Mircera®
ROCHE
Metoxipolietilenglicol-epoetina beta.
Agente estimulante de la eritropoyesis (AEE).
Composición.
Cada jeringa prellenada monodosis de 0,3 ml contiene 30 mg (a una concentración de 100 mg/ml) de metoxipolietilenglicol-epoetina beta*, en un excipiente compuesto por fosfato monosódico monohidratado 0,414 mg, sulfato de sodio anhidro 1,704 mg, manitol 9,000 mg, L-metionina 0,447 mg, poloxámero 188: 0,030 mg, ácido clorhídrico c.s.p. pH = 6,2, hidróxido de sodio c.s.p. pH= 6,2 y agua para inyectables c.s.p. 0,3 ml. Cada jeringa prellenada monodosis de 0,3 ml contiene 40 mg (a una concentración de 133 mg/ml) de metoxipolietilenglicol-epoetina beta*, en un excipiente compuesto por fosfato monosódico monohidratado 0,414 mg, sulfato de sodio anhidro 1,704 mg, manitol 9,000 mg, L-metionina 0,447 mg, poloxámero 188: 0,030 mg, ácido clorhídrico c.s.p. pH = 6,2, hidróxido de sodio c.s.p. pH= 6,2 y agua para inyectables c.s.p. 0,3 ml. Cada jeringa prellenada monodosis de 0,3 ml contiene 50 mg (a una concentración de 167 mg/ml) de metoxipolietilenglicol-epoetina beta*, en un excipiente compuesto por fosfato monosódico monohidratado 0,414 mg, sulfato de sodio anhidro 1,704 mg, manitol 9,000 mg, L-metionina 0,447 mg, poloxámero 188: 0,030 mg, ácido clorhídrico c.s.p. pH = 6,2, hidróxido de sodio c.s.p. pH= 6,2 y agua para inyectables c.s.p. 0,3 ml. Cada jeringa prellenada monodosis de 0,3 ml contiene 60 mg (a una concentración de 200 mg/ml) de metoxipolietilenglicol-epoetina beta*, en un excipiente compuesto por fosfato monosódico monohidratado 0,414 mg, sulfato de sodio anhidro 1,704 mg, manitol 9,000 mg, L-metionina 0,447 mg, poloxámero 188: 0,030 mg, ácido clorhídrico c.s.p. pH = 6,2, hidróxido de sodio c.s.p. pH= 6,2 y agua para inyectables c.s.p. 0,3 ml. Cada jeringa prellenada monodosis de 0,3 ml contiene 75 mg (a una concentración de 250 mg/ml) de metoxipolietilenglicol-epoetina beta*, en un excipiente compuesto por fosfato monosódico monohidratado 0,414 mg, sulfato de sodio anhidro 1,704 mg, manitol 9,000 mg, L-metionina 0,447 mg, poloxámero 188: 0,030 mg, ácido clorhídrico c.s.p. pH = 6,2, hidróxido de sodio c.s.p. pH= 6,2 y agua para inyectables c.s.p. 0,3 ml. Cada jeringa prellenada monodosis de 0,3 ml contiene 100 mg (a una concentración de 333 mg/ml) de metoxipolietilenglicol-epoetina beta*, en un excipiente compuesto por fosfato monosódico monohidratado 0,414 mg, sulfato de sodio anhidro 1,704 mg, manitol 9,000 mg, L-metionina 0,447 mg, poloxámero 188: 0,030 mg, ácido clorhídrico c.s.p. pH = 6,2, hidróxido de sodio c.s.p. pH= 6,2 y agua para inyectables c.s.p. 0,3 ml. Cada jeringa prellenada monodosis de 0,3 ml contiene 120 mg (a una concentración de 400 mg/ml) de metoxipolietilenglicol-epoetina beta*, en un excipiente compuesto por fosfato monosódico monohidratado 0,414 mg, sulfato de sodio anhidro 1,704 mg, manitol 9,000 mg, L-metionina 0,447 mg, poloxámero 188: 0,030 mg, ácido clorhídrico c.s.p. pH = 6,2, hidróxido de sodio c.s.p. pH= 6,2 y agua para inyectables c.s.p. 0,3 ml. Cada jeringa prellenada monodosis de 0,3 ml contiene 150 mg (a una concentración de 500 mg/ml) de metoxipolietilenglicol-epoetina beta*, en un excipiente compuesto por fosfato monosódico monohidratado 0,414 mg, sulfato de sodio anhidro 1,704 mg, manitol 9,000 mg, L-metionina 0,447 mg, poloxámero 188: 0,030 mg, ácido clorhídrico c.s.p. pH = 6,2, hidróxido de sodio c.s.p. pH= 6,2 y agua para inyectables c.s.p. 0,3 ml. Cada jeringa prellenada monodosis de 0,3 ml contiene 200 mg (a una concentración de 667 mg/ml) de metoxipolietilenglicol-epoetina beta*, en un excipiente compuesto por fosfato monosódico monohidratado 0,414 mg, sulfato de sodio anhidro 1,704 mg, manitol 9,000 mg, L-metionina 0,447 mg, poloxámero 188: 0,030 mg, ácido clorhídrico c.s.p. pH = 6,2, hidróxido de sodio c.s.p. pH= 6,2 y agua para inyectables c.s.p. 0,3 ml. Cada jeringa prellenada monodosis de 0,3 ml contiene 250 mg (a una concentración de 833 mg/ml) de metoxipolietilenglicol-epoetina beta*, en un excipiente compuesto por fosfato monosódico monohidratado 0,414 mg, sulfato de sodio anhidro 1,704 mg, manitol 9,000 mg, L-metionina 0,447 mg, poloxámero 188: 0,030 mg, ácido clorhídrico c.s.p. pH = 6,2, hidróxido de sodio c.s.p. pH= 6,2 y agua para inyectables c.s.p. 0,3 ml. Cada jeringa prellenada monodosis de 0,6 ml contiene 360 mg (a una concentración de 600 mg/ml) de metoxipolietilenglicol-epoetina beta*, en un excipiente compuesto por fosfato monosódico monohidratado 0,828 mg, sulfato de sodio anhidro 3,408 mg, manitol 18 mg, L-metionina 0,894 mg, poloxámero 188: 0,060 mg, ácido clorhídrico c.s.p. pH = 6,2, hidróxido de sodio c.s.p. pH = 6,2 y agua para inyectables c.s.p. 0,6 ml. La concentración indica la cantidad del componente proteínico de la molécula metoxipolietinglicol-epoetina beta, sin tener en cuenta la glicosilación. *El principio activo, metoxipolietilenglicol-epoetina beta, es un conjugado covalente de una proteína obtenida por tecnología del ADN recombinante a partir de las células de ovario de hámster chino y conjugada con un metoxipolietilenglicol (PEG) lineal. La potencia de metoxipolietilenglicol-epoetina beta no debe compararse con la de otra proteína, pegilada o no, del mismo grupo terapéutico. Para más información, véase Farmacología, Propiedades farmacodinámicas. La solución es transparente y de incolora a ligeramente amarillenta.
Farmacología.
Código ATC: B03XA03. Grupo farmacoterapéutico: Otros preparados antianémicos- Agente estimulante de la eritropoyesis (AEE). Propiedades farmacodinámicas: Mecanismo de acción: Mircera estimula la eritropoyesis al interaccionar con el receptor de eritropoyetina de las células progenitoras medulares. Metoxipolietilenglicol-epoetina beta, principio activo de Mircera, es un activador continuo del receptor de la eritropoyetina que presenta una actividad diferente a nivel del receptor en comparación con la eritropoyetina, que se caracteriza por una asociación más lenta y una disociación más rápida del receptor, una reducción de la actividad específica in vitro y un aumento en la actividad in vivo, así como un incremento de la vida media. La masa molecular media es de aproximadamente 60 kDa, de la que el componente proteínico más el componente glucídico representan aproximadamente 30 kDa. Efectos farmacodinámicos: La hormona natural eritropoyetina, factor principal para el crecimiento eritroide, se produce en los riñones y se libera al torrente circulatorio en respuesta a la hipoxia. En respuesta a la hipoxia, la hormona natural eritropoyetina interacciona con las células progenitoras eritroides incrementando la producción de eritrocitos. Eficacia clínica y seguridad: Los datos de los estudios de corrección en pacientes tratados una vez cada dos semanas y una vez cada cuatro semanas muestran que la tasa de respuesta de la hemoglobina en el grupo de Mircera al final del período de corrección fue mayor y equiparable a la de los comparadores. La mediana del tiempo hasta la respuesta fue de 43 días en el brazo de Mircera y de 29 días en el brazo comparador, con incrementos de hemoglobina en las seis primeras semanas, de 0,2 g/dl/semana y 0,3 g/dl/semana, respectivamente. Se realizaron cuatro ensayos controlados y aleatorizados en pacientes dializados que se estaban tratando con darbepoetina alfa o epoetina. Se distribuyó al azar a los pacientes de modo que continuaran con su tratamiento de ese momento o pasaran a recibir Mircera a fin de mantener niveles estables de hemoglobina. En el período de evaluación (semana 29 - 36), la media y la mediana del nivel de hemoglobina de los pacientes tratados con Mircera fueron prácticamente idénticas al valor basal de la misma. En un ensayo aleatorizado, doble-ciego, controlado con placebo, realizado en 4.038 pacientes con enfermedad renal crónica no sometidos a diálisis, con diabetes tipo 2 y niveles de hemoglobina ≤ 11 g/dl, los pacientes recibieron tratamiento bien con darbepoetina alfa para alcanzar niveles de hemoglobina de 13 g/dl o con placebo (véase Precauciones). El ensayo no cumplió su objetivo principal de demostrar una disminución en el riesgo de todas las causas de mortalidad, la morbilidad cardiovascular o enfermedad renal terminal. El análisis de los componentes individuales de la variable combinada mostró los siguientes hazard ratio (HR) (IC 95%): fallecimiento 1,05 (0,92; 1,21), infarto 1,92 (1,38; 2,68), insuficiencia cardíaca congestiva (ICC) 0,89 (0,74; 1,08), infarto de miocardio (IM) 0,96 (0,75; 1,23), hospitalización por isquemia del miocardio 0,84 (0,55; 1,27), enfermedad renal terminal 1,02 (0,87; 1,18). La eritropoyetina es un factor de crecimiento que estimula en forma primaria la producción de glóbulos rojos. Los receptores de eritropoyetina se encuentran también presentes en la superficie de algunas líneas celulares malignas. Se ha estudiado la sobrevida y la progresión tumoral en cinco grandes ensayos que incluyeron a 2.833 pacientes, de los cuales cuatro fueron doble-ciego controlados con placebo y uno fue abierto. En dos de ellos se incorporaron pacientes que estaban siendo tratados con quimioterapia. El nivel de hemoglobina que se quería alcanzar era > 13 g/dl en dos de los ensayos y de entre 12 y 14 g/dl en los otros tres. En el abierto no se observaron diferencias en la sobrevida global entre los pacientes tratados con eritropoyetina humana recombinante y el grupo control. En los cuatro controlados con placebo, el índice de riesgo (hazard ratio) para la sobrevida global osciló entre 1,25 y 2,47 a favor de los grupos control. En todos estos ensayos se ha observado un aumento inexplicable y estadísticamente significativo en pacientes que presentaban anemia asociada con diversos tipos frecuentes de cáncer y que recibieron eritropoyetina humana recombinante, en comparación con los grupos control. Las diferencias verificadas en la incidencia de trombosis y complicaciones relacionadas, entre los pacientes que recibieron eritropoyetina humana recombinante y que formaban parte del grupo control, no permiten explicar los resultados de sobrevida global observados en los ensayos. Asimismo, se ha realizado un análisis de datos a nivel de paciente, en más de 13.900 pacientes con cáncer (tratados con quimioterapia, radioterapia, quimiorradioterapia o sin tratamiento) que participaban en un total de 53 ensayos clínicos controlados que implicaban a varias epoetinas. En este meta-análisis se obtuvo un índice de riesgo (hazard ratio) para la sobrevida global de 1,06 a favor de los grupos control (IC 95%: 1,00; 1,12; 53 ensayos y 13.933 pacientes) y para los pacientes con cáncer que recibieron quimioterapia, el índice de riesgo (hazard ratio) para la sobrevida global fue de 1,04 (IC 95%: 0,97; 1,11; 38 ensayos y 10.441 pacientes). Por consiguiente, este meta-análisis además indica un aumento significativo del riesgo relativo de acontecimientos tromboembólicos en pacientes con cáncer tratados con eritropoyetina recombinante humana (véase Precauciones). En este análisis de datos no se han incluido pacientes tratados con Mircera. Mircera no está aprobado para el tratamiento de pacientes con anemia inducida por quimioterapia (véanse Indicaciones y Precauciones). Propiedades farmacocinéticas: La farmacocinética de la metoxipolietilenglicol-epoetina beta se estudió en voluntarios sanos y en pacientes anémicos con ERC, incluyendo pacientes dializados y otros no dializados. Las concentraciones séricas máximas de metoxipolietilenglicol-epoetina beta, tras su administración subcutánea a pacientes con ERC no dializados, se observaron a las 95 horas (mediana) después de su administración. La biodisponibilidad absoluta de metoxipolietilenglicol-epoetina beta tras su aplicación subcutánea fue del 54%. La vida media de eliminación terminal hallada fue de 142 horas en los pacientes con ERC no dializados. Las concentraciones séricas máximas de metoxipolietilenglicol-epoetina beta, tras su administración subcutánea a pacientes con ERC dializados, se observaron a las 72 horas (mediana) después de su administración. La biodisponibilidad absoluta de metoxipolietilenglicol-epoetina beta tras su aplicación subcutánea fue del 62% y la vida media de eliminación terminal observada fue de 139 horas, en los pacientes con ERC dializados. El clearance sistémico total, después de la administración intravenosa a pacientes con ERC dializados, fue de 0,494 ml/hora por kg. La vida media de eliminación después de la administración intravenosa de metoxipolietilenglicol-epoetina beta es de 134 horas. La comparación entre las concentraciones séricas de metoxipolietilenglicol-epoetina beta de 41 pacientes con ERC, medidas antes de la hemodiálisis y después de ésta, reveló que la hemodiálisis no modifica la farmacocinética de este medicamento. El análisis de 126 pacientes con ERC no mostró diferencias farmacocinéticas entre los pacientes dializados y los no dializados. En un ensayo en dosis única, después de la administración intravenosa, se observó que la farmacocinética de la metoxipolietilenglicol-epoetina beta en pacientes con insuficiencia hepática grave es similar a la observada en sujetos sanos (véase Dosificación). Datos preclínicos sobre seguridad: Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos, según los estudios convencionales de farmacotoxicología cardiovascular, toxicidad de dosis repetidas y toxicidad para la reproducción. El potencial carcinogénico de metoxipolietilenglicol-epoetina beta no se ha evaluado en estudios prolongados con animales. Esta sustancia no indujo una respuesta proliferativa de líneas de células tumorales no hematológicas in vitro. En un estudio de toxicidad a seis meses con ratas, no se observaron respuestas cancerígenas o mitógenas inesperadas en tejidos extrahematológicos. Además, en un estudio con diversos tejidos humanos, sólo se observó unión in vitro de metoxipolietilenglicol-epoetina beta a las células diana (células progenitoras de la médula ósea). No se ha detectado ninguna transferencia placentaria significativa de metoxi-polietilenglicol epoetina beta en las ratas y en los estudios con animales no se han apreciado efectos nocivos para el embarazo, el desarrollo embrionario fetal, el parto o la evolución posnatal. Sin embargo, se encontró un descenso reversible del peso fetal y una disminución del incremento ponderal posnatal en la descendencia, propios de este grupo terapéutico, después de administrar dosis que habían producido efectos farmacodinámicos maternos exagerados. El desarrollo físico, cognitivo o sexual de la descendencia de madres tratadas con metoxipolietilenglicol-epoetina beta durante la gestación y la lactancia no se vio alterado. Cuando se administró metoxipolietilenglicol-epoetina beta por vía subcutánea a machos y hembras de rata antes y durante el apareamiento, la función reproductora, la fertilidad y los parámetros espermáticos no se modificaron.
Indicaciones.
Tratamiento de la anemia sintomática asociada con la enfermedad renal crónica (ERC) en pacientes adultos (véase Farmacología, Propiedades farmacodinámicas).
Dosificación.
El tratamiento con Mircera se debe iniciar bajo la supervisión de un médico con experiencia en el manejo de pacientes con insuficiencia renal. Posología: Tratamiento de la anemia sintomática en pacientes adultos con enfermedad renal crónica: Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente. Mircera se puede administrar tanto por vía subcutánea como intravenosa con el fin de aumentar la concentración de hemoglobina hasta un máximo de 12 g/dl (7,45 mmol/l). En pacientes que no están sometidos a hemodiálisis es preferible utilizar la vía subcutánea para evitar la punción de venas periféricas. Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones se pueden observar valores individuales de hemoglobina superiores o inferiores a los niveles deseados. Esta variabilidad se debe controlar mediante ajuste de la dosis con el objeto de mantener los valores de hemoglobina dentro del intervalo entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,45 mmol/l). El nivel de hemoglobina no debe mantenerse en forma continuada por encima de 12 g/dl (7,45 mmol/l); más adelante se proporcionan instrucciones para adaptar en forma correcta las dosis cuando la concentración de hemoglobina sea superior a 12 g/dl (7,45 mmol/l). Debe evitarse un aumento de hemoglobina superior a 2 g/dl (1,24 mmol/l) durante un período de cuatro semanas. Si esto ocurre, se debe hacer un ajuste adecuado de la dosis según las instrucciones incluidas en este mismo ítem. Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis más baja autorizada de Mircera que permita un control óptimo de los síntomas de la anemia. Se recomienda vigilar la hemoglobina cada dos semanas hasta que se estabilice y luego, de manera periódica. Pacientes no tratados actualmente con un agente estimulante de la eritropoyesis (AEE): Para aumentar la hemoglobina por encima de 10 g/dl (6,21 mmol/l), la dosis inicial recomendada en pacientes no dializados es de 1,2 mg/kg de peso corporal, administrada una vez al mes, en una inyección única, por vía subcutánea. Alternativamente, en pacientes dializados y no dializados, se puede administrar una dosis inicial de 0,6 mg/kg de peso corporal una vez cada dos semanas, en una inyección única, utilizando tanto la vía intravenosa como la subcutánea. La dosis se puede elevar aproximadamente un 25% de la dosis anterior, si la tasa de aumento de la hemoglobina es inferior a 1,0 g/dl (0,621 mmol/l) durante un mes. Se pueden efectuar incrementos posteriores de aproximadamente el 25%, en intervalos mensuales, hasta alcanzar el valor deseado de hemoglobina para cada individuo. Si la tasa de aumento de la hemoglobina es mayor de 2 g/dl (1,24 mmol/l) en un mes o si el nivel de hemoglobina está aumentando y alcanzando 12 g/dl (7,45 mmol/l), la dosis se reducirá aproximadamente en un 25%. Si el nivel de hemoglobina continúa ascendiendo, se debe interrumpir el tratamiento hasta que comience a descender, momento en el que debe reanudarse con una dosis aproximadamente un 25% inferior a la dosis administrada previamente. Tras la interrupción de la dosis, se espera una disminución de la hemoglobina de 0,35 g/dl (0,22 mmol/l) a la semana aproximadamente. Los ajustes posológicos no se deben efectuar con una frecuencia mayor que mensual. Aquellos pacientes tratados una vez cada dos semanas cuya concentración de hemoglobina es superior a 10 g/dl (6,21 mmol/l), pueden recibir Mircera administrado una vez al mes, utilizando una dosis igual al doble de la administrada anteriormente cada dos semanas. Pacientes tratados actualmente con un AEE: Los pacientes tratados actualmente con un AEE pueden cambiar a Mircera, administrado una vez al mes en inyección única, intravenosa o subcutánea. La dosis inicial de Mircera se basará en la dosis semanal previa calculada de darbepoetina alfa o epoetina en el momento del cambio, tal y como se expone en la Tabla 1. Se aplicará la primera inyección cuando estuviera prevista la siguiente dosis de darbepoetina alfa o de epoetina administrada previamente.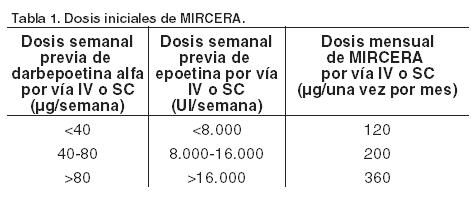
Si se precisara un ajuste posológico para mantener la concentración deseada de hemoglobina por encima de 10 g/dl (6,21 mmol/l), se podrá incrementar la dosis mensual en aproximadamente un 25%. Si la tasa del aumento de la hemoglobina es mayor de 2 g/dl (1,24 mmol/l) a lo largo de un mes o si el nivel de hemoglobina está aumentando y alcanzando 12 g/dl (7,45 mmol/l), se reducirá la dosis en un 25% aproximadamente. Si el nivel de hemoglobina continúa ascendiendo, se debe interrumpir el tratamiento hasta que comience a descender, momento en el que el tratamiento debe reanudarse con una dosis aproximadamente un 25% inferior a la dosis administrada previamente. Tras la interrupción de la dosis, se espera una disminución de la hemoglobina de 0,35 g/dl (0,22 mmol/l) a la semana aproximadamente. Los ajustes posológicos no se deben efectuar con una frecuencia mayor que mensual. Como la experiencia en el tratamiento de pacientes sometidos a diálisis peritoneal es limitada, en estos pacientes se recomienda vigilar el nivel de hemoglobina regularmente y cumplir rigurosamente los consejos sobre el ajuste posológico. Interrupción del tratamiento: El tratamiento con Mircera es habitualmente a largo plazo. Sin embargo, se podrá interrumpir en cualquier momento, si fuera necesario. Olvido de la dosis: Si se olvida una dosis de Mircera, la dosis omitida se administrará cuanto antes y la administración de Mircera se reanudará con la frecuencia posológica prescripta. Población pediátrica: Mircera no está recomendado para uso en niños y adolescentes menores de 18 años, debido a la ausencia de datos sobre seguridad y eficacia. Pacientes de edad avanzada: El 24% de los pacientes tratados con Mircera en los ensayos clínicos tenía una edad entre 65 y 74 años, mientras que el 20% tenía 75 o más años. Los pacientes de 65 o más años no precisan ningún ajuste posológico. Pacientes con insuficiencia hepática: En pacientes con insuficiencia hepática no es necesario ajustar la dosis inicial ni las pautas de modificación de dosis posteriores (véase Farmacología, Propiedades farmacocinéticas). Formas de administración: Mircera se puede administrar tanto por vía subcutánea como intravenosa. Se puede inyectar por vía subcutánea en el abdomen, en el brazo o en el muslo. Estos tres lugares de inyección son igualmente idóneos. Para consultar las instrucciones de administración del medicamento, véase Precauciones especiales de eliminación y otras manipulaciones.
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de sus excipientes. Hipertensión no controlada.
Reacciones adversas.
Resumen del perfil de seguridad: La base de datos de seguridad, procedente de los ensayos clínicos, contiene información sobre 3.042 pacientes con ERC, entre ellos 1.939 tratados con Mircera y 1.103 con otro AEE. Aproximadamente el 6% de los pacientes tratados con Mircera puede sufrir reacciones adversas. Entre éstas la notificada con mayor frecuencia fue la hipertensión (frecuente). Lista tabulada de reacciones adversas: En la Tabla 2, las reacciones adversas se ordenan según el sistema de clasificación por órganos y sistemas MedDRA y por las categorías de frecuencias. Estas están definidas de la siguiente manera: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1.000 a < 1/100); raras (≥ 1/10.000 a < 1/1.000); muy raras ( < 1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).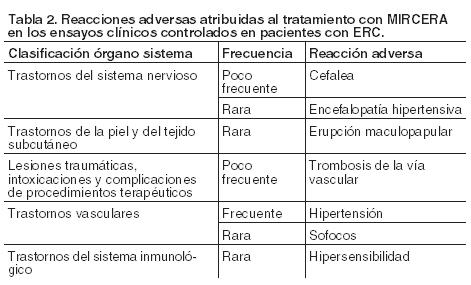
Todas las demás reacciones adversas atribuidas a Mircera fueron notificadas con frecuencia rara y, en su mayoría, fueron de intensidad leve a moderada. Estos efectos adversos se correspondieron con la comorbilidad conocida de esta población. Los resultados obtenidos en un ensayo clínico controlado realizado con epoetina alfa o darbepoetina alfa, evidenciaron que la incidencia de infarto es frecuente. Alteraciones de laboratorio: Durante el tratamiento con Mircera se observó en los ensayos clínicos un ligero descenso del recuento plaquetario, que se mantuvo dentro de los límites normales. Se ha notado un recuento plaquetario inferior a 100 x 109/l en el 7% de los pacientes tratados con Mircera y en el 4% de los que recibieron otros AEEs. Experiencia poscomercialización: Se han notificado espontáneamente, con una frecuencia no conocida, reacciones de hipersensibilidad, incluyendo casos de reacción anafiláctica. Al igual que otros AEEs, en el período poscomercialización se han informado con una frecuencia no conocida, casos de trombosis, incluyendo embolia pulmonar (véase Precauciones). Se han notificado espontáneamente, con una frecuencia no conocida, casos de trombocitopenia. Se ha comunicado, con frecuencia no conocida, aplasia eritrocitaria pura (AEP) mediada por anticuerpos neutralizantes antieritropoyetina. Cuando se diagnostique AEP, los pacientes deben interrumpir el tratamiento con Mircera, y no sustituirlo por otra eritropoyetina recombinante (véase Precauciones). Se reportaron casos de Síndrome de Stevens-Johnson/necrólisis epidérmica tóxica. Comunicación de reportes de reacciones adversas Es importante comunicar las presuntas reacciones adversas después de la autorización del medicamento. Esto permite la monitorización continua de la relación riesgo/beneficio. Se solicita a los profesionales de la salud informar de cualquier sospecha de eventos adversos asociados con el uso de Mircera® al Área de Farmacovigilancia de Roche al siguiente teléfono 0800-77-ROCHE (76243). En forma alternativa, esta información puede ser reportada ante ANMAT. Ante cualquier inconveniente con el producto, el paciente puede llenar la ficha que está en la Página Web de la ANMAT: http://www.anmat.gov.ar/farmacovigilancia/Notificar.asp o llamar a ANMAT responde al 0800-333-1234.
Precauciones.
No se ha establecido la seguridad y eficacia del tratamiento con Mircera en otras indicaciones, incluida la anemia en pacientes con cáncer. Tratamiento suplementario con hierro: Se recomienda el tratamiento suplementario con hierro de todos los pacientes con valores de ferritina sérica inferiores a 100 microgramos/l o con una saturación de transferrina por debajo del 20%. Para garantizar una eritropoyesis eficaz, hay que evaluar el estado del hierro en todos los pacientes antes y durante el tratamiento. Falta de respuesta al tratamiento con Mircera: La falta de respuesta al tratamiento con Mircera debe seguirse de un estudio de los factores causales. Las carencias de hierro, ácido fólico o vitamina B12 reducen la eficacia de los AEEs y, en consecuencia, deben corregirse. Las infecciones intercurrentes, los episodios inflamatorios o traumáticos, las pérdidas hemáticas ocultas, la hemólisis, la intoxicación grave por aluminio, las enfermedades hematológicas subyacentes o la mielofibrosis también pueden comprometer la respuesta eritropoyética. Se debe considerar la realización de un recuento de reticulocitos como parte del estudio. Si se descartan todos los trastornos mencionados y el paciente experimenta una caída brusca de la hemoglobina, asociada con reticulocitopenia y anticuerpos antieritropoyetina, debe considerarse la realización de un examen de la médula ósea para excluir el diagnóstico de aplasia eritrocitaria pura (AEP). Si se diagnostica una AEP, se debe suspender el tratamiento con Mircera y no se debe cambiar a los pacientes a ningún otro AEE. Aplasia eritrocitaria pura: Se ha notificado aplasia eritrocitaria pura causada por anticuerpos antieritropoyetina en relación con el uso de AEEs, incluido Mircera. Se ha comprobado que estos anticuerpos muestran reacción cruzada con todos los AEEs y, por lo tanto, si se sospecha o se confirma que los pacientes presentan anticuerpos contra la eritropoyetina no debe cambiarse su tratamiento a Mircera (véase Reacciones adversas). Aplasia eritrocitaria pura en pacientes con Hepatitis C: Se debe suspender inmediatamente el tratamiento con epoetina, y realizar ensayos de anticuerpos antieritropoyetina si se produce una disminución paradójica de la hemoglobina y se desarrolla anemia severa asociada a bajos recuentos de reticulocitos. En pacientes con hepatitis C tratados con interferón y ribavirina se han notificado casos cuando las epoetinas se han utilizado concomitantemente. Las epoetinas no están aprobadas en el tratamiento de la anemia vinculada con la hepatitis C. Concentración de hemoglobina: En pacientes con enfermedad renal crónica, la concentración de hemoglobina en la fase de mantenimiento no debe exceder el límite superior del rango recomendado en Dosificación durante un período de tiempo prolongado. En ensayos clínicos se observó un aumento del riesgo de fallecimiento, de acontecimientos cardiovasculares graves incluyendo trombosis o acontecimientos cerebrovasculares incluido el infarto, cuando se administraron AEEs con el fin de alcanzar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l) (véase Reacciones adversas). En ensayos clínicos controlados no se han observado beneficios significativos atribuibles a la administración de epoetinas cuando se incrementaba la concentración de hemoglobina por encima del nivel necesario para controlar los síntomas de anemia y evitar las transfusiones sanguíneas. No se ha establecido la seguridad y eficacia del tratamiento con Mircera en pacientes con hemoglobinopatías, crisis epilépticas, hemorragia o antecedentes recientes de hemorragia con necesidad de transfusiones o con niveles plaquetarios mayores de 500 x 109/l. Por lo tanto, se debe tener precaución en estos casos. Control de la presión arterial: Como ocurre con otros AEEs, la presión arterial puede elevarse durante el tratamiento con Mircera. Hay que controlar adecuadamente la presión arterial de todos los pacientes, antes, al comienzo y durante el tratamiento con Mircera. Si resultara difícil estabilizarla con medicamentos o dieta, se deberá reducir la dosis o interrumpir la administración (véase Dosificación). Efectos sobre el crecimiento tumoral: Mircera, como otros AEEs, es un factor de crecimiento que estimula sobre todo la producción de eritrocitos. Los receptores de eritropoyetina se pueden expresar en la superficie de diversas células tumorales. Como ocurre con todos los factores de crecimiento, cabe la posibilidad de que los AEEs puedan estimular el crecimiento de cualquier tipo de tumor maligno. En dos ensayos clínicos controlados, donde se administraron epoetinas a pacientes con diversos tipos de cáncer, entre ellos cáncer de cabeza y cuello, y cáncer de mama, se observó un exceso inexplicable de mortalidad. Uso incorrecto de Mircera: El uso incorrecto de Mircera por personas sanas puede motivar un aumento excesivo de hemoglobina. Puede estar asociado con complicaciones cardiovasculares con peligro para la vida. Trazabilidad de Mircera: Para mejorar la trazabilidad de los AEEs, se debe registrar (o identificar) claramente el nombre comercial del AEE administrado en la historia clínica del paciente. Contenido de sodio: Este medicamento contiene menos de 1 mmol (23 mg) de sodio por ml, por lo que se considera esencialmente "exento de sodio". Efectos sobre la capacidad para conducir y utilizar máquinas: Mircera posee un efecto nulo o insignificante sobre la capacidad para conducir y utilizar máquinas. Fertilidad, embarazo y lactancia: Embarazo: No existen datos sobre el empleo de Mircera en mujeres embarazadas. Los estudios con animales no indican que existan efectos nocivos directos para el embarazo, el desarrollo embrionario o fetal, el parto o la evolución posnatal, pero indican una reducción reversible del peso fetal relacionado con este grupo terapéutico (véase Farmacología, Datos preclínicos sobre seguridad). Se debe tener precaución cuando se prescriba a mujeres embarazadas. Lactancia: Se desconoce si Mircera se excreta en la leche humana. Un estudio con animales ha mostrado excreción de metoxipolietilenglicol-epoetina beta en la leche materna. La decisión de continuar o interrumpir la lactancia o de continuar o interrumpir Mircera se tomará sopesando las ventajas de la lactancia para el bebé y los beneficios del tratamiento con Mircera para la madre. Fertilidad: Los estudios en animales no han mostrado evidencia de alteración de la fertilidad (véase Farmacología, Datos preclínicos sobre seguridad). Se desconoce el riesgo potencial en seres humanos.
Interacciones.
No se han realizado estudios de interacciones. No existen pruebas de que Mircera altere el metabolismo de otros medicamentos.
Incompatibilidades.
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
Conservación.
Las jeringas prellenadas deben conservarse en heladera entre 2° y 8° C. No congelar. Conservar las jeringas prellenadas en el embalaje exterior para proteger su contenido de la luz. El usuario final podrá sacar el medicamento de la heladera y conservarlo a temperatura ambiente (nunca por encima de 30° C) durante un único período de 1 mes. Una vez fuera de la heladera, el producto deberá utilizarse dentro de este lapso. Naturaleza y contenido del envase: Jeringa prellenada (vidrio Tipo I), con pistón laminado (goma bromobutílica) y protector (goma bromobutílica) y una aguja. Envase unitario. Precauciones especiales de eliminación y otras manipulaciones: La jeringa prellenada está lista para su uso. La jeringa prellenada estéril no contiene conservantes y debe utilizarse sólo para una inyección. Sólo se debe administrar una dosis por jeringa prellenada. Únicamente se deberán inyectar las soluciones transparentes, de incoloras a ligeramente amarillentas, que estén exentas de partículas visibles. No agitar. Dejar que la jeringa prellenada alcance la temperatura ambiente antes de inyectar. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local. Este medicamento no debe ser utilizado después de la fecha de vencimiento indicada en el envase. Este medicamento debe ser usado exclusivamente bajo prescripción y vigilancia médica y no puede repetirse sin nueva receta médica. Mantenga los medicamentos fuera del alcance de los niños.
Sobredosificación.
El margen terapéutico de Mircera es amplio. Cuando se inicie el tratamiento, deberá tenerse en cuenta la capacidad individual de respuesta. La sobredosis puede producir manifestaciones farmacodinámicas exageradas, por ejemplo eritropoyesis excesiva. Si los niveles de hemoglobina son muy elevados, se interrumpirá temporalmente el tratamiento con Mircera (véase Dosificación). Si estuviera clínicamente indicado, se puede realizar una flebotomía. Ante la eventualidad de una sobredosificación concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Dr. Ricardo Gutiérrez: 4962-6666/2247; Policlínico Dr. G.A. Posadas: 4654-6648; 4658-7777; Hospital General de Niños Dr. Pedro de Elizalde: 4300-2115; 4363-2100/2200 Interno 6217.
Presentación.
Jeringa prellenada monodosis con 30 mg/0,3 ml: envase con 1. Jeringa prellenada monodosis con 40 mg/0,3 ml: envase con 1. Jeringa prellenada monodosis con 50 mg/0,3 ml: envase con 1. Jeringa prellenada monodosis con 60 mg/0,3 ml: envase con 1. Jeringa prellenada monodosis con 75 mg/0,3 ml: envase con 1. Jeringa prellenada monodosis con 100 mg/0,3 ml: envase con 1. Jeringa prellenada monodosis con 120 mg/0,3 ml: envase con 1. Jeringa prellenada monodosis con 150 mg/0,3 ml: envase con 1. Jeringa prellenada monodosis con 200 mg/0,3 ml: envase con 1. Jeringa prellenada monodosis con 250 mg/0,3 ml: envase con 1. Jeringa prellenada monodosis con 360 mg/0,6 ml: envase con 1.
Revisión.
Julio 2015. Aprobación: 23/02/2016. Disp. ANMAT N° 1.740 (RI+EMA+ANMAT C004/13+Shpe+CDS: 8.0C).