mediadores predominantes del rechazo inmunológico. En modelos de primates no humanos de trasplante renal, belatacept como monoterapia prolongó la sobrevida del injerto y disminuyó la producción de anticuerpos anti-donante en comparación con el vehículo. Farmacodinámica: El bloqueo de la coestimulación mediada por belatacept da como resultado la inhibición de la producción de citoquinas por parte de las células T requerida para la producción de anticuerpos antígeno-específicos por parte de las células B. En los ensayos clínicos, se observaron mayores reducciones de la inmunoglobulina media (IgG, IgM e IgA) desde la condición basal hasta el Mes 6 y el Mes 12 postrasplante en los pacientes tratados con belatacept en comparación con los pacientes tratados con ciclosporina. En un análisis exploratorio de subconjuntos, se observó una tendencia hacia concentraciones decrecientes de IgG con mayores concentraciones mínimas de belatacept en el Mes 6. Además, en este análisis exploratorio de subconjuntos, entre los pacientes tratados con belatacept con PTLD del SNC, infecciones del SNC incluida PML, otras infecciones serias y malignidades, se observó una mayor incidencia de concentraciones de IgG por debajo del límite inferior del rango normal ( < 694 mg/dl) en el Mes 6 que en aquellos pacientes que no experimentaron estos eventos adversos. Esta observación fue más pronunciada con la dosis de belatacept mayor a la recomendada. También se observó una tendencia similar para los pacientes tratados con ciclosporina con infecciones serias y malignidades. Sin embargo, no se sabe con certeza si existe alguna relación causal entre una concentración de IgG por debajo del nivel inferior del rango normal y estos eventos adversos, ya que el análisis puede haber estado viciado por otros factores (por ejemplo, una edad superior a los 60 años, recepción de un riñón donado según criterios expandidos, exposición a agentes de depleción de linfocitos), los cuales también se asociaron con un nivel de IgG por debajo del límite inferior del rango normal en el Mes 6 en estos ensayos. Farmacocinética: La Tabla 5 resume los parámetros farmacocinéticos de belatacept en adultos sanos después de una infusión intravenosa única de 10 mg/kg, y en receptores de trasplante renal después de una infusión intravenosa de 10 mg/kg en la Semana 12 y después de una infusión intravenosa de 5 mg/kg cada 4 semanas en el Mes 12 postrasplante o más adelante.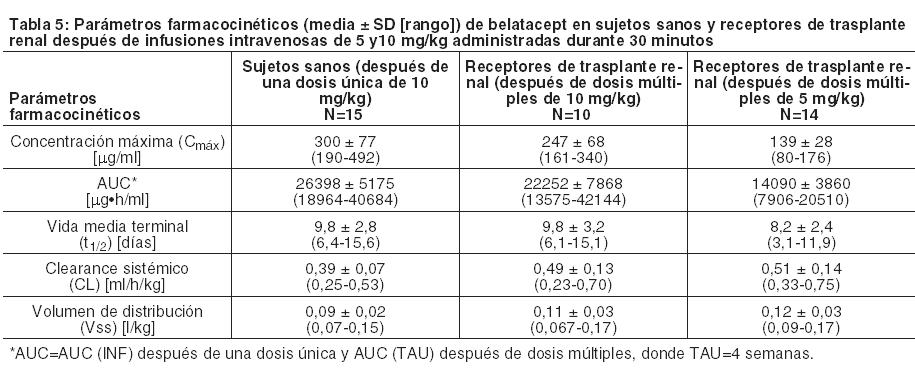
En individuos sanos, la farmacocinética de belatacept fue lineal, y la exposición al belatacept aumentó en forma proporcional después de una dosis única por infusión intravenosa de 1 a 20 mg/kg. La farmacocinética de belatacept en pacientes con trasplante renal de novo y sujetos sanos es comparable. Luego del régimen de dosis recomendado, la concentración sérica media de belatacept alcanzó el estado estacionario para la Semana 8 en la fase inicial después del trasplante, y para el Mes 6 durante la fase de mantenimiento. Luego de una infusión intravenosa mensual de 10 mg/kg y de 5 mg/kg, hubo una acumulación sistémica de aproximadamente 20% y 10% de belatacept en receptores de trasplante renal, respectivamente. Sobre la base del análisis farmacocinético de población de 924 pacientes con trasplante renal hasta 1 año después del trasplante, la farmacocinética de belatacept fue similar en diferentes períodos de tiempo posteriores al trasplante. En los ensayos clínicos, la concentración mínima de belatacept se mantuvo consistentemente desde el Mes 6 hasta los 3 años después del trasplante. Los análisis farmacocinéticos de población en pacientes con trasplante renal mostraron una tendencia hacia un mayor clearance de belatacept al aumentar el peso corporal. La edad, el sexo, la raza, la función renal (medida por el índice de filtración glomerular [GFR] calculado), la función hepática (medida en función de la albúmina), la diabetes y la diálisis concomitante no afectaron el clearance de belatacept. Interacciones medicamentosas: Micofenolato mofetilo: En un subestudio farmacocinético de los Estudios 1 y 2, se midieron las concentraciones plasmáticas de MPA en 41 pacientes que recibieron dosis fijas de MMF de 500 a 1500 mg dos veces por día con 5 mg/kg de NULOJIX o ciclosporina. Los valores medios de Cmáx y AUC0-12 de MPA normalizados por dosis fueron aproximadamente 20% y 40% mayores, respectivamente, con la coadministración de NULOJIX que con la coadministración de ciclosporina [véase Interacciones medicamentosas]. Sustratos del citocromo P450: El potencial de NULOJIX para alterar las concentraciones sistémicas de fármacos que son sustratos de CYP450 fue investigado en individuos sanos luego de la administración de un cóctel de fármacos de prueba administrados en forma concomitante y luego de 3 y 7 días de una dosis intravenosa única de 10 mg/kg de NULOJIX. NULOJIX no alteró la farmacocinética de fármacos que son sustratos de CYP1A2 (cafeína), CYP2C9 (losartán), CYP2D6 (dextrometorfán), CYP3A (midazolam) y CYP2C19 (omeprazol) [véase Interacciones medicamentosas].
Toxicología.
Carcinogénesis, mutagénesis y deterioro de la fertilidad: No se realizaron estudios de carcinogenicidad con belatacept. Sin embargo, se realizó un estudio de carcinogenicidad en murinos con abatacept (un análogo más activo en los roedores) para determinar el potencial carcinogénico del bloqueo de CD28. Las inyecciones subcutáneas semanales de 20, 65 ó 200 mg/kg de abatacept se asociaron con un aumento en la incidencia de linfomas malignos (con todas las dosis) y tumores de la glándula mamaria (dosis intermedia y alta en hembras) a exposiciones clínicamente relevantes. Los ratones de este estudio se infectaron con virus endógenos de leucemia murina y de tumor mamario de ratón, los cuales se asocian con un aumento de la incidencia de linfomas y tumores de glándula mamaria, respectivamente, en ratones inmunodeprimidos. Aunque se desconoce la relevancia precisa de estos hallazgos para el uso clínico de NULOJIX, se reportaron casos de PTLD (una proliferación premaligna o maligna de linfocitos B) en los ensayos clínicos. Las pruebas de genotoxicidad no son necesarias para los agentes terapéuticos de proteínas; por lo tanto, no se realizaron estudios de genotoxicidad con belatacept. Belatacept no tuvo efectos adversos en la fertilidad de ratas hembras y machos con dosis de hasta 200 mg/kg por día (25 veces la exposición con la MRHD). Toxicología y/o farmacología en animales: Abatacept, una proteína de fusión que se diferencia de belatacept por 2 aminoácidos, se une a los mismos ligandos (CD80/CD86) y bloquea la coestimulación de células T como belatacept, pero es más activo que belatacept en roedores. Por lo tanto, las toxicidades identificadas con abatacept en roedores pueden predecir los efectos adversos en humanos tratados con belatacept. Los estudios realizados en ratas expuestas a abatacept han demostrado anormalidades en el sistema inmunológico, que incluyen una baja incidencia de infecciones que conducen a la muerte (observadas en ratas jóvenes y ratas preñadas), así como autoinmunidad de tiroides y páncreas (observada en ratas expuestas in utero, así como en ratas jóvenes o adultas). Los estudios de abatacept en ratones y monos adultos, así como los de belatacept en monos adultos, no han demostrado hallazgos similares. La mayor susceptibilidad a las infecciones oportunistas observada en ratas jóvenes probablemente esté asociada con la exposición al abatacept antes del completo desarrollo de respuestas inmunológicas de memoria. En ratas preñadas, la mayor susceptibilidad a infecciones oportunistas puede deberse a las interrupciones inherentes en la inmunidad que se producen en las ratas durante la última etapa de la preñez/lactancia. Se han observado infecciones relacionadas con NULOJIX en ensayos clínicos con humanos [véase Advertencias]. La administración de abatacept a ratas se asoció con un significativo descenso de las células T regulatorias (de hasta un 90%). La deficiencia de células T regulatorias en humanos se ha asociado con autoinmunidad. La ocurrencia de eventos de autoinmunidad en los ensayos clínicos centrales fue infrecuente. Sin embargo, no puede excluirse la posibilidad de que los pacientes que reciben NULOJIX desarrollen autoinmunidad (o que los fetos expuestos a NULOJIX in utero desarrollen autoinmunidad). En un estudio de toxicidad de 6 meses realizado con belatacept en monos cynomolgus que recibieron dosis semanales de hasta 50 mg/kg (6 veces la exposición con la MRHD) y en un estudio de toxicidad de 1 año con abatacept en monos cynomolgus adultos que recibieron dosis semanales de hasta 50 mg/kg, no se observaron toxicidades significativas relacionadas con el fármaco. Los efectos farmacológicos reversibles consistieron en disminuciones transitorias mínimas de IgG sérica y depleción linfoide entre mínima y severa de centros germinales en el bazo y/o los ganglios linfáticos. Luego de 5 dosis (10 mg/kg ó 50 mg/kg, una vez por semana durante 5 semanas) de administración sistémica, no se detectó belatacept en el tejido cerebral de monos cynomolgus sanos normales. El número de células que expresan antígenos clase II del complejo mayor de histocompatibilidad (MHC) (potencial marcador de la activación de células inmunes) en el cerebro aumentó en monos que recibieron belatacept en comparación con el control de vehículo. Sin embargo, la distribución de algunas otras células que expresan CD68, CD20, CD80 y CD86, típicamente expresadas en células positivas clase II del MHC, no se vio alterada, y no hubo otros cambios histológicos en el cerebro. Se desconoce la relevancia clínica de estos hallazgos.
Indicaciones.
Adultos receptores de trasplante renal: NULOJIX® (belatacept) está indicado para la profilaxis del rechazo de órganos en pacientes adultos receptores de un trasplante renal. NULOJIX debe utilizarse en combinación con inducción con basiliximab, micofenolato mofetilo y corticosteroides. Limitaciones de uso: NULOJIX sólo debe usarse en pacientes que son EBV seropositivos [véase Contraindicaciones y Advertencias]. No se ha establecido el uso de NULOJIX para la profilaxis del rechazo de órganos en órganos trasplantados distintos del riñón [véase Advertencias].
Dosificación.
Posología en adultos receptores de trasplante renal: NULOJIX debe administrarse en combinación con inducción con basiliximab, micofenolato mofetilo (MMF) y corticosteroides. En los ensayos clínicos, la mediana (percentiles 25 - 75) de las dosis de corticosteroides disminuyó gradualmente hasta aproximadamente 15 mg (10-20 mg) por día durante las primeras 6 semanas y permaneció en aproximadamente 10 mg (5-10 mg) por día durante los primeros 6 meses post-trasplante. La utilización de corticosteroides debe ser consistente con la experiencia en ensayos clínicos con NULOJIX [véase Advertencias y Estudios Clínicos]. Debido a un mayor riesgo de trastorno linfoproliferativo postrasplante (PTLD) que involucra fundamentalmente el sistema nervioso central (SNC), leucoencefalopatía multifocal progresiva (PML) e infecciones serias del SNC, no se recomienda la administración de dosis más altas de las recomendadas ni una posología más frecuente de NULOJIX [véase Advertencias y Reacciones Adversas]. NULOJIX es para usar únicamente como infusión intravenosa. Los pacientes no requieren medicación previa antes de la administración de NULOJIX. Las instrucciones posológicas se proporcionan en la Tabla 1. La dosis de la infusión total de NULOJIX se debe basar en el peso corporal real del paciente en el momento del trasplante, y no se debe modificar durante el transcurso de la terapia, a menos que se produzca un cambio del peso corporal superior al 10%. La dosis prescrita de NULOJIX debe poder dividirse uniformemente por 12,5 mg para poder preparar la dosis con precisión usando la solución reconstituida y la jeringa descartable libre de silicona proporcionada. Los incrementos divisibles uniformemente son 0; 12,5; 25; 37,5; 50; 62,5; 75; 87,5; y 100. Por ejemplo: Un paciente pesa 64 kg. La dosis es de 10 mg por kg. Dosis calculada: 64 kg x 10 mg por kg = 640 mg. Las dosis más próximas divisibles uniformemente por 12,5 mg por debajo y por encima de 640 mg son 637,5 mg y 650 mg. La dosis más próxima a 640 mg es 637,5 mg. Por lo tanto, la dosis prescrita real para el paciente debe ser 637,5 mg.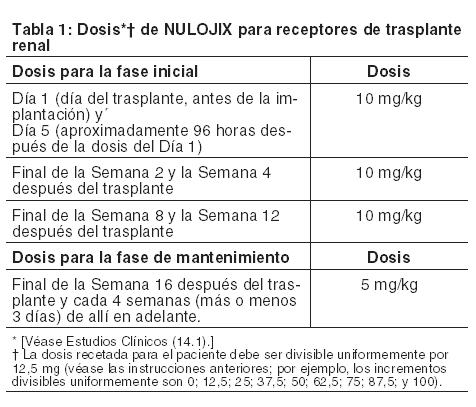
Instrucciones de Preparación y Administración: NULOJIX es para usar únicamente como infusión intravenosa. Precaución: NULOJIX se debe reconstituir/preparar usando sólo la jeringa descartable libre de silicona proporcionada con cada vial. Si la jeringa descartable libre de silicona se cae o se contamina, desecharla y usar una nueva. Preparación para la administración: Calcular la cantidad de viales de NULOJIX requeridos para obtener la dosis total de la infusión. Cada vial contiene 250 mg de belatacept como polvo liofilizado. Reconstituir el contenido de cada vial de NULOJIX con 10,5 mL de un diluyente adecuado, usando la jeringa descartable libre de silicona proporcionada con cada vial y una aguja de calibre 18 a 21. Los diluyentes adecuados incluyen agua estéril para uso inyectable (SWFI), cloruro de sodio al 0,9% (NS) o dextrosa en agua al 5% (D5W). Nota: Si el polvo de NULOJIX es reconstituido accidentalmente usando una jeringa distinta de la proporcionada, la solución puede desarrollar algunas partículas traslúcidas. Descartar toda solución preparada usando jeringas siliconadas. Para reconstituir el polvo de NULOJIX, retirar la tapa del vial y limpiar la parte superior con un trozo de algodón embebido en alcohol. Insertar la aguja en el vial por el centro del tapón de goma y dirigir el chorro del diluyente (10,5 mL de SWFI, NS o D5W) hacia la pared de vidrio del vial. Para minimizar la formación de espuma, rotar el vial e invertirlo con movimientos rotatorios suaves hasta que el contenido se disuelva completamente. Evitar la agitación prolongada o vigorosa. No agitar. La solución reconstituida contiene una concentración de belatacept de 25 mg/mL, y debe ser entre transparente y levemente opalescente, y entre incolora y de color amarillo pálido. No usar si se observan partículas opacas, decoloración u otras partículas extrañas. Calcular el volumen total de la solución de NULOJIX de 25 mg/mL reconstituida que se requiere para proporcionar la dosis total de la infusión. Volumen de solución de NULOJIX de 25 mg/mL (en mL) = Dosis prescrita (en mg) ÷ 25 mg/mL. Antes de la administración intravenosa, el volumen requerido de la solución reconstituida de NULOJIX debe diluirse adicionalmente con un líquido para infusión adecuado (NS o D5W). Si NULOJIX fue reconstituido con: SWFI, debe diluirse adicionalmente con NS o D5W. NS, debe diluirse adicionalmente con NS. D5W, debe diluirse adicionalmente con D5W. De una bolsa o un frasco para infusión del tamaño adecuado, retirar un volumen de líquido de infusión que sea igual al volumen de la solución reconstituida de NULOJIX necesario para suministrar la dosis prescrita. Con la misma jeringa descartable libre de silicona utilizada para la reconstitución, retirar la cantidad necesaria de solución de belatacept del vial, inyectarla en una bolsa o un frasco para infusión y rotar este último suavemente para mezclar. La concentración final de belatacept en una bolsa o un frasco para infusión debe oscilar entre 2 mg/mL y 10 mg/mL. Generalmente, un volumen de infusión de 100 mL será apropiado para la mayoría de los pacientes y las dosis, pero se puede usar un volumen de infusión total entre 50 mL y 250 mL. Toda porción de so