LYRICA
ASPEN PHARMA
Antiepiléptico.
Composición.
Cada cápsula contiene: 25 mg: Pregabalina 25 mg. Lactosa monohidrato 35 mg. Almidón de maíz 20 mg. Talco 20 mg. 50 mg: Pregabalina 50 mg. Lactosa monohidrato 70 mg. Almidón de maíz 40 mg. Talco 40 mg. 75 mg: Pregabalina 75 mg. Lactosa monohidrato 8,25 mg. Almidón de maíz 8,375 mg. Talco 8,375 mg. 150 mg: Pregabalina 150 mg. Lactosa monohidrato 16,5 mg. Almidón de maíz 16,75 mg. Talco 16,75 mg. 300 mg: Pregabalina 300 mg. Lactosa monohidrato 33 mg. Almidón de maíz 33,5 mg. Talco 33,5 mg.
Farmacología.
Propiedades Farmacodinámicas: El principio activo, pregabalina, es un análogo del ácido gamma-aminobutírico (GABA) (ácido (S)-3- (aminometil)-5-metilhexanoico). Mecanismo de acción: La pregabalina se une a una subunidad auxiliar (proteína a-2 -) de los canales de calcio voltaje dependientes en el Sistema Nervioso Central, desplazando potencialmente a [3H]-gabapentina. Experiencia clínica: Dolor neuropático: Se ha demostrado la eficacia en estudios en neuropatía diabética, neuralgia post-herpética y lesión de la médula espinal. No se ha estudiado la eficacia en otros modelos de dolor neuropático. La pregabalina se ha estudiado en 10 estudios clínicos controlados con una duración de hasta 13 semanas y dos administraciones al día (DVD) y con una duración de hasta 8 semanas y tres administraciones al día (TVD). En términos generales, los perfiles de seguridad y eficacia para los regímenes posológicos de dos y tres veces al día fueron similares. En ensayos clínicos de hasta 12 semanas de duración en dolor neuropático periférico y central, se observó una reducción del dolor a la primera semana de tratamiento y se mantuvo a lo largo del período de tratamiento. En ensayos clínicos controlados en dolor neuropático periférico, el 35% de los pacientes tratados con pregabalina y el 18% de los pacientes tratados con placebo experimentaron una mejoría de un 50% en la escala de dolor. En el caso de los pacientes que no experimentaron somnolencia, dicha mejoría se observó en un 33% de los pacientes tratados con pregabalina y en un 18% de los pacientes tratados con placebo. En el caso de los pacientes que experimentaron somnolencia, los porcentajes que respondieron fueron del 48% para pregabalina y 16% para placebo. En el estudio clínico controlado en dolor neuropático central, el 22% de los pacientes tratados con pregabalina y el 7% de los pacientes tratados con placebo tuvieron una mejoría del 50% en la escala de dolor. Epilepsia: La pregabalina se ha estudiado en 3 estudios clínicos controlados con una duración de hasta 12 semanas tanto con dos (DVD) como con tres administraciones al día (TVD). En términos generales, los perfiles de seguridad y eficacia para los regímenes posológicos de dos y tres veces al día fueron similares. Se observó una reducción en la frecuencia de las crisis a la primera semana de tratamiento. Población pediátrica: No se ha establecido la eficacia ni la seguridad de pregabalina como tratamiento complementario para la epilepsia en pacientes pediátricos de menos de 12 años y adolescentes. Los acontecimientos adversos observados en un estudio de farmacocinética y tolerabilidad en el que participaron pacientes de entre 3 meses y 16 años de edad (n = 65) con crisis de epilepsia parciales, fueron similares a los observados en los adultos. Los resultados de un estudio de 12 semanas controlado con placebo de 295 pacientes pediátricos entre 4 y 16 años y otro estudio de 14 días, controlado con placebo, de 175 pacientes pediátricos de 1 mes hasta 4 años de edad, realizados para evaluar la eficacia y seguridad de la pregabalina como tratamiento complementario para el tratamiento de crisis de inicio parcial, y un estudio de seguridad, abierto, de 1 año de duración en 54 pacientes pediátricos de entre 3 meses y 16 años de edad con epilepsia indican que los acontecimientos adversos de pirexia e infecciones respiratorias altas se observaron con mayor frecuencia que en los estudios en pacientes adultos con epilepsia (ver Posologia y forma de administracion, Reacciones adversas y Propiedades farmacocinéticas). En el estudio de 12 semanas controlado con placebo, los pacientes pediátricos (de 4 a 16 años de edad) fueron asignados a pregabalina de 2,5 mg/kg/día (máximo, 150 mg/día) o a pregabalina de 10 mg/ kg/día (máximo, 600 mg/día) o placebo. El porcentaje de sujetos con al menos un 50% de reducción en las crisis de inicio parciales en comparación con el inicio, fue 40,6% de los sujetos tratados con pregabalina 10 mg/kg/día (p = 0,0068 versus placebo), 29,1% de los sujetos tratados con 2,5 mg de pregabalina/kg/día (p = 0.2600 versus placebo) y 22.6% de los que recibieron placebo. En el estudio de 14 días controlado con placebo, los pacientes pediátricos (de 1 mes hasta menos de 4 años de edad) recibieron 7 mg/kg/día de pregabalina, 14 mg/kg/día de pregabalina o placebo. La mediana de la frecuencia de las crisis en 24 horas al inicio y en la visita final fue de 4,7 y de 3,8 para 7 mg/kg/día de pregabalina, 5,4 y 1,4 para 14 mg/kg/día de pregabalina y 2,9 y 2,3 para placebo. Pregabalina 14 mg/kg/día redujo significativamente la frecuencia de inicio de las crisis parciales transformadas logarítmicamente en comparación con placebo (p = 0,0223); pregabalina 7 mg/kg/día no mostró mejoría en comparación con placebo. Monoterapia (pacientes recientemente diagnosticados): La pregabalina se ha estudiado en un ensayo clínico controlado de 56 semanas de duración y con una dosificación de dos veces al día (DVD). La pregabalina no alcanzó la ausencia de inferioridad de la lamotrigina con base en el criterio de valoración de 6 meses sin convulsiones. Se encontró que, de manera similar, la pregabalina y la lamotrigina fueron inocuas y bien toleradas. Trastorno de ansiedad generalizada: La pregabalina se ha evaluado en 6 estudios controlados de 4-6 semanas de duración, un estudio en ancianos de 8 semanas de duración y un estudio a largo plazo de prevención de recaídas con una fase doble ciego de prevención de recaídas de 6 meses de duración. En la primera semana se observó un alivio de los síntomas del TAG como se reflejó en la Escala de Valoración de la Ansiedad de Hamilton (HAM-A). En los estudios clínicos controlados (4-8 semanas de duración) el 52% de los pacientes tratados con pregabalina y el 38% de los que recibieron placebo mejoraron la puntuación total de la HAM-A en al menos un 50% desde la visita basal hasta la finalización del estudio. En estudios clínicos controlados, una mayor proporción de pacientes tratados con pregabalina, en comparación con aquellos tratados con placebo, notificó visión borrosa, evento que en la mayoría de los casos se resolvió al continuar con el tratamiento. Se realizaron pruebas oftalmológicas (incluyendo pruebas de agudeza visual, pruebas de campo visual y examen fundoscópico en pupila dilatada) a más de 3.600 pacientes como parte de los estudios clínicos controlados. En dichos pacientes la agudeza visual se redujo en un 6,5% de los pacientes tratados con pregabalina frente al 4,8% de los pacientes tratados con placebo. Se detectaron alteraciones del campo visual en el 12,4% de los pacientes tratados con pregabalina frente al 11,7% de los pacientes tratados con placebo. Se observaron cambios fundoscópicos en el 1,7% de los pacientes tratados con pregabalina frente al 2,1% de los pacientes tratados con placebo. Fibromialgia: La eficacia de Lyrica para el tratamiento de la fibromialgia se determinó en un estudio multicéntrico, doble ciego, controlado con placebo, de 14 semanas de duración (F1) y en un estudio aleatorizado de retiro de la droga, de 6 meses de duración (F2). En los estudios F1 y F2 participaron pacientes a los cuales se les diagnosticó la fibromialgia usando los criterios del Colegio Estadounidense de Reumatología [American College of Rheumatology (ACR)] (antecedentes de dolor diseminado durante 3 meses y dolor presente en 11 o más de 18 puntos específicos de dolor a la palpación). Los estudios demostraron una reducción del dolor en la escala analógica visual. Además, la mejoría se demostró en base a la evaluación global del paciente (PGIC), y en el cuestionario de impacto de la fibromialgia [Fibromialgia Impact Questionnaire (FIQ)]. Estudio F1: En este estudio de 14 semanas de duración se compararon dosis diarias de 300 mg, 450 mg y 600 mg de Lyrica con un placebo. Los pacientes fueron incluidos con un puntaje medio mínimo del dolor basal mayor o igual a 4 en una escala de 11 puntos de calificación numérica del dolor y una puntuación mayor o igual a 40 mm en la escala analógica visual del dolor de 100 mm (VAS). El puntaje medio del dolor basal en este estudio fue de 6,7. Los pacientes que respondieron al placebo en la fase inicial de una semana, no fueron aleatorizados a las fases posteriores del estudio. El 64% de los pacientes aleatorizados al tratamiento con Lyrica completó el estudio. No hubo evidencias de un mayor efecto sobre los puntajes del dolor con la dosis de 600 mg diarios en comparación con la dosis de 450 mg diarios, pero sí hubo evidencias de reacciones adversas dependientes de la dosis (ver Reacciones adversas). En algunos pacientes el dolor se redujo ya a partir de la Semana 1, y dicha reducción persistió durante todo el estudio. Estudio F2: En este estudio aleatorizado de retiro de la droga se comparó a Lyrica con un placebo. Durante una fase abierta de optimización de la dosis de 6 semanas de duración, se ajustó la dosis a un total diario de 300 mg, 450 mg, o 600 mg. Se consideró que el paciente respondía si tenía tanto: 1) una reducción de al menos un 50% del dolor (VAS) como, 2) si calificaba su mejoría general en la PGIC como "mejor" o "mucho mejor. A los pacientes que respondieron al tratamiento se los aleatorizó posteriormente a la fase de tratamiento doble ciego para recibir la dosis alcanzada en la fase abierta o el placebo. Los pacientes recibieron tratamiento durante un máximo de 6 meses a partir de la aleatorización. La eficacia se evaluó en función del tiempo transcurrido hasta la pérdida de la eficacia terapéutica, definida como 1) una reducción de menos del 30% del dolor (VAS) respecto del nivel basal del tratamiento abierto durante dos visitas consecutivas de la fase doble ciego, o 2) empeoramiento de los síntomas de la FM que requiriera un tratamiento alternativo. El 54% de los pacientes soportaron la titulación hasta una dosis eficaz y tolerable de Lyrica durante la fase abierta de 6 semanas. De los pacientes que ingresaron a la fase de tratamiento aleatorio que continuaron el tratamiento con Lyrica, el 38% de los pacientes completó las 26 semanas de tratamiento en comparación con el 19% de los pacientes tratados con el placebo. Cuando se consideró el regreso del dolor o el retiro a causa de los eventos adversos como la pérdida de la respuesta (LTR), el tratamiento con Lyrica produjo un mayor tiempo hasta la pérdida de la respuesta terapéutica que el tratamiento con el placebo. El 53% de los sujetos tratados con pregabalina, en comparación con el 33% de los pacientes tratados con el placebo, continuaron tomando la droga del estudio y mantuvieron la respuesta terapéutica hacia la Semana 26 del estudio. El tratamiento con Lyrica también tuvo como resultado un tiempo más prolongado hasta la pérdida de la respuesta en base al FIQ, y un tiempo más prolongado hasta la pérdida de la evaluación general del estado del paciente, medida con la PGIC. Propiedades Farmacocinéticas: Los parámetros farmacocinéticos de pregabalina en el estado estable son similares en voluntarios sanos, pacientes con epilepsia recibiendo fármacos antiepilépticos y pacientes con dolor crónico. Absorción: La pregabalina se absorbe rápidamente cuando se administra en ayunas, alcanzando concentraciones plasmáticas máximas una hora tras la administración tanto de dosis única como de dosis múltiples. La biodisponibilidad oral de pregabalina se estima que es ≥ 90% y es independiente de la dosis. Tras la administración repetida, el estado estable se alcanza dentro de 24 a 48 horas. La velocidad de absorción de pregabalina disminuye cuando se administra con alimentos, produciéndose un descenso en la Cmax de aproximadamente un 25-30% y un retraso en el Tmax hasta aproximadamente 2,5 horas. Sin embargo, la administración de pregabalina junto con alimentos no tiene ningún efecto clínicamente significativo sobre el grado de absorción de pregabalina. Distribución: En estudios preclínicos, se ha visto que la pregabalina atraviesa la barrera hematoencefálica en ratones, ratas y monos. Se ha visto que la pregabalina atraviesa la placenta en ratas y está presente en la leche de ratas lactantes. En humanos, el volumen de distribución aparente de la pregabalina tras la administración oral es de aproximadamente 0,56 l/kg. La pregabalina no se une a las proteínas plasmáticas. Biotransformación: La pregabalina sufre un metabolismo insignificante en humanos. Tras una dosis de pregabalina marcada isotópicamente, aproximadamente el 98% de la radioactividad recuperada en la orina procedía de pregabalina inalterada. El derivado N-metilado de pregabalina, metabolito principal de ésta encontrado en orina, representó el 0,9% de la dosis. En estudios preclínicos, no hubo indicios de que el S-enantiómero de pregabalina se racemice al R-enantiómero. Eliminación: La pregabalina se elimina del sistema circulatorio principalmente mediante excreción renal como fármaco inalterado. La vida media de eliminación promedio de pregabalina es de 6,3 horas. El clearance plasmático y renal de la pregabalina son directamente proporcionales al clearance de creatinina (ver Alteración de la función renal). Es necesario un ajuste de la dosis en pacientes con la función renal alterada o en hemodiálisis (ver Posología y forma de administración - Tabla 1). Linealidad / no linealidad: La farmacocinética de pregabalina es lineal en el rango de dosis diaria recomendada. La variabilidad farmacocinética interindividual de pregabalina es baja ( < 20%). La farmacocinética de dosis múltiples es predecible a partir de los datos obtenidos con dosis única. Por tanto, no es necesario llevar un monitoreo de rutina de las concentraciones plasmáticas de pregabalina. Farmacocinética en grupos especiales de pacientes: Sexo: Los ensayos clínicos indican que el sexo no tiene influencia clínicamente significativa sobre las concentraciones plasmáticas de pregabalina. Alteración de la función renal: El clearance de pregabalina es directamente proporcional al clearance de creatinina. Además, la pregabalina se elimina del plasma de forma eficaz mediante hemodiálisis (tras una sesión de hemodiálisis de 4 horas, las concentraciones plasmáticas de pregabalina se reducen aproximadamente al 50%). Dado que la eliminación por vía renal es la principal vía de eliminación, en pacientes con insuficiencia renal es necesaria una reducción de la dosis y una dosis complementaria tras la hemodiálisis (ver Posología y Forma de Administración, ver Tabla 1). Alteración de la función hepática: No se han llevado a cabo estudios de farmacocinética específicos en pacientes con función hepática alterada. Puesto que la pregabalina no sufre un metabolismo significativo y se excreta mayoritariamente como fármaco inalterado en orina, no es previsible que la alteración de la función hepática altere de forma significativa las concentraciones plasmáticas de pregabalina. Población pediátrica: En un estudio de farmacocinética y tolerabilidad se evaluó la farmacocinética de pregabalina en pacientes pediátricos con epilepsia (grupos de edad: de 1 a 23 meses, de 2 a 6 años, de 7 a 11 años y de 12 a 16 años) con concentraciones de dosis de 2,5, 5, 10 y 15 mg/kg/día. En general, el tiempo transcurrido hasta alcanzar la concentración plasmática máxima tras la administración oral de pregabalina a pacientes pediátricos en ayunas fue similar en todo el grupo de edad y se produjo entre 0,5 horas y 2 horas después de la dosis. Los parámetros de Cmax y AUC de pregabalina aumentaron de forma lineal con el aumento de la dosis en cada grupo de edad. El AUC fue un 30% menor en los pacientes pediátricos con un peso inferior a 30 kg debido a un mayor aclaramiento ajustado al peso corporal del 43% en estos pacientes en comparación con los pacientes con un peso ≥30 kg. La semivida terminal promedio de pregabalina fue, aproximadamente, de entre 3 y 4 horas en los pacientes pediátricos de hasta 6 años de edad, y de entre 4 y 6 horas en los de 7 años o más. El análisis de farmacocinética poblacional mostró que el aclaramiento de creatinina era una covariable significativa del aclaramiento de pregabalina oral, el peso corporal era una covariable significativa del volumen de distribución aparente de pregabalina oral, y que dichas relaciones eran similares en los pacientes pediátricos y adultos. No se ha estudiado la farmacocinética de pregabalina en pacientes de menos de 3 meses de edad (ver Posologia y forma de administracion, Reacciones adversas y Propiedades Farmacodinámicas). Ancianos: El clearance de pregabalina tiende a disminuir al aumentar la edad. Este descenso en el clearance de pregabalina oral está en relación con el descenso del clearance de creatinina asociado con el aumento de la edad. Podría requerirse una reducción de la dosis de pregabalina en pacientes que tengan la función renal alterada debido a la edad (ver Posología y forma de administración - Tabla 1). Madres lactantes: Se evaluó la farmacocinética de 150 mg de pregabalina administrados cada 12 horas (dosis diaria de 300 mg), en 10 mujeres lactantes, tras al menos 12 semanas después del parto. La lactancia tuvo un efecto nulo o pequeño sobre la farmacocinética de pregabalina. La pregabalina se excretó por la leche materna a concentraciones promedio, en estado estacionario, de aproximadamente el 76% de las presentes en el plasma materno. La dosis estimada para el lactante procedente de la leche materna (suponiendo un consumo medio de leche de 150 ml/kg/día) de las mujeres que reciben 300 mg/día o la dosis máxima de 600 mg/día sería de 0,31 o 0,62 mg/kg/día, respectivamente. Estas dosis estimadas son aproximadamente el 7% de la dosis materna diaria total, en mg/kg.
Indicaciones.
Dolor neuropático: Lyrica está indicado en el tratamiento del dolor neuropático periférico y central en adultos. Epilepsia: Lyrica está indicado en adultos como terapia adjunta de las crisis parciales con o sin generalización secundaria. Trastorno de ansiedad generalizada: Lyrica está indicado en el tratamiento del trastorno de ansiedad generalizada (TAG) en adultos. Fibromialgia: Lyrica está indicado en el tratamiento de la fibromialgia en adultos.
Dosificación.
El rango de dosis es de 150 a 600 mg al día, dividiendo su administración en dos o tres tomas. Lyrica se puede administrar con o sin alimentos. Lyrica se administra únicamente por vía oral. Dolor neuropático: El tratamiento con pregabalina se puede comenzar con una dosis de 150 mg al día, que se puede administrar dividida en dos o tres tomas. En función de la respuesta y tolerabilidad individual de cada paciente, la dosificación se puede incrementar hasta 300 mg al día después de un intervalo de 3 a 7 días, y si fuese necesario, hasta una dosis máxima de 600 mg al día después de un intervalo adicional de 7 días. Epilepsia: El tratamiento con pregabalina se puede iniciar con una dosis de 150 mg al día, que se puede administrar dividida en dos o tres tomas. En función de la respuesta y tolerabilidad individual de cada paciente, la dosis se puede incrementar a 300 mg al día después de una semana. La dosis máxima que se puede alcanzar, después de una semana adicional, es de 600 mg al día. Trastorno de ansiedad generalizada: El rango de dosis es de 150 a 600 mg al día, dividiendo su administración en dos o tres tomas. Se debe reevaluar de forma periódica la necesidad del tratamiento. El tratamiento con pregabalina se puede iniciar con una dosis de 150 mg al día. En función de la respuesta y tolerabilidad individual de cada paciente, la dosis se puede incrementar a 300 mg al día después de una semana. Tras una semana adicional, la dosis se puede incrementar a 450 mg al día. La dosis máxima que se puede alcanzar, después de una semana adicional, es de 600 mg al día. Fibromialgia: La dosis recomendada de Lyrica para el tratamiento de la fibromialgia es de 300 a 450 mg/día. El tratamiento deberá comenzar con una dosis de 75 mg dos veces por día (150 mg/día) y se puede incrementar a 150 mg dos veces por día (300 mg/día) en el intervalo de una semana en base a la eficacia y tolerabilidad. Los pacientes que no experimenten beneficio suficiente con 300 mg/día, se les puede incrementar la dosis a 225 mg dos veces por día (450 mg/día). Aunque Lyrica se estudió también a dosis de 600 mg/día, no hay evidencia de que esta dosis brinde un beneficio adicional y esta dosis no fue muy bien tolerada. No se recomienda el tratamiento con dosis mayores a 450 mg/día, dadas las reacciones adversas dosis-dependientes (ver Reacciones adversas). Dado que la pregabalina se elimina principalmente por excreción renal, la dosis debe ser ajustada en pacientes con función renal reducida (ver Pacientes con alteración de la función renal). Interrupción del tratamiento con pregabalina: De acuerdo con la práctica clínica actual, si se tiene que interrumpir el tratamiento con pregabalina, se deberá hacer de forma gradual durante un período mínimo de 1 semana independientemente de la indicación (ver Advertencias y precauciones especiales de uso y Reacciones adversas). Poblaciones especiales: Alteración de la función renal: La pregabalina se elimina del sistema circulatorio principalmente por excreción renal como fármaco inalterado. Dado que el clearance plasmático de pregabalina es directamente proporcional al clearance de creatinina (ver Propiedades Farmacocinéticas), la reducción de la dosis en pacientes con función renal alterada se deberá realizar de forma individualizada de acuerdo al clearance de creatinina (Ccr), tal como se indica en la Tabla 1, que se ha determinado usando la fórmula siguiente:
La pregabalina se elimina del plasma de forma eficaz mediante hemodiálisis (50% del fármaco en 4 horas). En pacientes sometidos a hemodiálisis, se debe ajustar la dosis diaria de pregabalina según su función renal. Además de la dosis diaria, después de cada sesión de 4 horas de hemodiálisis se debe administrar de forma inmediata una dosis complementaria (ver Tabla 1).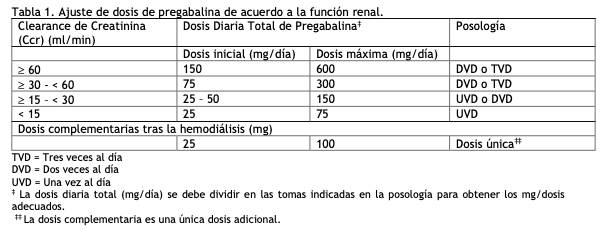
Alteración de la función hepática: No se requiere ajuste de la dosis en pacientes con la función hepática alterada (ver Propiedades Farmacocinéticas). Población pediátrica: No se ha establecido la seguridad y eficacia de Lyrica en niños menores de 12 años ni en adolescentes (de 12 a 17 años de edad). Los datos actualmente disponibles están descriptos en las secciones de Reacciones adversas y Propiedades Farmacocinéticas/Farmacodinámicas, sin embargo no se puede hacer una recomendación posológica. Uso en ancianos: Los pacientes ancianos pueden precisar una reducción de la dosis de pregabalina debido a la disminución de la función renal (ver Uso en pacientes con alteración de la función renal).
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes.
Reacciones adversas.
El programa clínico de pregabalina incluyó a más de 8900 pacientes que fueron expuestos a pregabalina, de los que más de 5600 participaron en ensayos doble ciego controlados con placebo. Las reacciones adversas comunicadas con más frecuencia fueron mareos y somnolencia. Generalmente, las reacciones adversas fueron de intensidad leve a moderada. En todos los estudios controlados, la tasa de abandono a causa de reacciones adversas fue del 12% para pacientes que estaban recibiendo pregabalina y del 5% para pacientes que recibieron placebo. Las reacciones adversas que con más frecuencia dieron lugar a una interrupción del tratamiento en los grupos tratados con pregabalina fueron mareos y somnolencia. En la tabla 2 siguiente se relacionan todas las reacciones adversas, que tuvieron lugar con una incidencia superior a la detectada con placebo y en más de un paciente, ordenadas por sistema y frecuencia [muy frecuentes (≥ 1/10), frecuentes (≥1/100, ≤1/10), poco frecuentes (≥1/1.000 y ≤1/100), raras (≥1/10.000 a ≤1/1000), muy raras (≤1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles)]. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Las reacciones adversas enumeradas también pueden estar relacionadas con la enfermedad subyacente y/o con la medicación que se administra concomitantemente. En el tratamiento del dolor neuropático central debido a lesión de la médula espinal se incrementó la incidencia de eventos adversos en general, efectos adversos a nivel del SNC y especialmente somnolencia (ver Advertencias). Las reacciones adversas adicionales notificadas durante la experiencia post-comercialización se incluyen en la siguiente tabla en cursiva.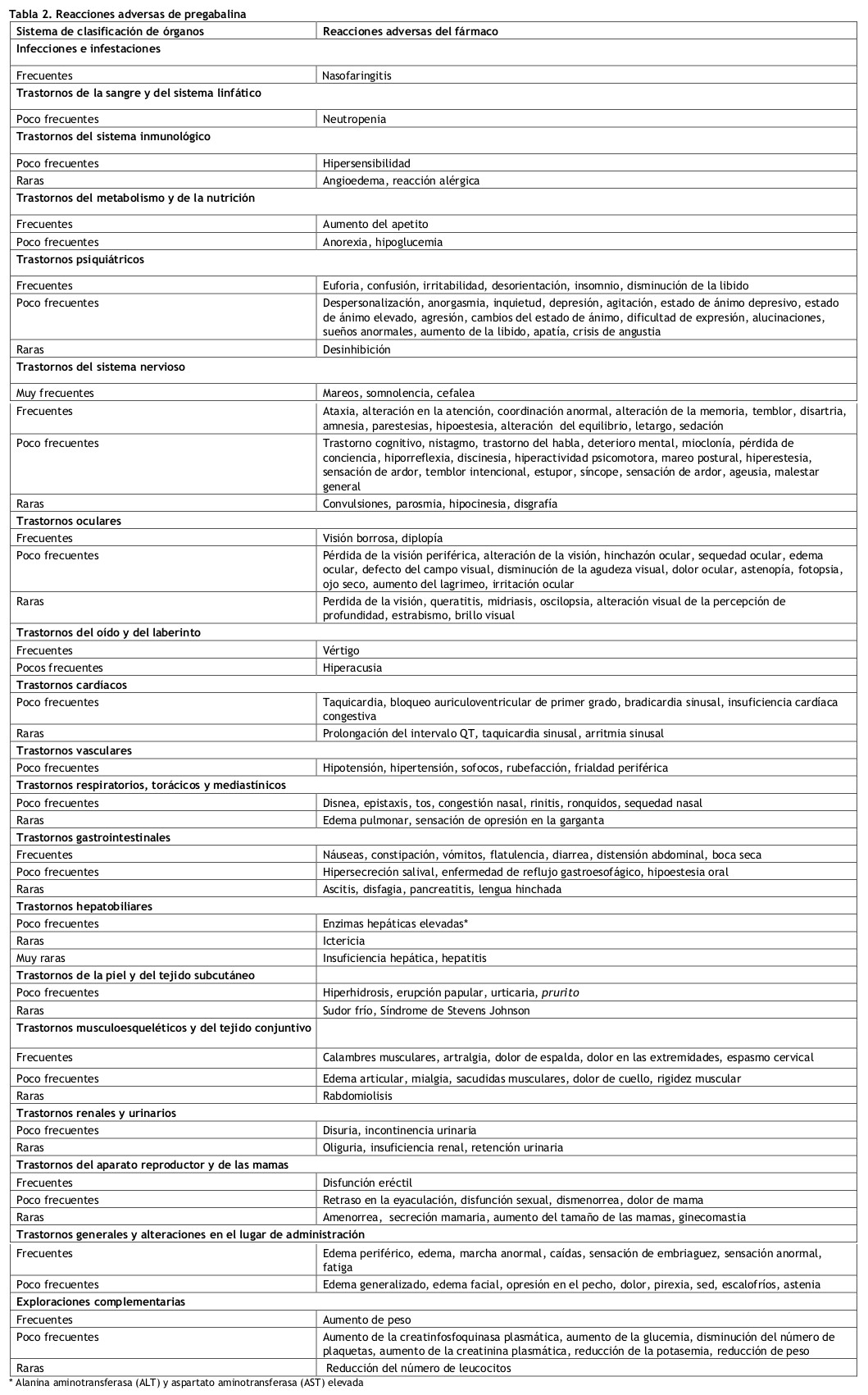
En algunos pacientes se han observado síntomas de retirada tras la interrupción del tratamiento con pregabalina tanto a corto como a largo plazo. Se han mencionado los siguientes eventos: insomnio, dolor de cabeza, náuseas, ansiedad, diarrea, síndrome gripal, convulsiones, nerviosismo, depresión, dolor, hiperhidrosis y mareos, sugestivos de dependencia física. Se debe informar al paciente sobre esto al inicio del tratamiento. Con respecto a la interrupción del tratamiento de pregabalina a largo plazo, los datos sugieren que la incidencia y gravedad de los síntomas de retirada pueden estar relacionadas con la dosis. Población pediátrica: El perfil de seguridad de pregabalina observado en cuatro estudios en pacientes pediátricos con crisis de epilepsia parciales con o sin generalización secundaria (estudio de seguridad y eficacia en 12 semanas en pacientes de 4 a 16 años de edad, n=295; estudio de eficacia y seguridad de 14 días en pacientes de 1 mes hasta menos de 4 años de edad, n=175; estudio de farmacocinética y tolerabilidad, n = 65; y un estudio de seguimiento de la seguridad, abierto, de 1 año de duración, n = 54) fue similar al observado en los estudios en pacientes adultos con epilepsia. Las reacciones adversas más comunes observadas en este estudio de 12 semanas de tratamiento con pregabalina fueron somnolencia, pirexia, infecciones del tracto respiratorio superior, aumento del apetito, aumento de peso y nasofaringitis. Los acontecimientos adversos más frecuentes observados en el estudio de 14 días con el tratamiento de pregabalina fueron somnolencia, infección de las vías respiratorias superiores y pirexia (ver Posología y forma de administración y Propiedades Farmacocinéticas/ Farmacodinámicas). Estudios controlados en fibromialgia: Reacciones adversas que motivaron la discontinuación: En estudios clínicos de pacientes con fibromialgia, el 19% de los pacientes tratados con pregabalina (150-600 mg/día) y 10% de los pacientes con placebo, discontinuaron el estudio prematuramente debido a las reacciones adversas. En el grupo tratado con pregabalina, las reacciones adversas más comunes que motivaron la discontinuación fueron: mareos (6%) y somnolencia (3%). En comparación, < 1% de los pacientes con placebo discontinuaron el estudio debido a mareos y somnolencia. Otras razones que motivaron la discontinuación de los estudios, que ocurrieron con mayor frecuencia en el grupo tratado con pregabalina que en el de placebo, fueron: fatiga, cefaleas, trastorno del equilibrio y aumento de peso. Cada una de estas reacciones adversas llevó a discontinuar el estudio en aproximadamente el 1% de los pacientes. En estudios clínicos controlados de Lyrica en fibromialgia, 106 pacientes fueron de 65 años de edad o mayores. A pesar de que el perfil de reacciones adversas fue similar en los dos grupos de edad, las siguientes reacciones adversas neurológicas fueron más frecuentes en pacientes de 65 años de edad o mayores: mareos, visión borrosa, trastorno del equilibrio, temblores, estado de confusión, coordinación anormal y letargia. Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas de acuerdo con las regulaciones locales.
Advertencias.
Intolerancia a la lactosa: Lyrica contiene lactosa. Los pacientes con problemas hereditarios raros de intolerancia a la galactosa, con deficiencia de Lapp lactasa o con mala absorción de glucosa-galactosa, no deben tomar este medicamento. Pacientes diabéticos: De acuerdo a la práctica clínica actual, ciertos pacientes diabéticos que ganen peso durante el tratamiento con pregabalina, pueden precisar un ajuste de la medicación hipoglucemiante. Reacciones de hipersensibilidad: Se han reportado durante la etapa posterior a la comercialización, reacciones de hipersensibilidad incluyendo casos de angioedema. La pregabalina debe suspenderse inmediatamente si se presentan síntomas de angioedema, tal como edema facial, perioral, o de las vías respiratorias superiores. Mareos, somnolencia, pérdida de la conciencia, confusión y alteración de la función mental: El tratamiento con pregabalina se ha asociado a mareos y somnolencia, lo cual podría incrementar los casos de lesiones accidentales (caídas) en la población anciana. También ha habido reportes post- comercialización de pérdida de la conciencia, confusión, y alteración de la función mental. Por tanto, se debe aconsejar a los pacientes que tengan precaución hasta que se familiaricen con los efectos potenciales del fármaco. Efectos relacionados con la visión: En estudios clínicos controlados, una mayor proporción de pacientes tratados con pregabalina, en comparación con aquellos tratados con placebo, notificó visión borrosa, evento que en la mayoría de los casos se resolvió al continuar con el tratamiento. En los estudios clínicos en los que se llevaron a cabo pruebas oftalmológicas, la incidencia de disminución de la agudeza visual y alteración del campo visual fue mayor en pacientes tratados con pregabalina que en aquellos tratados con placebo; la incidencia de cambios fundoscópicos fue mayor en pacientes tratados con placebo (ver Carácterísticas farmacológicas, Propiedades Farmacodinámicas). Durante el período posterior a la comercialización también se han notificado reacciones adversas visuales incluyendo pérdida de visión, visión borrosa u otros cambios de agudeza visual, muchos de los cuales fueron transitorios. La suspensión del tratamiento con pregabalina puede resolver o mejorar estos síntomas visuales. Insuficiencia renal: Se han notificado casos de insuficiencia renal que revirtieron con la interrupción del tratamiento con pregabalina. Retiro de la medicación antiepiléptica concomitante: No hay datos suficientes que permitan suprimir la medicación antiepiléptica concomitante, tras alcanzar el control de las crisis con pregabalina en el tratamiento combinado, para lograr la monoterapia con pregabalina. Síntomas de retirada: En algunos pacientes se han observado síntomas de retirada tras la interrupción del tratamiento con pregabalina tanto a corto como a largo plazo. Se han mencionado los siguientes eventos: insomnio, dolor de cabeza, náuseas, ansiedad, diarrea, síndrome gripal, nerviosismo, depresión, dolor, convulsiones, hiperhidrosis y mareos, sugestivos de dependencia física. Se debe informar al paciente sobre esto al inicio del tratamiento. Durante el tratamiento con pregabalina, o al poco tiempo de interrumpir el tratamiento con pregabalina, pueden aparecer convulsiones, incluyendo estatus epiléptico y convulsiones de tipo gran mal. Con respecto a la interrupción del tratamiento de pregabalina a largo plazo, se sugiere que la incidencia y gravedad de los síntomas de retirada pueden estar relacionadas con la dosis. Insuficiencia cardíaca congestiva: En algunos pacientes tratados con pregabalina se han recibido reportes posteriores a la comercialización de insuficiencia cardíaca congestiva. Estas reacciones se observan sobre todo en pacientes ancianos (mayores de 65 años) con función cardiovascular comprometida y tratados con pregabalina en la indicación de tratamiento del dolor neuropático. La pregabalina debe ser usada con cautela en este grupo de pacientes. Estas reacciones pueden revertir tras la suspensión del tratamiento. Tratamiento del dolor neuropático central debido a lesión de la médula espinal: En el tratamiento del dolor neuropático central debido a lesión de la médula espinal se incrementó la incidencia de eventos adversos en general, eventos adversos a nivel del SNC y especialmente somnolencia. Esto puede atribuirse a un efecto aditivo debido a la medicación concomitante (ej. agentes antiespasmódicos) necesaria para esta patología. Este hecho debe tenerse en cuenta cuando se prescriba pregabalina en estos casos. Ideación y comportamientos suicidas: Resultados de un estudio sugieren un aumento del riesgo de ideas o comportamientos suicidas en los pacientes tratados con drogas antiepilépticas (DAEs). Se realizó una evaluación de 199 estudios clínicos controlados para evaluar la incidencia de comportamiento e ideación suicida en pacientes en tratamiento con DAEs (11 diferentes drogas antiepilépticas). Estos estudios evaluaron la eficacia de diferentes drogas antiepilépticas en tratamiento de epilepsia y alteraciones psiquiátricas (trastorno bipolar, depresión y ansiedad) y otras condiciones. Los pacientes aleatorizados a alguna de las drogas antiepilépticas tuvieron casi el doble del riesgo de tener ideación o comportamiento suicida comparados con los pacientes aleatorizados al grupo placebo (riesgo relativo ajustado 1.8, 95% IC:1,2; 2,7). El número de casos de suicidio dentro de estos estudios es muy pequeño para permitir estimar cualquier conclusión sobre el efecto de las DAEs sobre el suicidio consumado. Las indicaciones para las cuales se prescriben DAEs comprenden patologías que en sí mismas se asocian a un riesgo creciente de morbilidad y mortalidad, de ideas y de comportamiento suicida. Los pacientes, sus cuidadores y las familias deben ser informados del potencial aumento de riesgo de tener ideas y comportamientos suicidas y se debe aconsejar sobre la necesidad de estar alerta ante la aparición o el empeoramiento de los síntomas de depresión, cualquier cambio inusual en humor o comportamiento, o la aparición de ideas y comportamientos suicidas. Disminución de la funcionalidad del tracto gastrointestinal inferior: Durante el período posterior a la comercialización se han notificado casos relacionados con la disminución de la funcionalidad del tracto gastrointestinal inferior (ej. obstrucción intestinal, íleo paralítico, estreñimiento) al administrarse pregabalina conjuntamente con medicamentos con potencial para producir estreñimiento, como los analgésicos opioides. En caso de que se vayan a administrar en combinación pregabalina y opioides, debe considerarse la utilización de medidas para evitar el estreñimiento (especialmente en mujeres y pacientes de edad avanzada). Uso incorrecto potencial de abuso o dependencia: Se han notificado casos de uso incorrecto, abuso o dependencia. Se debe tener precaución en pacientes con antecedentes de abuso de drogas, y los pacientes han de ser monitoreados para detectar síntomas de uso incorrecto, abuso o dependencia con pregabalina (se han notificado casos de tolerancia, aumento de la dosis, búsqueda compulsiva de drogas). Encefalopatía: Se han notificado casos de encefalopatía, mayoritariamente en pacientes con enfermedades subyacentes que podrían haber provocado la encefalopatía. Interacción con otros medicamentos y otras formas de interacción: Dado que la pregabalina se excreta principalmente inalterada en orina, experimenta un metabolismo insignificante en humanos ( < 2% de la dosis recuperada en orina en forma de metabolitos), no inhibe el metabolismo de fármacos in vitro y no se une a proteínas plasmáticas, no es probable que produzca interacciones farmacocinéticas o sea susceptible a las mismas. Estudios in vivo y análisis farmacocinético de la población: En consecuencia, en los estudios in vivo, no se observaron interacciones farmacocinéticas relevantes desde el punto de vista clínico entre pregabalina y fenitoína, carbamazepina, ácido valproico, lamotrigina, gabapentina, lorazepam, oxicodona o etanol. El análisis farmacocinético de la población indicó que los hipoglucemiantes orales, diuréticos, insulina, fenobarbital, tiagabina y topiramato no presentaban un efecto clínicamente importante sobre el clearance de pregabalina. Anticonceptivos orales, noretisterona y/o etinilestradiol: La administración de pregabalina junto con anticonceptivos orales como noretisterona y/o etinilestradiol, no influye en la farmacocinética en el estado de equilibrio de ninguna de estas sustancias. Influencia del SNC en medicamentos: La pregabalina puede potenciar los efectos del etanol y lorazepam. En estudios clínicos controlados, las dosis múltiples orales de pregabalina administrada junto con oxicodona, lorazepam o etanol no produjeron efectos clínicamente importantes sobre la respiración. En la experiencia post- comercialización, existen reportes de insuficiencia respiratoria y coma en pacientes que toman pregabalina y otros medicamentos depresores del Sistema Nervioso Central. La pregabalina parece tener un efecto aditivo en la alteración de la función cognitiva y motora causada por oxicodona. Interacciones y pacientes de edad avanzada: No se realizaron estudios farmacodinámicos específicos de interacción en voluntarios ancianos. Los estudios de interacciones se han realizado sólo en adultos. Carcinogénesis, mutagénesis y deterioro de la fertilidad: En los estudios convencionales sobre farmacología de seguridad en animales, la pregabalina fue bien tolerada a dosis clínicamente relevantes. En estudios de toxicidad de dosis repetidas en ratas y monos se observaron efectos en el SNC, incluyendo hipoactividad, hiperactividad y ataxia. Se observó un aumento en la incidencia de atrofia retiniana, observada frecuentemente en ratas albinas ancianas, tras la exposición a largo plazo de pregabalina a exposiciones ≥5 veces la exposición media en humanos a la dosis clínica máxima recomendada. La pregabalina no fue teratogénica ni en ratones ni en ratas ni en conejos. Sólo hubo toxicidad fetal en ratas y conejos a exposiciones lo suficientemente por encima de la exposición en humanos. En estudios de toxicidad prenatal/postnatal, la pregabalina indujo toxicidad en el desarrollo de las crías en ra tas a exposiciones > 2 veces la exposición máxima recomendada en el hombre. Únicamente se observaron efectos adversos sobre la fertilidad en ratas macho y hembra a exposiciones por encima de la dosis terapéutica. Los efectos adversos sobre los órganos reproductores masculinos y sobre el esperma fueron de carácter reversible y únicamente se produjeron