MYVITLA
TAKEDA ARG.
Antineoplásico.
Venta bajo receta archivada.
Composición.
Cada Cápsula dura de 2,3 mg de MYVITLA contiene: Citrato de Ixazomib 3,29 mg (equivalente a Ixazomib 2,30 mg). Excipientes cs. Cada Cápsula dura de 3 mg de MYVITLA contiene: Citrato de Ixazomib 4,30 mg (equivalente a Ixazomib 3,00 mg). Excipientes cs. Cada Cápsula dura de 4 mg de MYVITLA contiene: Citrato de Ixazomib 5,70 mg (equivalente a Ixazomib 4,00 mg). Excipientes cs.
Indicaciones.
MYVITLA está indicado en combinación con lenalidomida y dexametasona para el tratamiento de pacientes con mieloma múltiple que han recibido al menos un tratamiento previo.
Dosificación.
MYVITLA en combinación con lenalidomida y dexametasona: La dosis inicial recomendada de MYVITLA es una cápsula de 4 mg administrada oralmente una vez a la semana en los días 1, 8 y 15 de un ciclo de tratamiento de 28 días. La dosis inicial recomendada de lenalidomida es de 25 mg administrados diariamente en los días 1 al 21 de un ciclo de tratamiento de 28 días. La dosis inicial recomendada de dexametasona es de 40 mg administrados en los días 1, 8, 15 y 22 de un ciclo de tratamiento de 28 días. Programa De Dosificación: MYVITLA tomado con lenalidomida y dexametasona: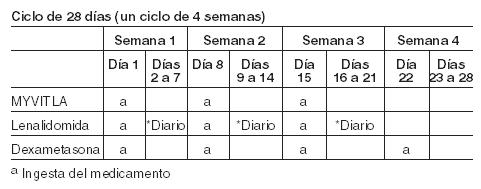
Para obtener información adicional sobre la lenalidomida y dexametasona, referirse a la información de dichos productos. Antes de iniciar un nuevo ciclo de tratamiento: El recuento absoluto de neutrófilos debe ser ≥ 1,000/mm3. El recuento de plaquetas debe ser ≥ 75,000/mm3. Las toxicidades no hematológicas deben, a criterio del médico, recuperarse en la condición inicial del paciente o ≤ Grado 1. El tratamiento debe continuar hasta la evolución de la enfermedad o una toxicidad inaceptable. Dosis Aplazadas u Olvidadas: En caso de que una dosis de MYVITLA sea olvidada o se aplace, debe tomarse sólo si la siguiente dosis programada es ≥ 72 horas de distancia. Una dosis olvidada no debe tomarse dentro de las 72 horas de la siguiente dosis programada. No se debe tomar una dosis doble para compensar la dosis olvidada. Si el paciente vomita después de tomar una dosis, no debe repetirla, sino que debe reanudarla en el momento de tomar la próxima dosis. Modificaciones De La Dosis: Los pasos de reducción de la dosis de MYVITLA se presentan en la Tabla 3 y las indicaciones de modificación se proporcionan en la Tabla 4.
Se recomienda un enfoque de modificación de dosis alternado para el MYVITLA y la lenalidomida debido a las toxicidades de trombocitopenia y sarpullido que se sobreponen. Para estas toxicidades el primer paso de la modificación de dosis es suspender/reducir la lenalidomida. Consulte los pasos para la reducción de dosis para estas toxicidades y neutropenia en la ficha técnica de la lenalidomida.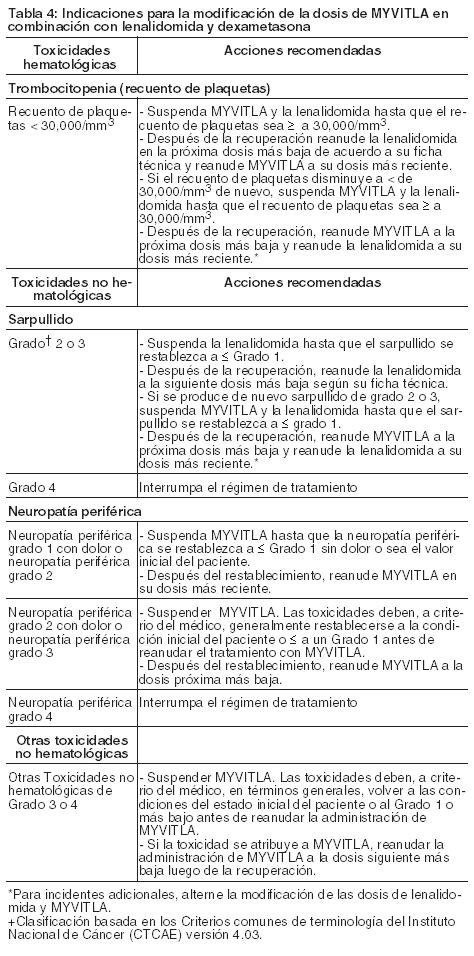
Medicamentos Concomitantes: La profilaxis antiviral debe ser considerada en pacientes que están siendo tratados con Ixazomib para disminuir el riesgo de reactivación de herpes zóster. Los pacientes incluidos en los estudios con Ixazomib quienes recibieron profilaxis antiviral tenían una menor incidencia de la infección de herpes zóster en comparación con los pacientes que no recibieron profilaxis. Poblaciones Especiales De Pacientes: Ancianos: Con base en los resultados de un análisis farmacocinético (PK) de la población, no es necesario un ajuste de la dosis de MYVITLA para pacientes mayores de 65 años. En estudios de MYVITLA, no hubo diferencias clínicamente significativas en la seguridad y eficacia entre pacientes menores de 65 años y en pacientes de 65 años de edad o mayores. Población Pediátrica: No se han establecido la seguridad ni la eficacia de MYVITLA en adolescentes menores de 18 años. No hay datos disponibles. Insuficiencia Hepática: No se requiere un ajuste de dosis del MYVITLA para pacientes con insuficiencia renal leve (bilirrubina total ≤ al límite superior al normal (LSN) y asparto aminotransferasa (AST) > al LSN o bilirrubina total > a 1- 1.5 x LSN y cualquier AST) con base en los resultados de un análisis farmacocinético de la población. Se recomienda una dosis más baja de una cápsula de 3 mg para pacientes con insuficiencia hepática moderada (bilirrubina total > a 1.5 a 3 x LSN) o severa (bilirrubina total > a 3 x LSN) con base en los resultados de un estudio PK. Insuficiencia Renal: No se requiere un ajuste de dosis de MYVITLA para pacientes con insuficiencia renal leve o moderada (depuración de creatinina ≥ 30 ml/min) con base en los resultados de un análisis PK de la población. Se recomienda una dosis más baja de una cápsula de 3 mg para pacientes con insuficiencia renal severa (depuración de creatinina < a 30 ml/min) o enfermedad renal en etapa terminal (ERT) que requiere diálisis con base en los resultados de un estudio PK. MYVITLA no es dializable y por lo tanto puede ser administrado sin importar el tiempo de diálisis. Consulte el resumen de las características de la lenalidomida para las recomendaciones de la dosificación en pacientes con insuficiencia renal. Método De Administración: MYVITLA debe tomarse una vez a la semana el mismo día y aproximadamente a la misma hora durante las tres primeras semanas de un ciclo de cuatro semanas. MYVITLA debe tomarse al menos una hora antes o al menos dos horas después de la comida. Se debe tragar la cápsula entera con agua. No se debe moler la cápsula, masticarse, ni abrirse.
Contraindicaciones.
No se conocen.
Reacciones adversas.
Estudios Clínicos: La población de seguridad del estudio clínico en Fase 3, aleatorizado, doble ciego, controlado con placebo, incluyó a 720 pacientes con mieloma múltiple en recaída y/o resistente al tratamiento, que recibieron MYVITLA en combinación con lenalidomida y dexametasona (régimen de MYVITLA; N=360) o placebo en combinación con lenalidomida y dexametasona (régimen de placebo; N=360). Las reacciones adversas descritas con más frecuencia ( > 20%) en los regímenes de MYVITLA y placebo fueron diarrea (42% vs. 36%), estreñimiento (34% vs. 25%), trombocitopenia (28% vs. 14%), neuropatía periférica (28% vs. 21%), náusea (26% vs 21%), edema periférico (25% vs. 18%), vómito (22% vs 11%) y dolor de espalda (21% vs. 16%). Se describieron reacciones adversas graves en ≥ 2% de los pacientes incluyendo a la trombocitopenia (2%) y diarrea (2%). Para cada reacción adversa, uno o más de los tres medicamentos fue suspendido en ≤ 1% de los pacientes en el régimen de MYVITLA. La siguiente convención se utiliza para la clasificación de la frecuencia de una reacción adversa al medicamento (RAM) y está basada en las directrices del Consejo de Organizaciones Internacionales de las Ciencias Médicas (CIOMS): muy frecuente (≥ 1/10); frecuente (≥ 1/100 a < 1/10); poco frecuente (≥ 1/1,000 a < 1/100); rara (≥ 1/10,000 a < 1/1,000) muy rara (≥ 1/10,000); desconocida (no se puede calcular a partir de los datos disponibles).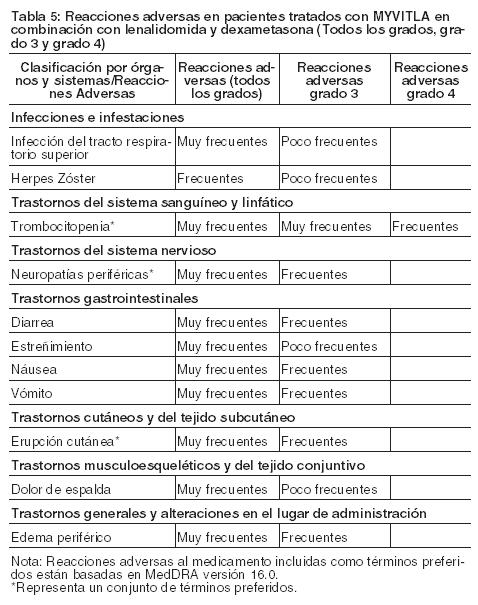
Erupción Cutánea: La erupción cutánea ocurrió en el 19% de los pacientes del régimen de MYVITLA comparado con el 11% de los pacientes en el régimen de placebo. La mayoría de los eventos de erupción cutánea fueron de grado 1 o grado 2. Se informó sobre una erupción cutánea de grado 3 en el 3% de pacientes en el régimen de MYVITLA comparado con el 1% de pacientes en el régimen de placebo y no hubo reacciones adversas serias de grado 4 o de erupción cutánea a través del estudio en Fase 3. El tipo de erupción cutánea más frecuente descrita en ambos regímenes fue el maculopapuloso y el macular. La erupción cutánea ocasionó la interrupción de uno o más de los tres medicamentos en < 1% de los pacientes en ambos regímenes. Se han dado informe de eventos cutáneos con lenalidomida y dexametasona. Neuropatía Periférica: La mayoría de reacciones adversas de neuropatía periférica fueron grado 1 y grado 2. Se notificaron reacciones adversas de grado 3 de neuropatía periférica en el 2% en ambos regímenes; no se presentaron reacciones adversas graves o grado 4. La reacción notificada con más frecuencia fue la neuropatía sensorial periférica (19% y 14% en el régimen con MYVITLA y placebo, respectivamente). No se notificó neuropatía motora periférica de manera frecuente en ninguno de los regímenes ( < 1%). La neuropatía periférica provocó la suspensión de uno o más de los tres medicamentos en el 1% de los pacientes en ambos regímenes. Otros Eventos Adversos: Fuera del estudio de Fase 3, raramente se notificaron los siguientes eventos adversos serios cuya causalidad no ha sido establecida: dermatosis neutrofílica febril aguda (Síndrome de Sweet), síndrome de Stevens Johnson, mielitis transversa, síndrome de encefalopatía posterior reversible, síndrome de lisis tumoral y púrpura trombocitopénica trombótica.
Advertencias.
Trombocitopenia: Se ha informado que se presenta trombocitopenia con MYVITLA con valores mínimos de plaquetas que ocurren típicamente entre los días 14 a 21 de cada ciclo de 28 días y el restablecimiento de los valores iniciales al inicio del siguiente ciclo. El tres por ciento de los pacientes bajo el régimen con MYVITLA y el 1% de los pacientes bajo el régimen con el placebo tuvieron un recuento de plaquetas ≥ 10,000/mm3 durante el tratamiento. Menos del 1% de los pacientes en ambos regímenes tuvo un recuento de plaquetas ≥ 5000/mm3 durante el tratamiento. La trombocitopenia dio lugar a la suspensión de uno o más de los tres medicamentos en < 1% de los pacientes en el régimen de MYVITLA y el 2% de los pacientes en el régimen de placebo. La trombocitopenia no ocasionó un aumento de los eventos hemorrágicos o transfusiones de plaquetas (ver Dosificación). Los recuentos de plaquetas se deben supervisar al menos una vez al mes durante el tratamiento con MYVITLA. Se debe considerar una supervisión más frecuente durante los primeros tres ciclos de acuerdo a la ficha técnica de la lenalidomida. La trombocitopenia se puede manejar con modificaciones de la dosis y transfusiones de plaquetas de acuerdo con los lineamientos médicos estándares. Efectos Secundarios Gastrointestinales: Se ha dado informe de diarrea, náuseas y vómitos con MYVITLA, a veces se requiere el uso de antieméticos y medicamentos antidiarreicos y atención de apoyo. La diarrea provocó la suspensión de uno o más de los tres medicamentos en el 1% de los pacientes en el régimen con MYVITLA y < 1% de los pacientes en el régimen con placebo. Se debe ajustar la dosis para síntomas graves (Grados 3 y 4) (ver Dosificación). Embarazo: MYVITLA puede causar daño fetal cuando se administra a una mujer embarazada basado en el mecanismo de acción y en los resultados en los animales. Las mujeres con potencial de quedar embarazadas deben ser advertidas de evitar quedar embarazada durante el tratamiento con MYVITLA. Si MYVITLA se utiliza durante el embarazo o si la paciente queda embarazada mientras está tomando MYVITLA, debe ser informada del riesgo potencial para el feto. Asesorar a las mujeres con potencial de quedar embarazadas que deben utilizar métodos anticonceptivos eficaces durante el tratamiento con MYVITLA y durante 90 días después de la última dosis. Las mujeres que usan anticonceptivos hormonales deben utilizar adicionalmente un método anticonceptivo de barrera.
Presentación.
Envases con 3 cápsulas duras.