MIRABET
ADIUM
Antiespasmódico urinario.
Composición.
Cada comprimido recubierto de liberación prolongada contiene: Mirabegron 25 mg y 50 mg.
Farmacología.
Mecanismo de acción: Mirabegron es un agonista del receptor adrenérgico (RA) beta-3 humano, como lo demuestran experimentos de laboratorio in vitro utilizando el RA beta-3 humano clonado. Mirabegron relaja el músculo liso detrusor durante la fase de almacenamiento del ciclo de llenado-vacío de la vejiga urinaria mediante la activación del RA beta-3, que aumenta la capacidad de la vejiga. Aunque mirabegron mostró una actividad intrínseca muy baja para los RA beta-1 y beta-2 humanos clonados, los resultados en humanos indican que la estimulación del RA beta-1 se produjo con una dosis de mirabegron de 200 mg. Efectos farmacodinámicos: Urodinámica: Los efectos de mirabegron sobre el flujo urinario máximo y la presión del detrusor al flujo máximo se evaluaron en un estudio urodinámico que incluyó a 200 pacientes masculinos con síntomas del tracto urinario inferior (STUI) y obstrucción del tracto de salida vesical. La administración de mirabegron una vez al día durante 12 semanas no afectó negativamente el caudal máximo medio ni la presión media del detrusor al caudal máximo en este estudio. No obstante, mirabegron debe administrarse con precaución a pacientes con obstrucción del tracto de salida vesical clínicamente significativa. Electrofisiología cardíaca: Se evaluó el efecto de dosis múltiples de mirabegron de 50 mg, 100 mg y 200 mg (cuatro veces la dosis máxima recomendada) una vez al día sobre el intervalo QTc en un estudio cruzado paralelo, aleatorizado, controlado con placebo y con tratamiento activo (moxifloxacina 400 mg), de cuatro brazos. en 352 sujetos sanos. En el estudio, con capacidad demostrada para detectar efectos pequeños, el límite superior del intervalo de confianza unilateral del 95% para el mayor QTc corregido con placebo y con base al inicio según el método de corrección individual (QTcI) fue inferior a 10 ms. Para el grupo de dosis de 50 mg de mirabegron (la dosis máxima aprobada), la diferencia media con respecto al placebo en el intervalo QTcI de 4 a 5 horas después de la dosis fue de 3,7 ms (límite superior del IC del 95 %: 5,1 ms). Para los grupos de dosis de mirabegron de 100 mg y 200 mg (dosis mayores que la dosis máxima aprobada y que resultan en múltiplos sustanciales de los niveles sanguíneos máximos anticipados con 50 mg), las diferencias medias con respecto al placebo en el intervalo QTcI de 4 a 5 horas después de la dosis fueron de 6,1 ms (superior límite del IC del 95 %: 7,6 ms) y 8,1 ms (límite superior del IC del 95 %: 9,8 ms), respectivamente. Con la dosis de 200 mg de mirabegron, en las mujeres, el efecto medio fue de 10,4 ms (límite superior del IC del 95 %: 13,4 ms). En este estudio exhaustivo del intervalo QT, mirabegron aumentó la frecuencia cardíaca en el ECG de forma dosis-dependiente. Los aumentos medios máximos desde el inicio en la frecuencia cardíaca para los grupos de dosis de 50 mg, 100 mg y 200 mg en comparación con el placebo fueron de 6,7 lpm, 11 lpm y 17 lpm, respectivamente. En los estudios de eficacia clínica y seguridad, el cambio desde el inicio en la frecuencia del pulso promedio para mirabegron 50 mg fue de aproximadamente 1 lpm. En este estudio exhaustivo del intervalo QT, mirabegron también aumentó la presión arterial de manera dependiente de la dosis (ver Efectos sobre la presión arterial). Efectos sobre la presión arterial: En un estudio de 352 sujetos sanos que evaluó el efecto de múltiples dosis diarias de 50 mg, 100 mg y 200 mg (cuatro veces la dosis máxima recomendada) de mirabegron durante 10 días en el intervalo QTc, el aumento medio máximo de la presión arterial sistólica en decúbito supino (PAS)/presión arterial diastólica (PAD) en la dosis máxima recomendada de 50 mg fue aproximadamente 4,0/1,6 mm Hg mayor que el placebo. Los aumentos promedio de 24 horas en la PAS en comparación con el placebo fueron de 3,0, 5,5 y 9,7 mm Hg con dosis de mirabegron de 50 mg, 100 mg y 200 mg, respectivamente. Los aumentos de la PAD también dependieron de la dosis, pero fueron menores que los de la PAS. En otro estudio en 96 sujetos sanos para evaluar el impacto de la edad en la farmacocinética de múltiples dosis diarias de 50 mg, 100 mg, 200 mg y 300 mg (seis veces la dosis máxima recomendada) de mirabegron durante 10 días, la PAS también aumentó de manera dependiente de la dosis. Los aumentos máximos medios de la PAS fueron aproximadamente 2,5; 4,5; 5,5 y 6,5 mmHg para exposiciones a mirabegron asociadas con dosis de 50 mg, 100 mg, 200 mg y 300 mg, respectivamente. En tres estudios de seguridad y eficacia, doble ciego, controlados con placebo, de 12 semanas de duración en pacientes con VH que recibieron mirabegron 25 mg, 50 mg o 100 mg (dos veces la dosis máxima recomendada) una vez al día, la media Se observaron aumentos de la PAS/PAD en comparación con el placebo de aproximadamente 0,5 -1 mm Hg. La PAS matutina aumentó al menos 15 mm Hg desde el inicio en el 5,3%; 5,1% y 6,7% de los pacientes que recibieron placebo, mirabegron 25 mg y mirabegron 50 mg, respectivamente. La PAD matutina aumentó al menos 10 mm Hg en el 4,6%; 4,1% y 6,6% de los pacientes que recibieron placebo, mirabegron 25 mg y mirabegron 50 mg, respectivamente. Tanto los aumentos de la PAS como de la PAD fueron reversibles al suspender el tratamiento. En un estudio de seguridad y eficacia, doble ciego, controlado con placebo, de 12 semanas de duración en pacientes con VH que recibieron 25 mg o 50 mg de mirabegron una vez al día coadministrado con 5 mg de solifenacina, no se observaron diferencias consistentes en la PAS/PAD media de 24 horas, en comparación con placebo, mirabegron o solifenacina en monoterapia según lo evaluado con monitorización ambulatoria de la presión arterial (MAPA) de 24 horas. Se observaron frecuencias similares de cambios categóricos para el tratamiento combinado versus placebo en la PAS/PAD media de 24 horas. Efecto sobre la presión intraocular (PIO): Mirabegron 100 mg una vez al día no aumentó la PIO en sujetos sanos después de 56 días de tratamiento. En un estudio de fase 1 que evaluó el efecto de mirabegron sobre la PIO mediante tonometría de aplanación de Goldmann en 310 sujetos sanos, una dosis de 100 mg de mirabegron no fue inferior al placebo para el criterio de valoración principal de la diferencia del tratamiento en el cambio medio desde el inicio hasta el día 56 en la PIO promedio de los sujetos; el límite superior del IC bilateral del 95% de la diferencia de tratamiento entre mirabegron 100 mg y placebo fue de 0,3 mm Hg. Farmacocinética: Absorción: Después de la administración oral de mirabegron en voluntarios sanos adultos, mirabegron se absorbió hasta alcanzar concentraciones plasmáticas máximas (Cmax) en aproximadamente 3,5 horas. La biodisponibilidad absoluta aumentó del 29% con una dosis de 25 mg al 35% con una dosis de 50 mg. La Cmax media y el AUC aumentaron más que la dosis proporcionalmente. Esta relación fue más evidente en dosis superiores a 50 mg. En la población general de hombres y mujeres, un aumento de 2 veces en la dosis de 50 mg a 100 mg de mirabegron aumentó la Cmax y el AUCtau aproximadamente 2,9 y 2,6 veces, respectivamente, mientras que un aumento de 4 veces en dosis de 50 a 200 mg de mirabegron aumentaron la Cmax y el AUCtau aproximadamente 8,4 y 6,5 veces. Las concentraciones en estado de equilibrio se alcanzaron dentro de los 7 días posteriores a la administración de una dosis diaria de mirabegron. Después de la administración una vez al día, la exposición plasmática de mirabegron en estado estacionario fue aproximadamente el doble que la observada después de una dosis única. La mediana de Tmax de mirabegron después de la administración oral de una dosis única de los comprimidos o la suspensión oral en pacientes pediátricos alimentados fue de 4 a 5 horas. El análisis farmacocinético poblacional predijo que la mediana de Tmax de mirabegron en estado estacionario fue de 34 horas. Efecto de los alimentos en adultos: No hubo diferencias clínicamente significativas en la farmacocinética de mirabegron cuando se administró con o sin alimentos en pacientes adultos. Efecto de los alimentos en pediatría: En el estado de ayuno, el AUC de mirabegron en estado estacionario aumentó en un 120% en relación con el estado de alimentación en pacientes pediátricos. La Cmax y el AUC en ayunas aumentaron en un 170% y un 80%, respectivamente, en comparación con el estado de alimentación después de la administración de mirabegron en voluntarios sanos. Distribución: Mirabegron se distribuye ampliamente en el cuerpo en pacientes adultos. El volumen de distribución en estado estacionario (Vss) es de aproximadamente 1.670 litros después de la administración intravenosa. Mirabegron se une (aproximadamente 71%) a las proteínas plasmáticas humanas y muestra una afinidad moderada por la albúmina y la glicoproteína ácida alfa-1. Mirabegron se distribuye a los eritrocitos. Según un estudio in vitro, las concentraciones de 14C-mirabegron en eritrocitos fueron aproximadamente 2 veces mayores que en plasma. El volumen de distribución de mirabegron fue relativamente grande en pacientes pediátricos (el rango de Vz/F promedio en estado de alimentación en pacientes pediátricos en todos los estudios: 4.895-13.726 litros) y aumentó con el aumento del peso corporal. Eliminación: La vida media de eliminación terminal (t1/2) de mirabegron es de aproximadamente 50 horas en pacientes adultos. La vida media de eliminación terminal promedio (t1/2) de mirabegron es de aproximadamente 26 a 31 horas en pacientes pediátricos. Metabolismo: Mirabegron se metaboliza a través de múltiples vías que implican desalquilación, oxidación, glucuronidación (directa) e hidrólisis de amida. Mirabegron es el principal componente circulante después de una dosis única de 14C-mirabegron. Se observaron dos metabolitos principales en el plasma humano y son glucurónidos de fase 2 que representan el 16 % y el 11 % de la exposición total, respectivamente. Estos metabolitos no son farmacológicamente activos frente al receptor adrenérgico beta-3. Aunque los estudios in vitro sugieren un papel de CYP2D6 y CYP3A4 en el metabolismo oxidativo de mirabegron, los resultados in vivo indican que estas isoenzimas desempeñan un papel limitado en la eliminación general. En sujetos sanos que eran genotípicamente metabolizadores lentos de CYP2D6, la Cmax media y el AUCtau fueron aproximadamente un 16% y un 17% más altos que en los metabolizadores rápidos de CYP2D6, respectivamente. Los estudios in vitro y ex vivo han demostrado la participación de las enzimas butilcolinesterasa, uridina difosfoglucuronosil-transferasa (UGT) y posiblemente alcohol deshidrogenasa en el metabolismo de mirabegron, además de CYP3A4 y CYP2D6. Excreción: El aclaramiento corporal total (Cltot) del plasma es de aproximadamente 57 l/h después de la administración intravenosa. El aclaramiento renal (ClR) es de aproximadamente 13 l/h, lo que corresponde a casi el 25 % del Cltot. La eliminación renal de mirabegron se produce principalmente mediante secreción tubular activa junto con filtración glomerular. La eliminación urinaria de mirabegron inalterado depende de la dosis y oscila entre aproximadamente el 6,0 % después de una dosis diaria de 25 mg y el 12,2 % después de una dosis diaria de 100 mg. Tras la administración de 160 mg de solución de 14C-mirabegron a voluntarios sanos, aproximadamente el 55 % de la dosis de radiactividad se recuperó en la orina y el 34 % en las heces. Aproximadamente el 25 % del mirabegron sin cambios se recuperó en la orina y el 0 % en las heces. El modelo farmacocinético poblacional predijo que el aclaramiento de mirabegron en pacientes pediátricos aumentaba con el peso corporal. Farmacocinética en poblaciones especiales: Pacientes geriátricos: Las Cmax y AUc de mirabegron después de dosis múltiples orales en pacientes ancianos (≥ 65 años) fueron similares a las de voluntarios más jóvenes (18 a 45 años). Pacientes pediátricos: En pacientes de 3 a 18 años de edad, no se predijo que la edad afectara los parámetros farmacocinéticos de mirabegron después de tener en cuenta las diferencias en el peso corporal. Género: La Cmax y el AUC de mirabegron fueron aproximadamente entre un 40% y un 50% más altos en mujeres que en hombres adultos. Cuando se corrigió por diferencias en el peso corporal, la exposición sistémica a mirabegron fue entre un 20 % y un 30 % mayor en las mujeres que en los hombres. El género no tiene un impacto significativo en la farmacocinética de mirabegron en la población pediátrica de 3 a 18 años de edad. Raza: La farmacocinética de mirabegron fue comparable entre caucásicos y negros afroamericanos. La comparación de estudios cruzados mostró que la exposición en sujetos japoneses fue mayor que en sujetos norteamericanos. Sin embargo, cuando la Cmax y el AUC se normalizaron para la dosis y el peso corporal, la diferencia fue menor. Pacientes con insuficiencia renal: Tras la administración de una dosis única de 100 mg de mirabegron en voluntarios adultos con insuficiencia renal leve (TFGe de 60 a 89 ml/min/1,73 m2 según lo estimado por MDRD), la Cmáx y el AUC medias de mirabegron aumentaron en un 6% y un 31% en relación con los voluntarios adultos con función renal normal. En voluntarios adultos con insuficiencia renal moderada (TFGe de 30 a 59 ml/min/1,73 m2), la Cmax y el AUC aumentaron en un 23% y un 66%, respectivamente. En voluntarios adultos con insuficiencia renal grave (TFGe de 15 a 29 ml/min/1,73 m2), los valores medios de Cmax y AUC fueron un 92 % y un 118 % más altos en comparación con los sujetos sanos con función renal normal. Mirabegron no se ha estudiado en pacientes adultos con enfermedad renal terminal (ESRD) (TFGe inferior a 15 ml/min/1,73 m2) ni en pacientes adultos que requieran diálisis. Pacientes con insuficiencia hepática: Después de la administración de una dosis única de 100 mg de mirabegron en voluntarios adultos con insuficiencia hepática leve (ChildPugh Clase A), la Cmáx y el AUC medias de mirabegron aumentaron en un 9% y un 19%, en relación con los voluntarios adultos con función hepática normal. En voluntarios adultos con insuficiencia hepática moderada (Child-Pugh Clase B), los valores medios de Cmax y AUC fueron un 175% y un 65% más altos. Mirabegron no se ha estudiado en pacientes adultos con insuficiencia hepática grave (Child-Pugh Clase C). Datos preclínicos sobre seguridad: Carcinogenicidad: Se realizaron estudios de carcinogenicidad a largo plazo en ratas y ratones a los que se les administró mirabegron por vía oral durante dos años. Las ratas macho recibieron dosis de 0; 12,5; 25 o 50 mg/kg/día y las ratas hembra y ratones de ambos sexos recibieron dosis de 0; 25; 50 o 100 mg/kg/día. Mirabegron no mostró potencial carcinogénico en exposiciones sistémicas (AUC) de 38 a 45 veces mayores que la MRHD en ratas y de 21 a 38 veces mayores que la MRHD en ratones que la exposición sistémica humana a la dosis de 50 mg. Mutagénesis: Mirabegron no fue mutagénico en el ensayo de mutación inversa bacteriana de Ames, no indujo aberraciones cromosómicas en linfocitos de sangre periférica humana en concentraciones que no fueron citotóxicas y no fue clastogénico en el ensayo de micronúcleos de rata. Trastornos de la fertilidad: Los estudios de fertilidad en ratas mostraron que mirabegron no tuvo ningún efecto sobre la fertilidad masculina o femenina en dosis no letales de hasta 100 mg/kg/día. Se estimó que las exposiciones sistémicas (AUC) a 100 mg/kg en ratas hembra eran 22 veces la MRHD en mujeres y 93 veces la MRHD en hombres.
Indicaciones.
Tratamiento sintomático de la urgencia, aumento de la frecuencia de micción y/o incontinencia de urgencia que puede producirse en pacientes adultos con síndrome de vejiga hiperactiva (VH).
Dosificación.
Posología La dosis inicial recomendada de mirabegron es de 25 mg por vía oral una vez al día. Si es necesario, se puede aumentar a la dosis máxima de 50 mg de mirabegron por vía oral, una vez al día, después de 4 a 8 semanas. Posología en poblaciones especiales Insuficiencia renal: La dosis recomendada de mirabegron (administrado por vía oral una vez al día) en pacientes adultos con insuficiencia renal se describe en la Tabla 1.
Insuficiencia hepática: La dosis recomendada de mirabegron (administrado por vía oral una vez al día) en pacientes adultos con insuficiencia hepática se describe en la Tabla 2.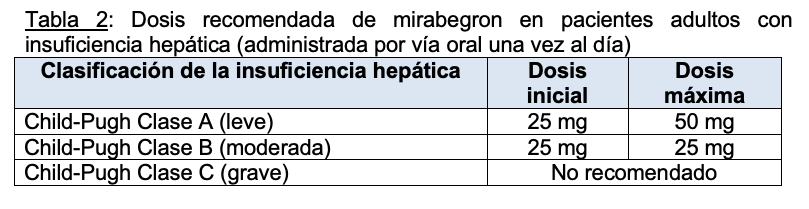
Forma de administración: Los comprimidos de mirabegron de deben tragar enteros, con agua. No se deben masticar, dividir ni triturar los comprimidos. Se pueden tomar con o sin alimentos. Dosis omitidas: Se debe indicar a los pacientes que tomen las dosis omitidas tan pronto como lo recuerden, a menos que hayan pasado más de 12 horas desde la dosis olvidada. Si han pasado más de 12 horas, se puede omitir la dosis olvidada y la siguiente dosis se debe tomar a la hora habitual.
Contraindicaciones.
Pacientes con reacciones de hipersensibilidad conocidas a mirabegron o a cualquier excipiente del producto.
Reacciones adversas.
Las siguientes reacciones adversas se analizan con más detalle en otras secciones del prospecto. Hipertensión. Retención urinaria. Angioedema Experiencia en ensayos clínicos: Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no se pueden comparar directamente con las tasas de los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica clínica. Según datos de la referencia, en tres estudios de seguridad y eficacia, doble ciego, controlados con placebo, de 12 semanas de duración en pacientes con VH, se evaluó la seguridad de mirabegron en 2736 pacientes. Uno de los estudios también incluyó un control activo. Para los estudios combinados 432 pacientes recibieron mirabegron 25 mg, 1375 recibieron mirabegron 50 mg y 929 recibieron mirabegron 100 mg una vez al día. En estos estudios, la mayoría de los pacientes eran caucásicos (94%) y mujeres (72%) con una edad media de 59 años (rango de 18 a 95 años). También se evaluó la seguridad de mirabegron en 1632 pacientes que recibieron mirabegron 50 mg una vez al día (n = 812 pacientes) o mirabegron 100 mg (n = 820 pacientes) en un estudio de seguridad de 1 año, aleatorizado, de dosis fija, doble ciego y controlado con activo en pacientes con VH. De estos pacientes, 731 recibieron mirabegron en un estudio anterior de 12 semanas. Un total de 1385 pacientes recibieron mirabegron de forma continua durante al menos 6 meses, 1311 pacientes recibieron mirabegron durante al menos 9 meses y 564 pacientes recibieron mirabegron durante al menos 1 año. Los eventos adversos más frecuentes (0,2%) que llevaron a la interrupción en los estudios de 12 semanas de duración, para la dosis de 25 mg o 50 mg fueron náuseas, dolor de cabeza, hipertensión, diarrea, constipación, mareos y taquicardia. Más de un paciente informó como eventos adversos graves la fibrilación auricular (0,2%) y el cáncer de próstata (0,1%) y en una tasa mayor que el placebo. La Tabla 3 enumera las reacciones adversas, derivadas de todos los eventos adversos, que se informaron en los estudios de 12 semanas de duración con una incidencia mayor que el placebo y en 1% o más de los pacientes tratados con mirabegron 25 mg o 50 mg una vez al día. Las reacciones adversas notificadas con más frecuencia (más del 2 % de los pacientes con mirabegron y más que el placebo) fueron hipertensión, nasofaringitis, infección del tracto urinario y dolor de cabeza.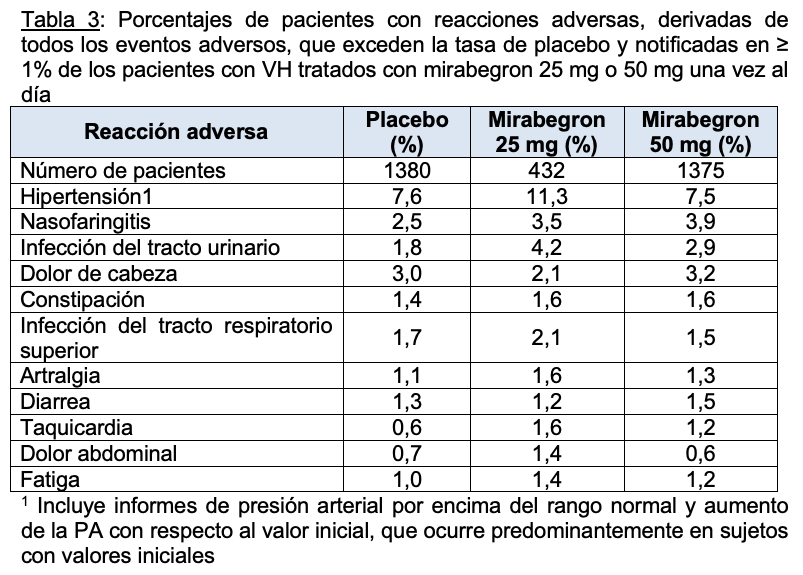
Otras reacciones adversas informadas por menos del 1% de los pacientes tratados con mirabegron en los estudios de 12 semanas de duración incluyeron: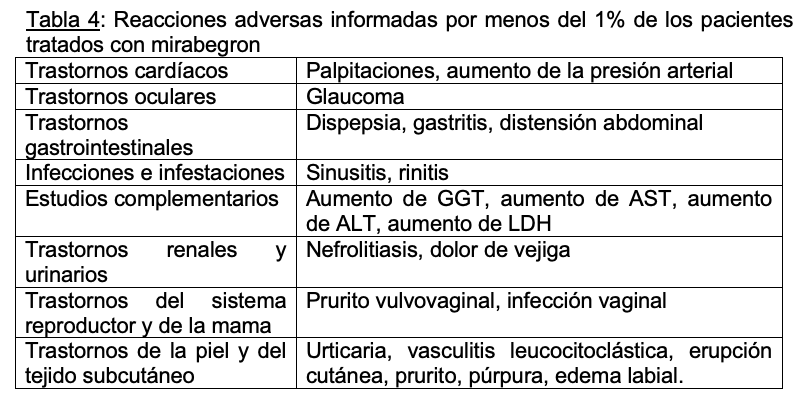
La Tabla 5 enumera las tasas de las reacciones adversas notificadas con más frecuencia, derivadas de todos los eventos adversos en pacientes tratados con mirabegron 50 mg durante hasta 52 semanas. Las reacciones adversas notificadas con mayor frecuencia ( > 3% de los pacientes con mirabegron) fueron hipertensión, infección del tracto urinario, dolor de cabeza y nasofaringitis.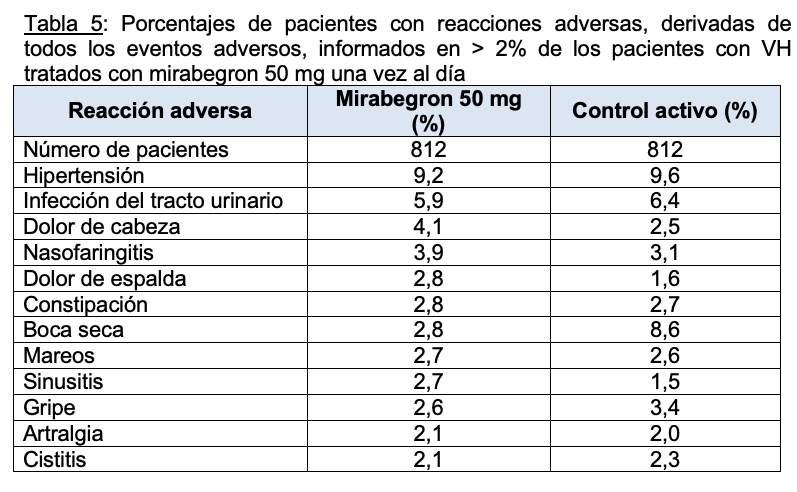
En el estudio de 52 semanas, en pacientes tratados con mirabegron 50 mg una vez al día, las reacciones adversas que llevaron a la interrupción informadas por más de 2 pacientes y a una tasa mayor que el control activo incluyeron: constipación (0,9%), dolor de cabeza (0,6%), mareos (0,5%), hipertensión (0,5%), ojos secos (0,4%), náuseas (0,4%), visión borrosa (0,4%) e infección del tracto urinario (0,4%). Los eventos adversos graves informados por al menos 2 pacientes y que excedieron el control activo incluyeron accidente cerebrovascular (0,4%) y osteoartritis (0,2%). La ALT/AST sérica aumentó con respecto al valor inicial más de 10 veces en 2 pacientes (0,3%) que tomaban mirabegron 50 mg; y estos marcadores posteriormente regresaron a los valores iniciales mientras ambos pacientes continuaron con mirabegron. En dicho estudio, el 0,1%, el 1,3% y el 0,5% de los pacientes tratados con mirabegron 50 mg, mirabegron 100 mg y control activo una vez al día informaron eventos adversos graves de neoplasia, respectivamente. Las neoplasias notificadas por 2 pacientes tratados con mirabegron 100 mg incluyeron cáncer de mama, neoplasia maligna de pulmón y cáncer de próstata. No se ha establecido una relación causal entre mirabegron y estas neoplasias notificadas. En un estudio clínico separado en Japón, se informó un solo caso de síndrome de Stevens-Johnson con aumento de ALT, AST y bilirrubina sérica en un paciente que tomaba mirabegron 100 mg, así como un medicamento a base de hierbas (Kyufu Gold). Experiencia posterior a la comercialización: Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de mirabegron. Debido a que estas reacciones son reportadas voluntariamente por una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al medicamento. Los siguientes eventos se han informado en asociación con el uso de mirabegron en la experiencia posterior a la comercialización a nivel mundial: Trastornos cardíacos: fibrilación auricular Trastornos gastrointestinales: náuseas, constipación, diarrea Trastornos del sistema nervioso: mareos, dolor de cabeza. Ha habido informes posteriores a la comercialización de confusión, alucinaciones, insomnio y ansiedad en pacientes que tomaban mirabegron. La mayoría de estos pacientes tenían condiciones médicas preexistentes o tomaban medicamentos concomitantes que pueden causar confusión, alucinaciones, insomnio y ansiedad. No se ha establecido una relación causal entre mirabegron y estos trastornos. Trastornos de la piel y del tejido subcutáneo: angioedema de la cara, labios, lengua y laringe, con o sin síntomas respiratorios; prurito Trastornos renales y urinarios: retención urinaria Notificación de sospechas de reacciones adversas: Notificación de Sospechas de Reacciones Adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Cualquier notificación de reacciones adversas, aunque estas sean conocidas, puede contribuir a detectar problemas relacionados con el uso de los medicamentos. Se invita a notificar las sospechas de reacciones adversas a través del Departamento de Farmacovigilancia del laboratorio vía email: fvigilancia@adium.com.ar o a través del siguiente enlace https://adium.creoscro.com/Safety/PublicGeneral y/o al Sistema Nacional de Farmacovigilancia al siguiente enlace: https://vigiflow-eforms.who-umc.org/ar/medicamentos
Precauciones.
Interacciones: Se realizaron estudios de interacción farmacológica en pacientes adultos para investigar el efecto de los fármacos coadministrados sobre la farmacocinética de mirabegron y el efecto de mirabegron sobre la farmacocinética de los fármacos coadministrados (p. ej., ketoconazol, rifampicina, succinato de solifenacina, tamsulosina y anticonceptivos orales). No se recomienda ningún ajuste de dosis cuando estos medicamentos se coadministran con mirabegron. Las siguientes son interacciones medicamentosas para las cuales se recomienda monitorear: Fármacos metabolizados por la enzima CYP2D6: Dado que mirabegron es un inhibidor moderado de CYP2D6, la exposición sistémica de los fármacos metabolizados por la enzima CYP2D6 aumenta cuando se coadministran con mirabegron. Por lo tanto, puede ser necesario un control adecuado y un ajuste de dosis cuando mirabegron se coadministre con estos medicamentos, especialmente con sustratos de CYP2D6 que tengan un índice terapéutico estrecho. Digoxina: Cuando se administraron en combinación, 100 mg de mirabegron aumentaron la Cmáx media de digoxina de 1,01 a 1,3 ng/ml (29 %) y el AUC de 16,7 a 19,3 ng.h/ml (27 %). La administración concomitante de 0,25 mg de digoxina con una combinación de 5 mg de solifenacina y 50 mg de mirabegron aumentó el AUCtau y la Cmáx de digoxina en aproximadamente un 10% y un 14%, respectivamente. Para los pacientes que inician una combinación de mirabegron y digoxina, inicialmente se debe considerar la dosis más baja de digoxina. Las concentraciones séricas de digoxina deben controlarse y usarse para ajustar la dosis de digoxina para obtener el efecto clínico deseado. Warfarina: La Cmáx media de warfarina S y R aumentó aproximadamente un 4% y el AUC aproximadamente un 9% cuando se administró como una dosis única de 25 mg después de dosis múltiples de 100 mg de mirabegron. Después de la administración de una dosis única de 25 mg de warfarina, mirabegron no tuvo ningún efecto sobre los criterios de valoración farmacodinámicos de warfarina, como la Razón Internacional Normalizada (RIN) y el tiempo de protrombina. Sin embargo, no se ha investigado completamente el efecto de mirabegron sobre dosis múltiples de warfarina y sobre los criterios de valoración farmacodinámicos de la warfarina, como el RIN y el tiempo de protrombina. Embarazo: No existen estudios con el uso de mirabegron en mujeres embarazadas o adolescentes que informen sobre el riesgo asociado al medicamento de defectos congénitos importantes, abortos espontáneos o resultados maternos o fetales adversos. La administración de mirabegron a animales preñados durante la organogénesis produjo variaciones esqueléticas reversibles (en ratas) a 22 veces (a través del AUC) la dosis humana máxima recomendada (MRHD) de 50 mg/día y una disminución del peso corporal fetal (en conejos) a 14 veces la dosis máxima recomendada en humanos (MRHD) de 50 mg/día. En exposiciones maternas tóxicas en ratas (96 veces MRHD), se observó una disminución del peso fetal y un aumento de la mortalidad fetal y, en conejos (36 veces MRHD), se observaron hallazgos cardíacos (cardiomegalia fetal y aorta fetal dilatada). Datos en animales: No se produjeron letalidad embriofetal ni anomalías morfológicas del desarrollo fetal en ratas preñadas después de la administración oral diaria de mirabegron durante el período de organogénesis (días 7 a 17 de gestación) en dosis de 0; 10; 30; 100 o 300 mg/kg. que se asociaron con exposiciones sistémicas (AUC) 0,1; 6; 22 y 96 veces la MRHD. Se observaron variaciones esqueléticas (costillas onduladas, osificación retardada) en fetos a dosis 22 veces superiores a la exposición sistémica en la MRHD y fueron reversibles durante el desarrollo. Las exposiciones 96 veces superiores a la MRHD fueron tóxicas para la madre (mortalidad, disminución del aumento de peso corporal) y se asociaron con una reducción del crecimiento fetal. Se trataron conejas preñadas con dosis orales diarias de mirabegron de 0; 3; 10 o 30 mg/kg/día durante el período de organogénesis (días 6 a 20 de gestación), lo que resultó en exposiciones plasmáticas de 0; 1; 14 o 36. veces la MRHD según el AUC. Con dosis de 10 mg/kg/día (14 veces la MRHD) y más, se redujo el peso corporal del feto. Con 30 mg/kg/día, se produjo toxicidad materna (aumento de la frecuencia cardíaca, mortalidad, reducción del aumento de peso corporal, reducción del consumo de alimentos) y se observaron muertes fetales, cardiomegalia fetal y dilatación de la aorta fetal a niveles de exposición sistémica (AUC) 36 veces mayores que los MRHD. En un estudio de desarrollo pre y posnatal, las ratas fueron tratadas con dosis orales diarias de mirabegron de 0; 10; 30 o 100 mg/kg/día (0; 1; 6 o 22 veces la MRHD) desde el día 7 de gestación hasta el día 20 después del nacimiento. Se observó una disminución del peso corporal materno junto con una disminución de la supervivencia de las crías en los primeros días después del nacimiento (92,7 % de supervivencia) en comparación con el grupo de control (98,8 % de supervivencia), con 100 mg/kg/día (22 veces la MRHD). El aumento de peso corporal de las crías se redujo hasta el día 7 posnatal, pero no se vio afectado durante el resto del período de lactancia. La exposición en el útero y durante la lactancia no afectó los hitos del desarrollo, el comportamiento o la fertilidad de la descendencia. No se observaron efectos con 30 mg/kg/día. Lactancia: No hay datos sobre la presencia de mirabegron en la leche humana, los efectos en el niño amamantado o los efectos en la producción de leche. Material relacionado con mirabegron estuvo presente en la leche de rata y en el estómago de cachorros lactantes luego de la administración de una dosis oral única de 10 mg/kg de mirabegron marcado con 14C a ratas lactantes. Cuando un fármaco está presente en la leche animal, es probable que esté presente en la leche humana. Se deben considerar los beneficios para el desarrollo y la salud de la lactancia materna junto con la necesidad clínica de la madre de mirabegron y cualquier posible efecto adverso en el niño amamantado debido a mirabegron o a la afección materna subyacente. Uso pediátrico: La seguridad y eficacia se han establecido únicamente para el tratamiento de la hiperactividad neurogénica del detrusor (HND) en pacientes pediátricos de 3 años de edad o más y que pesen 35 kg o más. Uso geriátrico: De 5.648 pacientes que recibieron mirabegron en monoterapia en los estudios de fase 2 y 3 para VH, 2.029 (35,9%) tenían 65 años de edad o más, y 557 (9,9%) tenían 75 años de edad o más. En estos estudios no se observaron diferencias generales en seguridad o eficacia entre pacientes menores de 65 años y aquellos de 65 años o más. Insuficiencia renal: Mirabegron no se ha estudiado en pacientes con enfermedad renal terminal (TFGe < 15 ml/min/1,73 m2) ni en pacientes que requieren hemodiálisis y, por lo tanto, no se recomienda su uso en estas poblaciones de pacientes. No es necesario ajustar la dosis en pacientes con insuficiencia renal leve o moderada (TFGe de 30 a 89 ml/min/1,73 m2). En pacientes adultos con insuficiencia renal grave (TFGe de 15 a 29 ml/min/1,73 m2), la dosis diaria de mirabegron no debe exceder los 25 mg. Insuficiencia hepática: Mirabegron no se han estudiado en pacientes con insuficiencia hepática grave (Child-Pugh Clase C) y, por lo tanto, no se recomienda su uso en esta población de pacientes. No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve (Child-Pugh Pugh Clase A). En pacientes adultos con insuficiencia hepática moderada (Child-Pugh Clase B), la dosis diaria de mirabegron no debe exceder los 25 mg.
Advertencias.
Aumentos de la presión arterial: Mirabegron puede aumentar la presión arterial. Se recomiendan determinaciones periódicas de la presión arterial, especialmente en pacientes hipertensos. Mirabegron no se recomienda para su uso en pacientes con hipertensión grave no controlada (definida como presión arterial sistólica mayor o igual a 180 mm Hg y/o presión arterial diastólica mayor o igual a 110 mm Hg). En dos estudios aleatorios, controlados con placebo, con voluntarios adultos sanos, mirabegron se asoció con aumentos relacionados con la dosis en la presión arterial en posición supina. En estos estudios, con la dosis máxima recomendada de 50 mg, el aumento máximo medio de la presión arterial sistólica/diastólica fue aproximadamente 3,5/1,5 mm Hg mayor que el placebo. Por el contrario, en pacientes adultos con VH en ensayos clínicos, mirabegron, tomado como monoterapia o en combinación con solifenacina 5 mg, el aumento medio de la presión arterial sistólica y diastólica con la dosis máxima recomendada de mirabegron de 50 mg fue aproximadamente de 0,5 a 1 mm Hg mayor que el placebo. Con poca frecuencia se informó un empeoramiento de la hipertensión preexistente en pacientes que tomaban mirabegron. Retención urinaria en pacientes con obstrucción de salida de la vejiga y en pacientes que toman medicamentos antagonistas muscarínicos para la VH: En pacientes que toman mirabegron, se ha informado que ocurre retención urinaria en pacientes con obstrucción de salida de la vejiga y en pacientes que toman medicamentos antagonistas muscarínicos para el tratamiento de VHA. Un estudio clínico controlado de seguridad en pacientes con obstrucción de salida de la vejiga no demostró un aumento de la retención urinaria en pacientes tratados con mirabegron; sin embargo, mirabegron aún debe administrarse con precaución a pacientes con obstrucción de salida de la vejiga clínicamente significativa. Por ejemplo, controle a estos pacientes para detectar signos y síntomas de retención urinaria. Mirabegron también debe administrarse con precaución a pacientes que toman medicamentos antagonistas muscarínicos para el tratamiento de la VH, incluyendo solifenacina. Angioedema: Se ha informado angioedema de la cara, labios, lengua y/o laringe con mirabegron. En algunos casos, el angioedema ocurrió después de la primera dosis; sin embargo, se han informado casos que ocurrieron horas después de la primera dosis o después de múltiples dosis. El angioedema, asociado con la inflamación de las vías respiratorias superiores, puede poner en peligro la vida. Si se produce afectación de la lengua, la hipofaringe o la laringe, suspenda de inmediato el tratamiento con mirabegron y proporcione la terapia adecuada y/o las medidas necesarias para garantizar una vía aérea permeable. Pacientes que toman medicamentos metabolizados por CYP2D6: Dado que mirabegron es un inhibidor moderado de CYP2D6, la exposición sistémica a los sustratos de CYP2D6 aumenta cuando se coadministra con mirabegron. Por lo tanto, puede ser necesario un control adecuado y un ajuste de dosis, especialmente con fármacos de índice terapéutico estrecho metabolizados por CYP2D6.
Conservación.
Mantener a temperatura ambiente preferentemente entre 15 y 30 °C.
Sobredosificación.
Mirabegron se ha administrado a voluntarios sanos en dosis únicas de hasta 400 mg. Con esta dosis, los eventos adversos informados incluyeron palpitaciones (1 de 6 sujetos) y aumento de la frecuencia del pulso que excedió los 100 latidos por minuto (3 de 6 sujetos). Múltiples dosis de mirabegron de hasta 300 mg diarios durante 10 días mostraron aumentos en la frecuencia del pulso y la presión arterial sistólica cuando se administraron a voluntarios sanos. El tratamiento de la sobredosis debe ser sintomático y de apoyo. En caso de sobredosis, se recomienda monitorizar la frecuencia del pulso, la presión arterial y el ECG. Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los centros de toxicología: Hospital de Pediatría Ricardo Gutiérrez: Teléfono: (011) 4962-6666/2247. Hospital A. Posadas Teléfono: (011) 4654-6648/4658-7777. Centro de Asistencia Toxicológica de La Plata Teléfono: (0221) 451-5555. Optativamente otros centros de intoxicaciones.
Presentación.
Envases conteniendo 30 comprimidos recubiertos de liberación prolongada de 25 mg y 50 mg.