LIPITOR
ASPEN PHARMA
Hipolipemiante. Inhibidor de la HMG-CoA reductasa.
Composición.
Cada comprimido recubierto de LIPITOR 10 mg contiene: Atorvastatina cálcica 10,85 mg. Excipientes: carbonato de calcio 33 mg; celulosa microcristalina 60 mg; lactosa monohidrato 32,8 mg; croscarmelosa sódica 9 mg; polisorbato 80 0,60 mg; hidroxipropilcelulosa 3 mg; estearato de magnesio 0,75 mg; simeticona emulsionada 0,03 mg; cera candelilla 0,08 mg; hidroxipropilmetilcelulosa 2,955 mg; polietilenglicol 8000 0,845 mg; dióxido de titanio 0,085 mg; talco 0,585 mg. Cada comprimido recubierto de LIPITOR 20 mg contiene: Atorvastatina cálcica 21,69 mg. Excipientes: carbonato de calcio 66 mg; celulosa microcristalina 120 mg; lactosa monohidrato 65,61 mg; croscarmelosa sódica 18 mg; polisorbato 80 1,20 mg; hidroxipropilcelulosa 6 mg; estearato de magnesio 1,50 mg; simeticona emulsionada 0,06 mg; cera candelilla 0,16 mg; hidroxipropilmetilcelulosa 5,911 mg; polietilenglicol 8000 1,69 mg; dióxido de titanio 0,17 mg; talco 1,169 mg. Cada comprimido recubierto de LIPITOR 40 mg contiene: Atorvastatina cálcica 43,38 mg. Excipientes: carbonato de calcio 132 mg; celulosa microcristalina 240 mg; lactosa monohidrato 131,22 mg; croscarmelosa sódica 36 mg; polisorbato 80 2,40 mg; hidroxipropilcelulosa 12 mg; estearato de magnesio 3 mg; simeticona emulsionada 0,12 mg; cera candelilla 0,32 mg; hidroxipropilmetilcelulosa 11,822 mg; polietilenglicol 8000 3,379 mg; dióxido de titanio 0,34 mg; talco 2,339 mg. Cada comprimido recubierto de LIPITOR 80 mg contiene: Atorvastatina cálcica 86,76 mg. Excipientes: carbonato de calcio 264 mg; celulosa microcristalina 480 mg; lactosa monohidrato 262,44 mg; croscarmelosa sódica 72 mg; polisorbato 80 4,80 mg; hidroxipropilcelulosa 24 mg; estearato de magnesio 6 mg; Opadry White YS-1-7040-A 35,76 mg; emulsión de simeticona 0,24 mg.
Farmacología.
Descripción: LIPITOR (Atorvastatina cálcica) es un agente sintético que reduce los lípidos. Atorvastatina cálcica es un inhibidor de la 3-hidroxi-3-metilglutaril-coenzima A (HMG-CoA) reductasa. Esta enzima cataliza la conversión de la HMG-CoA en mevalonato, un paso temprano y velocidad- limitante de la biosíntesis del colesterol. Atorvastatina cálcica es [R-(R*, R*)]-2-(4-fluorofenil)-, d-dihidroxi-5-(1-metiletil)-3-fenil-4- [(fenilamina) carbonil]-1H-pirrol-1-ácido heptanoico, sal cálcica (2:1) trihidrato. La fórmula empírica de atorvastatina cálcica es (C33H34FN2O5)2Ca•3H2O y su peso molecular es 1209,42. Su fórmula estructural es la siguiente: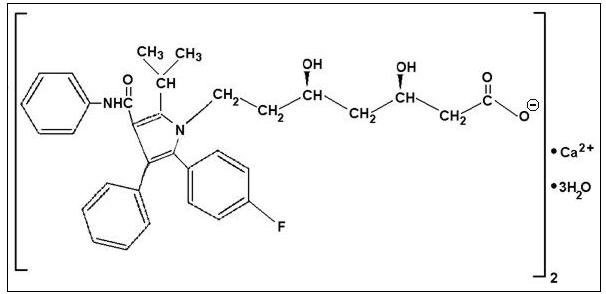
Atorvastatina cálcica es un polvo cristalino blanco o blancuzco que es insoluble en soluciones acuosas de pH ≤ 4. Atorvastatina cálcica es muy ligeramente soluble en agua destilada, buffer fosfato pH 7,4 y acetonitrilo, ligeramente soluble en etanol, y libremente soluble en metanol. Acción Farmacológica: Mecanismos de acción: Atorvastatina cálcica es un inhibidor competitivo y selectivo de la HMG-CoA reductasa, la enzima que limita la velocidad de conversión de 3-hidroxi-3-metilglutaril-Coenzima A en mevalonato, un precursor de los esteroles, incluido el colesterol. En animales, LIPITOR reduce los niveles de colesterol en el plasma y los niveles de lipoproteínas al inhibir la HMG-CoA reductasa y la síntesis del colesterol en el hígado y al aumentar el número de receptores LDL hepáticos en la superficie celular para aumentar la captación y catabolismo de LDL. LIPITOR también reduce la producción de LDL y el número de partículas de LDL. Farmacodinamia: Atorvastatina cálcica, así como también algunos de sus metabolitos, son farmacológicamente activos en el hombre. El hígado es el primer sitio de acción y el principal lugar de síntesis del colesterol y de depuración del LDL. La dosificación del medicamento se asocia mejor con la reducción del colesterol LDL que la concentración sistémica del medicamento. La individualización de la dosis de la droga debe basarse en la respuesta terapéutica (ver Posologia y modo de administracion). Farmacocinética: Absorción: Atorvastatina cálcica se absorbe rápidamente después de su administración oral; las concentraciones plasmáticas máximas ocurren en el término de una a dos horas. El grado de absorción aumenta en proporción a la dosis de atorvastatina cálcica. La biodisponibilidad absoluta de atorvastatina cálcica (droga principal) es aproximadamente del 14% y la biodisponibilidad sistémica de la actividad inhibitoria de la HMG-CoA reductasa es aproximadamente del 30%. La disponibilidad sistémica baja se atribuye a una depuración presistémica en la mucosa gastrointestinal y/o a un metabolismo de primer paso hepático. Aunque la comida disminuye el alcance y grado de absorción de la droga en un 25% y 9%, respectivamente, cuando se mide por medio de la Cmax y ABC, la reducción del C-LDL es similar cuando la atorvastatina cálcica se administra con o sin comidas. Las concentraciones plasmáticas de atorvastatina cálcica son menores (aproximadamente 30% para la Cmax y ABC) después de la administración vespertina comparada con la administración matinal. Sin embargo, la reducción del C-LDL es la misma independientemente de la hora del día en que se administre el medicamento (ver Posologia y modo de administracion). Distribución: El volumen medio de distribución de atorvastatina cálcica es de aproximadamente 381 litros. Atorvastatina cálcica se une ≥ 98% a las proteínas del plasma. La relación sangre/plasma de aproximadamente 0,25 indica una pobre penetración de la droga en los glóbulos rojos. Sobre la base de las observaciones en ratas, atorvastatina cálcica parece ser secretada en la leche materna (ver Contraindicaciones). Metabolismo: Atorvastatina cálcica es extensamente metabolizada a derivados orto- y para- hidroxilados y varios productos de beta-oxidación. La inhibición in vitro de HMG-CoA reductasa por los metabolitos orto- y para-hidroxilados es equivalente a la de atorvastatina cálcica. Aproximadamente el 70% de la actividad inhibitoria circulante sobre la HMG-CoA reductasa se atribuye a los metabolitos activos. Los estudios in vitro indican la importancia del citocromo P450 3A4 en el metabolismo de la atorvastatina cálcica, de acuerdo con los aumentos de las concentraciones plasmáticas de atorvastatina cálcica en el hombre después de una administración conjunta con eritromicina, un conocido inhibidor de esta isoenzima (ver Precauciones, Interacción con otras drogas). En animales, el orto-hidroximetabolito es posteriormente glucuronizado. Eliminación: Atorvastatina cálcica y sus metabolitos son eliminados principalmente en la bilis después de su metabolismo hepático y/o extrahepático, sin embargo, la droga no parece sufrir recirculación enterohepática. La vida media de eliminación plasmática de atorvastatina cálcica en el hombre es de aproximadamente 14 horas, pero la vida media de la actividad inhibitoria sobre HMG-CoA reductasa es de 20 a 30 horas debido a la contribución de los metabolitos activos. Menos del 2% de una dosis de atorvastatina cálcica se recupera en la orina después de la administración oral. Poblaciones Especiales: Geriatría: Las concentraciones plasmáticas de atorvastatina cálcica son mayores (aproximadamente 40% para la Cmax y 30% para el ABC) en individuos mayores sanos (edad 65 años) que en adultos jóvenes. Los datos clínicos indican un grado mayor de disminución del C-LDL con cualquier dosis de la droga en la población de pacientes mayores en comparación con los adultos jóvenes. Pediatría: El clearance oral aparente de la atorvastatina en sujetos pediátricos parecía similar al de los adultos cuando se comparó alométricamente por peso corporal, ya que el peso corporal era la única covariable significativa en el modelo de farmacocinética de la población de atorvastatina, que incluían datos de pacientes con HF heterocigota pediátricos (de 10 a 17 años, N=29), en un estudio abierto de 8 semanas de duración. Sexo: Las concentraciones plasmáticas de atorvastatina cálcica en mujeres difieren en comparación a las observadas en los hombres (aproximadamente 20% mayores para la Cmax y 10% menores para el ABC); sin embargo, no hay diferencias clínicamente significativas en la reducción del C-LDL con LIPITOR entre hombres y mujeres. Insuficiencia Renal: La enfermedad renal no afecta las concentraciones plasmáticas de atorvastatina cálcica o la disminución del C-LDL; por lo que no es necesario el ajuste de dosis en pacientes con insuficiencia renal (ver Posologia y modo de administracion). Hemodiálisis: Aunque no se han realizado estudios en pacientes con enfermedad renal terminal, la hemodiálisis no aumenta significativamente la depuración de atorvastatina cálcica debido a que la droga se encuentra extensamente unida a las proteínas plasmáticas. Insuficiencia Hepática: Las concentraciones plasmáticas de atorvastatina cálcica aumentan notablemente en pacientes con hepatopatía alcohólica crónica. La Cmaxy el ABC son cuatro veces mayores en pacientes con enfermedad Childs-Pugh A. En pacientes con la enfermedad Childs- Pugh B la Cmax aumenta aproximadamente 16 veces y el ABC aumenta 11 veces (ver Contraindicaciones).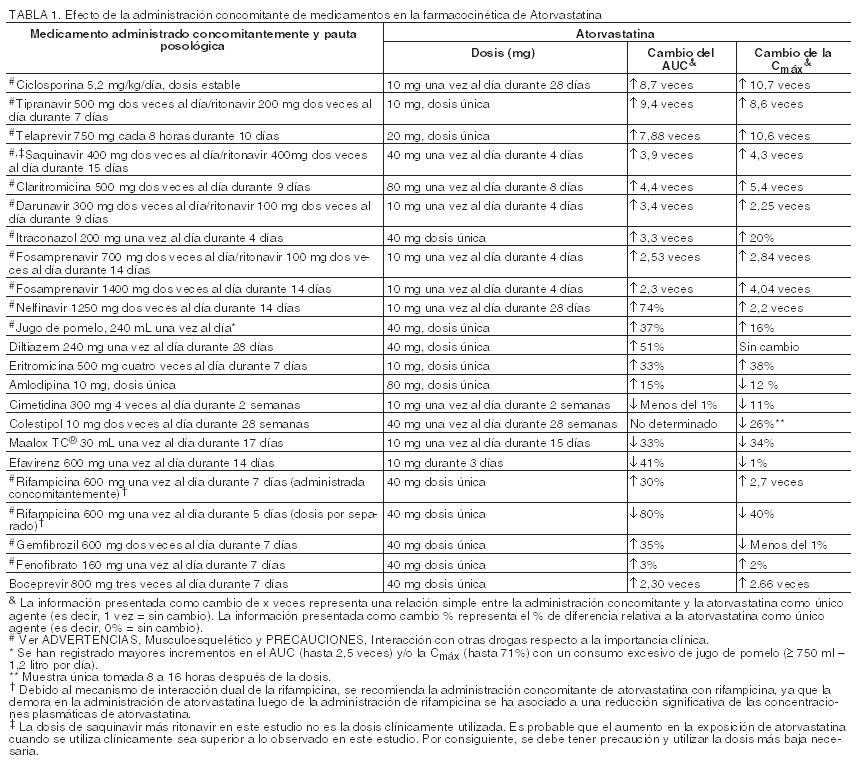
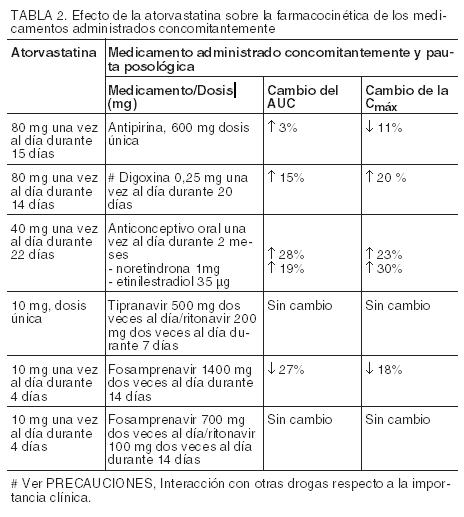
Estudios clínicos: Prevención de la enfermedad cardiovascular: En el estudio ASCOT (Anglo-Scandinavian Cardiac Outcomes Trial), se evaluó el efecto de LIPITOR en la enfermedad coronaria fatal y no fatal en 10.305 pacientes hipertensos de entre 40 y 80 años de edad (media de 63 años), sin infarto de miocardio (IM) previo y con niveles de colesterol total (C-Total) ≤251 mg/dL (6,5 mmol/L). Además, todos los pacientes presentaban al menos tres de los siguientes factores de riesgo cardiovasculares: sexo masculino (81,1%), edad > 55 años (84,5%), tabaquismo (33,2%), diabetes (24,3%), historial de enfermedad coronaria (EC) en un familiar de primer grado (26%), C-Total: C-HDL > 6 (14,3%), enfermedad vascular periférica (5,1%), hipertrofia ventricular izquierda (14,4%), evento cerebrovascular previo (9,8%), anormalidad específica del ECG (14,3%), proteinuria/albuminuria (62,4%). En este estudio doble ciego controlado con placebo, los pacientes recibieron tratamiento antihipertensivo (PA objetivo < 140/90 mmHg para los pacientes no diabéticos; < 130/80 mmHg para los pacientes diabéticos) y se asignaron a uno de dos grupos de tratamiento con LIPITOR a dosis de 10 mg diarias (n=5.168) o placebo (n=5.137), utilizando un método adaptativo de covariable que tomó en cuenta la distribución de nueve características iniciales de los pacientes ya inscriptos y minimizó el desequilibrio de esas características entre los grupos. Se realizó el seguimiento de los pacientes durante un promedio de 3,3 años. El efecto de la dosis de 10 mg/día de LIPITOR sobre los niveles lipídicos fue similar al observado en estudios clínicos anteriores. LIPITOR redujo significativamente el índice de eventos coronarios [ya sea enfermedad coronaria fatal (46 eventos en el grupo de placebo frente a 40 eventos en el grupo de LIPITOR) o IM no fatal (108 eventos en el grupo de placebo frente a 60 eventos en el grupo de LIPITOR)], obteniendo una reducción del riesgo relativo del 36% [(basado en incidencias del 1,9% para LIPITOR frente al 3,0% para el placebo), p=0,0005 (ver la Figura 1)]. La reducción del riesgo fue coherente independientemente de la edad, el tabaquismo, la obesidad o la presencia de disfunción renal. El efecto de LIPITOR se observó independientemente de los niveles iniciales de C-LDL. Debido a la pequeña cantidad de eventos, los resultados para las mujeres no son concluyentes.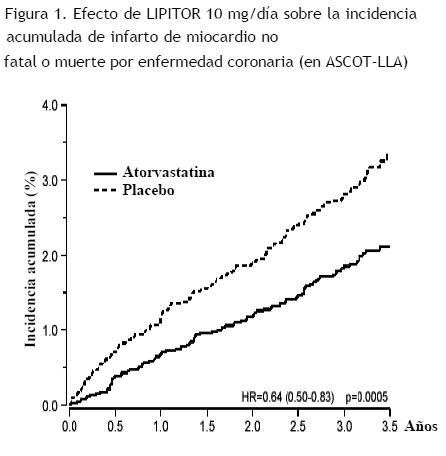
LIPITOR también redujo significativamente el riesgo relativo de procedimientos de revascularización en un 42% (incidencia del 1,4% para LIPITOR y 2,5% para placebo). Aunque la reducción de los accidentes cerebrovasculares fatales y no fatales no alcanzó un nivel de significancia predefinido (p=0,01), se observó una tendencia favorable con una reducción del 26% del riesgo relativo (incidencias del 1,7% en el caso de LIPITOR y del 2,3% en el caso del placebo). No se observó una diferencia significativa entre los grupos de tratamiento con respecto a la muerte por causas cardiovasculares (p=0,51) o causas no cardiovasculares (p=0,17). En el estudio CARDS (Collaborative Atorvastatin Diabetes Study), se evaluó el efecto de LIPITOR sobre el punto final primario de enfermedad cardiovascular (ECV) en 2.838 sujetos (94% blancos, 68% varones) de entre 40 y 75 años de edad con diabetes de tipo 2 según los criterios de la OMS, sin historial previo de enfermedad cardiovascular y con C-LDL ≤ 160 mg/dL, y TG ≤ 600 mg/dL. Además de diabetes, los sujetos presentaban uno o más de los siguientes factores de riesgo: tabaquismo actual (23%), hipertensión (80%), retinopatía (30%), microalbuminuria (9%) o macroalbuminuria (3%). No se incluyeron en el estudio pacientes en hemodiálisis. En este estudio clínico multicéntrico, doble ciego y controlado con placebo, los sujetos se asignaron aleatoriamente para recibir LIPITOR a dosis de 10 mg por día (1.429) o placebo (1.411) a razón de 1:1, y se realizó un seguimiento promedio de 3,9 años. El punto final primario fue la aparición de cualquiera de los principales eventos cardiovasculares: infarto de miocardio, muerte por EC aguda, angina inestable, revascularización coronaria o accidente cerebrovascular. El análisis primario fue el tiempo transcurrido hasta la primera aparición del punto final primario. Las características iniciales de los sujetos fueron las siguientes: edad media 62 años; HbA1c media 7,7%; mediana de C-LDL 120 mg/dL; mediana de C-Total 207 mg/dL; mediana de TG 151 mg/dL; mediana de C-HDL 52 mg/dL. El efecto de LIPITOR 10 mg/día sobre los niveles lipídicos fue similar al observado en estudios clínicos anteriores. LIPITOR redujo significativamente el índice de los principales eventos cardiovasculares (que constituyeron el punto final combinado) (83 eventos en el grupo de LIPITOR frente a 127 eventos en el grupo de placebo), obteniendo una reducción del riesgo relativo del 37%, cociente de riesgos instantáneos (hazard ratio, HR) de 0,63; IC del 95% (0,48; 0,83) (p=0,001) (ver la Figura 2). El efecto de LIPITOR se observó independientemente de la edad, el sexo o los niveles lipídicos iniciales. LIPITOR redujo significativamente el riesgo de accidente cerebrovascular en un 48% (21 eventos en el grupo de LIPITOR frente a 39 eventos en el grupo de placebo), cociente de riesgos instantáneos (hazard ratio, HR) de 0,52; IC del 95% (0,31; 0,89) (p=0,016) y redujo el riesgo de IM en un 42% (38 eventos en el grupo de LIPITOR frente a 64 eventos en el grupo de placebo), cociente de riesgos instantáneos (hazard ratio, HR) de 0,58; IC del 95,1% (0,39; 0,86) (p=0,007). No se observaron diferencias significativas entre los grupos de tratamiento con respecto a la angina, los procedimientos de revascularización y la muerte por EC aguda. Se produjeron 61 muertes en el grupo de LIPITOR frente a 82 muertes en el grupo de placebo [cociente de riesgos instantáneos (hazard ratio, HR) de 0,73; p=0,059].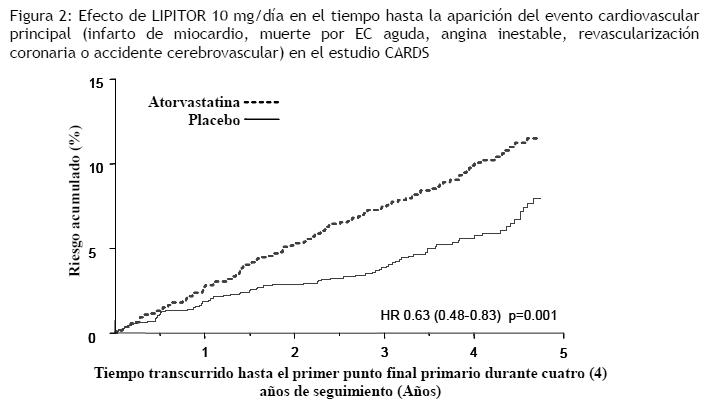
En el estudio TNT (Treating to New Targets), se evaluó el efecto de LIPITOR 80 mg/día en comparación con LIPITOR 10 mg/día sobre la reducción de eventos cardiovasculares en 10.001 sujetos (94% blancos, 81% varones, 38% ≥65 años) con cardiopatía coronaria clínicamente evidente, que habían alcanzado el nivel objetivo de C-LDL < 130 mg/dL luego de completar un período de preinclusión a rótulo abierto y de ocho semanas con LIPITOR 10 mg/día. Los sujetos se asignaron aleatoriamente a grupos de 10 mg/día o de 80 mg/día de LIPITOR y se realizó un seguimiento promedio de 4,9 años. El punto final primario fue el tiempo transcurrido hasta la aparición de cualquiera de los siguientes eventos cardiovasculares principales (ECVP): muerte por EC, infarto de miocardio no fatal, paro cardíaco recuperado y accidente cerebrovascular fatal y no fatal. Los niveles medios de C-LDL, C-Total, TG, colesterol no HDL y HDL a las 12 semanas fueron de 73, 145, 128, 98 y 47 mg/dL durante el tratamiento con dosis de 80 mg de LIPITOR, y 99, 177, 152, 129 y 48 mg/dL durante el tratamiento con dosis de 10 mg de LIPITOR. El tratamiento con LIPITOR 80 mg/día redujo significativamente la tasa de ECVP (434 eventos en el grupo de 80 mg/día contra 548 eventos en el grupo de 10 mg/día), con una reducción del riesgo relativo del 22%, cociente de riesgos instantáneos (hazard ratio, HR) de 0,78; IC del 95% (0,69; 0,89), p=0,0002 (ver la Figura 3 y la Tabla 3). La reducción total del riesgo fue coherente independientemente de la edad ( < 65, ≥65) o el sexo.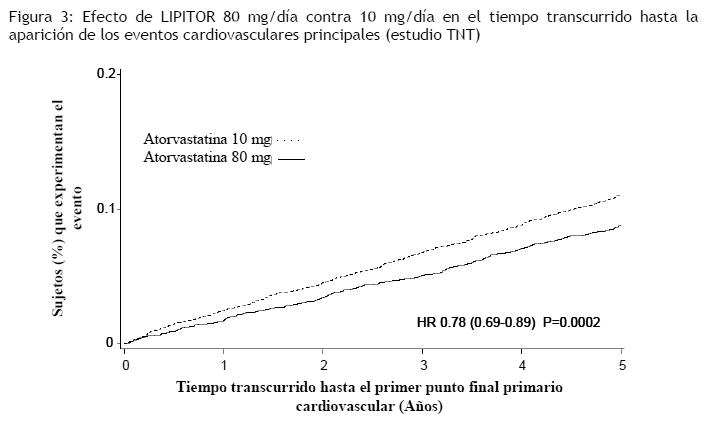
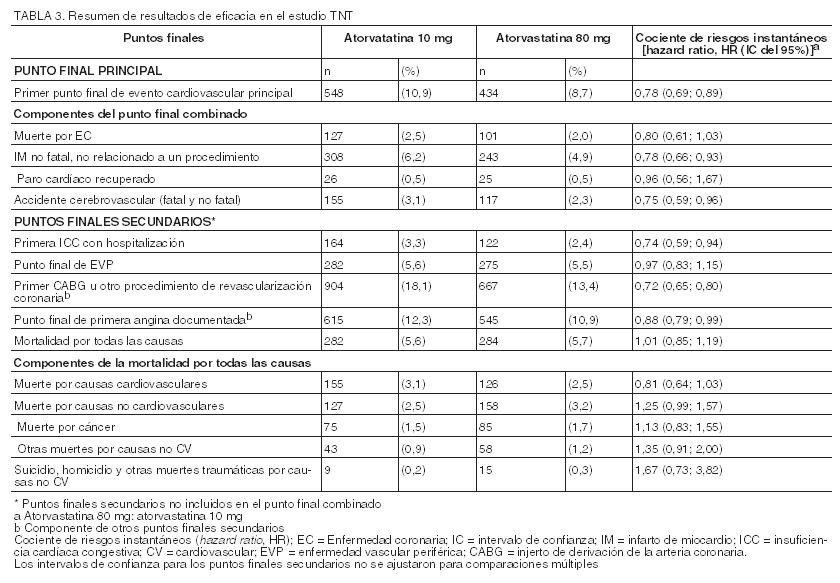
Entre los eventos que constituyeron el punto final de eficacia principal, el tratamiento con LIPITOR 80 mg/día redujo significativamente el índice de IM no fatal, no relacionado a un procedimiento, y de accidente cerebrovascular fatal y no fatal, pero no el índice de muerte por EC o de paro cardíaco recuperado (Tabla 3). Entre los puntos finales secundarios predefinidos, el tratamiento con LIPITOR 80 mg/día redujo significativamente el índice de revascularización coronaria, angina y hospitalización a causa de insuficiencia cardíaca, pero no el de enfermedad vascular periférica. La reducción en la tasa de ICC con hospitalización sólo se observó en el 8% de los pacientes con historial previo de ICC. No se observó una diferencia significativa entre los grupos de tratamiento con respecto a la mortalidad por todas las causas (Tabla 3). Las proporciones de sujetos que sufrieron muerte por causas cardiovasculares, incluidos los componentes de muerte por EC y derrame cerebral fatal, fueron numéricamente menores en el grupo de tratamiento con LIPITOR 80 mg que en el grupo de LIPITOR 10 mg. Las proporciones de sujetos que sufrieron muerte por causas no cardiovasculares fueron numéricamente mayores en el grupo de tratamiento con LIPITOR 80 mg que en el de LIPITOR 10 mg. En el estudio IDEAL (Incremental Decrease in Endpoints Through Aggressive Lipid Lowering Study) se comparó el tratamiento con LIPITOR 80 mg/día con el tratamiento con simvastatina 20-40 mg/día en 8.888 sujetos de hasta 80 años de edad con historial de EC para evaluar si se podía alcanzar una reducción del riesgo cardiovascular. Los pacientes fueron mayormente varones (81%), blancos (99%), con una edad promedio de 61,7 años y colesterol LDL promedio de 121,5 mg/dL al momento de la aleatorización; el 76% ya estaba recibiendo tratamiento con estatina. En este estudio prospectivo, aleatorizado, abierto, con evaluación ciega de los puntos finales (PROBE, por sus siglas en inglés), sin período de preinclusión, se realizó un seguimiento de los sujetos durante un promedio de 4,8 años. Los niveles medios de C-LDL, C-Total, TG, colesterol no HDL y HDL a las 12 semanas fueron de 78, 145, 115, 45, y 100 mg/dL durante el tratamiento con dosis de 80 mg de LIPITOR, y 105, 179, 142, 47, y 132 mg/dL durante el tratamiento con dosis de 20-40 mg de simvastatina. No se observaron diferencias significativas entre los grupos de tratamiento con respecto al punto final primario, la tasa del primer evento coronario principal (EC fatal, IM no fatal y paro cardíaco recuperado): 411 (9,3%) en el grupo de LIPITOR 80 mg/día frente a 463 (10,4%) en el grupo de simvastatina 20-40 mg/día, cociente de riesgos instantáneos (hazard ratio, HR) de 0,89; IC del 95% (0,78; 1,01), p=0,07. No se observó una diferencia significativa entre los grupos de tratamiento con respecto a la mortalidad por todas las causas: 366 (8,2%) en el grupo de LIPITOR 80 mg/día contra 374 (8,4%) en el grupo de simvastatina 20-40 mg/día. Las proporciones de sujetos que sufrieron muerte por causas cardiovasculares y no cardiovasculares fueron similares en el grupo de LIPITOR 80 mg y en el grupo de simvastatina 20-40 mg. Aterosclerosis: En el estudio REVERSAL (Reversing Atherosclerosis with Aggressive Lipid-Lowering) se evaluó el efecto sobre la aterosclerosis coronaria de una reducción intensiva de lípidos con 80 mg de atorvastatina y de una reducción estándar de lípidos con 40 mg de pravastatina en pacientes con enfermedad coronaria, mediante ultrasonografía intravascular (USIV) realizada durante la angiografía. En este estudio aleatorizado, doble-ciego, multicéntrico y controlado se realizó una USIV a 502 pacientes antes de iniciar el tratamiento y otra a los 18 meses. En el grupo de atorvastatina (n=253), no hubo progresión de la aterosclerosis. El porcentaje medio de cambio en el volumen total de ateroma (el criterio primario del estudio) respecto a los valores iniciales fue de -0,4% (p=0,98) en el grupo de atorvastatina y de +2,7% (p=0,001) en el grupo de pravastatina (n=249). Estos efectos de atorvastatina fueron estadísticamente significativos (p=0,02) cuando se compararon con los de pravastatina. En este estudio no se investigó el efecto del tratamiento hipolipemiante intensivo sobre puntos finales cardiovasculares (por ejemplo, necesidad de revascularización, infarto de miocardio no fatal, muerte por causa coronaria). En el grupo de atorvastatina, el C-LDL se redujo hasta una media de 2,04 mmol/L ± 0,8 (78,9 mg/dL ± 30) desde el valor inicial de 3,98 mmol/L ± 0,7 (150 mg/dL ± 28) y en el grupo de pravastatina, el C-LDL se redujo hasta una media de 2,85 mmol/L ± 0,7 (110 mg/dL ± 26) desde el valor inicial de 3,89 mmol/L ± 0,7 (150 mg/dL ± 26) (p < 0,0001). La atorvastatina también redujo de forma significativa la media de colesterol total un 34,1% (pravastatina: -18,4%, p < 0,0001), los niveles medios de TG un 20% (pravastatina: -6,8%, p < 0,0009), y la media de apolipoproteína B un 39,1% (pravastatina: -22,0%, p < 0,0001). La atorvastatina aumentó la media de C-HDL un 2,9% (pravastatina: +5,6%, p=NS). Hubo una reducción media del 36,4% de la PCR en el grupo de atorvastatina frente al 5,2% en el grupo de pravastatina (p < 0,0001). Los resultados de este estudio se obtuvieron con la dosis de 80 mg. Por lo tanto, no pueden extrapolarse a las dosis menores. Los perfiles de seguridad y tolerancia de los dos tratamientos fueron comparables. En este estudio no se investigó el efecto del tratamiento hipolipemiante intensivo sobre puntos finales cardiovasculares mayores. Por consiguiente, se desconoce la importancia clínica de los resultados de estas imágenes sobre la prevención primaria y secundaria de eventos cardiovasculares. Síndrome coronario agudo: En el estudio MIRACL (), se evaluó el efecto de 80 mg de atorvastatina en 3.086 pacientes (atorvastatina n=1.538; placebo n=1.548) con síndrome coronario agudo (infarto de miocardio sin onda Q o angina inestable). El tratamiento se inició durante la fase aguda posterior a la hospitalización y se prolongó durante un período de 16 semanas. El tratamiento con 80 mg/día de atorvastatina aumentó el tiempo hasta la aparición del punto final primario combinado, definido como muerte por cualquier causa, infarto no fatal, paro cardíaco recuperado, o angina de pecho con evidencia de isquemia de miocardio que precisa hospitalización, indicando una reducción del riesgo del 16% (p=0,048). Esto se debió fundamentalmente a una reducción del 26% en las re-hospitalizaciones por angina de pecho, con evidencia de isquemia de miocardio (p=0,018). Los otros puntos finales secundarios no alcanzaron significancia estadística en sí mismos (global: Placebo: 22,2%, Atorvastatina: 22,4%). El perfil de seguridad de atorvastatina en el estudio MIRACL, fue congruente con el descrito en Reacciones adversas. Accidente cerebrovascular recurrente: En el Estudio SPARCL (Stroke Prevention by Aggressive Reduction in Cholesterol Levels), se evaluó el efecto de 80 mg diarios de atorvastatina o placebo sobre el accidente cerebrovascular en 4.731 pacientes que habían tenido un accidente cerebrovascular o un ataque isquémico transitorio (AIT) dentro de los 6 meses precedentes y no tenían historia de enfermedad cardíaca coronaria. Los pacientes eran en un 60% varones, de 21 a 92 años de edad (edad promedio 63 años) y tenían un C-LDL promedio de 133 mg/dL (3,4 mmol/L). El C-LDL durante el tratamiento con atorvastatina fue de 73 mg/dL (1,9 mmol/L) y de 129 mg/dL (3,3 mmol/L) durante el tratamiento con placebo. La mediana de la duración del seguimiento fue de 4,9 años. La atorvastatina, en dosis de 80 mg, redujo el riesgo del punto final primario de accidente cerebrovascular fatal o no fatal en 15% [cociente de riesgos instantáneos (hazard ratio, HR) de 0,85; IC del 95%, 0,72-1,00; p=0,05 ó 0,84; IC del 95%, 0,71-0,99; p=0,03 después de ajustar por factores iniciales] en comparación con placebo. La mortalidad por cualquier causa fue del 9,1% (216/2.365) para la atorvastatina en comparación con el 8,9% (211/2.366) para el placebo. En los análisis retrospectivos (post-hoc), 80 mg de atorvastatina redujeron la incidencia de accidente cerebrovascular isquémico (218/2.365; 9,2% versus 274/2.366; 11,6%, p=0,01) y aumentaron la incidencia de accidente cerebrovascular hemorrágico (55/2.365; 2,3% versus 33/2.366; 1,4%, p=0,02), en comparación con placebo. El riesgo de accidente cerebrovascular hemorrágico aumentó en los pacientes que ingresaron en el estudio con accidente cerebrovascular hemorrágico anterior [7/45 para atorvastatina frente a 2/48 para placebo; cociente de riesgos instantáneos (hazard ratio, HR) de 4,06; IC del 95%, 0,84-19,57), y el riesgo de accidente cerebrovascular isquémico fue similar entre los grupos (3/45 para atorvastatina frente a 2/48 para placebo; cociente de riesgos instantáneos (hazard ratio, HR) de 1,64; IC del 95%, 0,27 - 9,82]. El riesgo de accidente cerebrovascular hemorrágico aumentó en los pacientes que ingresaron en el estudio con infarto lacunar anterior [20/708 para atorvastatina frente a 4/701 para placebo; cociente de riesgos instantáneos (hazard ratio, HR) de 4,99; IC del 95%, 1,71 - 14,61], pero el riesgo de accidente cerebrovascular isquémico también fue menor en estos pacientes [79/708 para atorvastatina frente a 102/701 para placebo; cociente de riesgos instantáneos (hazard ratio, HR) de 0,76; IC del 95%, 0,57 - 1,02]. Es posible que aumente el riesgo neto de accidente cerebrovascular en pacientes con infarto lacunar anterior que reciben atorvastatina 80 mg/día. La mortalidad por cualquier causa fue del 15,6% (7/45) para atorvastatina frente al 10,4% (5/48) para el subgrupo de pacientes con accidente cerebrovascular hemorrágico anterior. La mortalidad por cualquier causa fue del 10,9% (77/708) para atorvastatina frente al 9,1% (64/701) para el placebo en el subgrupo de pacientes con infarto lacunar anterior. Hiperlipidemia y dislipidemia mixta: LIPITOR reduce el C-Total, el colesterol LDL, el colesterol VLDL, la apo B y los TG, y aumenta el C-HDL en pacientes con hiperlipidemia (heterocigótica familiar y no familiar) y dislipidemia mixta (tipos IIa y IIb de Fredrickson). La respuesta al tratamiento se observa dentro del plazo de dos semanas, y la respuesta máxima se suele alcanzar a las cuatro semanas y se mantiene durante el tratamiento crónico. LIPITOR resulta efectivo en una amplia variedad de poblaciones de pacientes con hiperlipidemia, con o sin hipertrigliceridemia, en hombres y mujeres, y en ancianos. En dos estudios multicéntricos, del tipo dosis/respuesta y controlados con placebo en pacientes con hiperlipidemia, la administración de dosis únicas de LIPITOR durante seis semanas redujo significativamente el C-Total, el colesterol LDL, la apo B y los TG. (Los resultados agrupados se presentan en la Tabla 4.)
En pacientes con hiperlipoproteinemia tipos IIa y IIb de Fredrickson agrupados de 24 estudios controlados, las variaciones porcentuales medias (percentil 25 y 75) respecto de los valores basales en el C-HDL con LIPITOR 10, 20, 40, y 80 mg fueron 6,4 (-1,4; 14), 8,7 (0; 17), 7,8 (0; 16) y 5,1 (-2,7; 15), respectivamente. Asimismo, el análisis de los datos agrupados demostró disminuciones coherentes y significativas en el C-Total, el colesterol LDL, los TG, el C-Total/C- HDL y el C-LDL/C-HDL. LIPITOR se comparó con otras estatinas en tres estudios multicéntricos doble ciego en pacientes con hiperlipidemia. Luego de la aleatorización, los pacientes recibieron tratamiento durante 16 semanas con LIPITOR 10 mg por día o una dosis fija del agente de comparación (Tabla 5).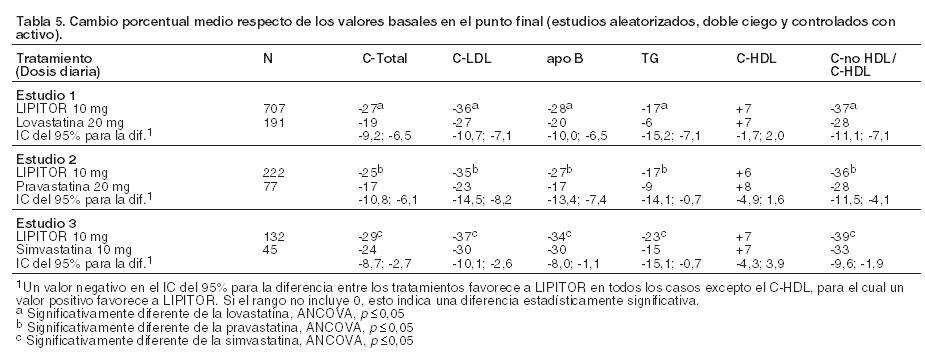
No se conoce el impacto en los resultados clínicos de las diferencias en los efectos de alteración de lípidos entre los tratamientos presentados en la Tabla 5. La tabla 5 no contiene datos que comparen los efectos de LIPITOR 10 mg con dosis más altas de lovastatina, pravastatina y simvastatina. Los medicamentos comparados en los estudios resumidos en la tabla no son necesariamente intercambiables. Hipertrigliceridemia En la siguiente tabla (Tabla 6) se muestra la respuesta a LIPITOR de 64 pacientes con hipertrigliceridemia aislada (tipo IV de Fredrickson) tratados en varios ensayos clínicos. Para los pacientes tratados con LIPITOR, el nivel basal mediano (mín., máx.) de TG fue 565 (267-1502).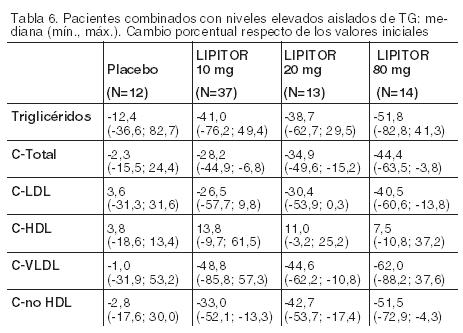
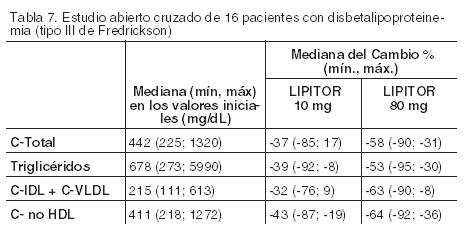
Hipercolesterolemia familiar homocigótica: En un estudio sin grupo de control concurrente, 29 pacientes de entre 6 y 37 años con hipercolesterolemia familiar homocigótica recibieron dosis diarias máximas de 20 a 80 mg de LIPITOR. La reducción media del C-LDL en este estudio fue del 18%. Veinticinco pacientes con una reducción en el colesterol LDL presentaron una respuesta media del 20% (rango del 7% al 53%, mediana del 24%); los cuatro pacientes restantes presentaron incrementos en el colesterol LDL del 7% al 24%. En cinco de los 29 pacientes se observó una ausencia de la función del receptor LDL. De los cinco, dos pacientes tenían además una derivación porto-cava y no presentaron una reducción significativa del colesterol LDL. Los tres pacientes restantes que carecían de receptores presentaron una reducción media del colesterol LDL del 22%. Hipercolesterolemia familiar heterocigótica en pacientes pediátricos: En un estudio doble ciego controlado con placebo y seguido de una etapa abierta, 187 niños y niñas postmenárquicas, entre 10 y 17 años de edad (edad media 14,1 años), con hipercolesterolemia familiar (HF) heterocigótica o hipercolesterolemia grave, fueron asignados aleatoriamente a uno de dos tratamientos con LIPITOR (n=140) o placebo (n=47) durante 26 semanas, y luego todos recibieron LIPITOR durante 26 semanas. Los requisitos para la inclusión en el estudio eran 1) un nivel inicial de colesterol LDL ≥ 190 mg/dL o 2) un nivel inicial de colesterol LDL ≥ 160 mg/dL, y un historial familiar positivo de HF o enfermedad cardiovascular prematura documentada en un familiar de primer o segundo grado. El valor basal medio de colesterol LDL fue 218,6 mg/dL (rango: 138,5-385,0 mg/dL) en el grupo de LIPITOR en comparación con 230,0 mg/dL (rango: 160,0-324,5 mg/dL) en el grupo de placebo. La dosis de LIPITOR (una vez al día) fue de 10 mg durante las primeras cuatro semanas y escalonada a 20 mg si el nivel de colesterol LDL era > 130 mg/dL. La cantidad de pacientes tratados con LIPITOR que requirió un ajuste ascendente de la dosis a 20 mg luego de la cuarta semana durante la etapa de doble ciego fue 78 (55,7%). LIPITOR redujo significativamente los niveles plasmáticos de C-Total, colesterol LDL, triglicéridos y apolipoproteína B durante la etapa de doble ciego de 26 semanas de duración (ver la Tabla 8).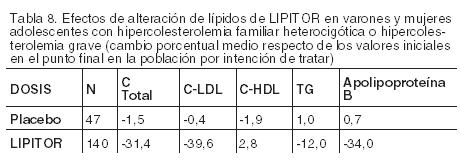
El valor medio alcanzado de colesterol LDL fue 130,7 mg/dL (rango: 70,0 - 242,0 mg/dL) en el grupo de LIPITOR en comparación con 228,5 mg/dL (rango: 152,0-385,0 mg/dL) en el grupo de placebo durante la etapa de doble ciego de 26 semanas de duración. La atorvastatina también se estudió en un ensayo abierto de tres años sin control que incluía 163 pacientes con HF heterocigota de 10 a 15 años de edad (82 niños y 81 niñas). Todos los pacientes tenían un diagnóstico clínico de HF heterocigota confirmado por análisis genético (si no ya confirmado por antecedentes familiares). Aproximadamente el 98% eran caucásicos y menos del 1% eran negros o asiáticos. La media de LDL-C en la línea de base fue de 232 mg/dL. La dosis inicial de atorvastatina fue de 10 mg una vez al día y las dosis se ajustaron para alcanzar un objetivo de LDL-C < 130 mg/dl. Las reducciones en el C-LDL desde la línea de base fueron generalmente consistentes entre los grupos de edad dentro del ensayo, así como con estudios clínicos previos en ensayos controlados con placebo tanto en adultos como en pediatría. No se ha establecido la eficacia a largo plazo del tratamiento con LIPITOR en la niñez para reducir la morbilidad y la mortalidad en la adultez.
Indicaciones.
La terapia con agentes que modifican los lípidos debe considerarse como parte de una intervención de los múltiples factores de riesgo en individuos con un elevado riesgo de contraer enfermedad vascular aterosclerótica debida a la hipercolesterolemia. Una dieta restringida en grasas y colesterol debe complementarse con agentes que modifican los lípidos sólo cuando no se alcancen los efectos necesarios con la dieta y otras medidas no farmacológicas. Prevención de la Enfermedad Cardiovascular en adultos: En pacientes adultos sin enfermedad cardiovascular clínicamente evidente, pero con múltiples factores de riesgo para enfermedad coronaria (EC), tales como edad, tabaquismo, hipertensión, C-HDL bajo, o una historia familiar de enfermedad coronaria temprana, LIPITOR está indicado para: Reducir el riesgo de infarto de miocardio. Reducir el riesgo de accidente cerebrovascular. Reducir el riesgo de procedimientos de revascularización y angina. En pacientes adultos con diabetes tipo 2 y sin enfermedad cardiovascular clínicamente evidente, pero con múltiples factores de riesgo para enfermedad coronaria, tales como retinopatía, albuminuria, tabaquismo o hipertensión, LIPITOR está indicado para: Reducir el riesgo de infarto de miocardio. Reducir el riesgo de accidente cerebrovascular. En pacientes adultos con enfermedad cardiovascular clínicamente evidente, en síndromes coronarios agudos o en accidente cerebrovascular reciente, LIPITOR está indicado para: Reducir el riesgo de infarto de miocardio no fatal. Reducir el riesgo de accidente cerebrovascular fatal o no fatal. Reducir el riesgo de procedimientos de revascularización. Reducir el riesgo de hospitalización en pacientes con Insuficiencia Cardíaca. Congestiva. Reducir el riesgo de angina. Hiperlipidemia: 1. Como un complemento de la dieta para reducir los niveles elevados de colesterol total (C-Total), colesterol LDL (C-LDL), apolipoproteína B (apo B) y los niveles de triglicéridos (TG) y para aumentar el colesterol HDL (C-HDL) en pacientes adultos con hipercolesterolemia primaria (heterocigota familiar y no familiar) y dislipidemia mixta (Fredrickson Tipo IIa y IIb). 2. Como un complemento de la dieta para el tratamiento de pacientes adultos con niveles séricos elevados de triglicéridos (TG) (Fredrickson Tipo IV). 3. Para el tratamiento de pacientes adultos con disbetalipoproteinemia (Fredrickson Tipo III) que no respondieron adecuadamente a la dieta. 4. Para reducir el C-Total y el C-LDL en pacientes con hipercolesterolemia familiar (HF) homocigota como un complemento de otros tratamientos para reducir los lípidos (por ej. aféresis de LDL) o si dichos tratamientos no se encuentran disponibles. 5. Como un complemento de la dieta para reducir los niveles de colesterol total (C-Total), colesterol LDL (C-LDL) y apolipoproteína B (apo B) en pacientes, de entre 10 y 17 años de edad, con hipercolesterolemia familiar (HF) heterocigota, que aún después de recibir un tratamiento adecuado o dieta presenten los siguientes parámetros: a. Colesterol LDL remanente ≥ 190 mg/dL o b. Colesterol LDL remanente ≥ 160 mg/dL y Exista historia familiar de enfermedad cardiovascular prematura o Cuando dos o más riesgos de accidente cerebrovascular están presentes en pacientes pediátricos. Limitaciones de uso: LIPITOR no ha sido bien estudiado en condiciones donde la principal anormalidad de lipoproteínas es la elevación de quilomicrones (Fredrickson Tipo I y V).
Dosificación.
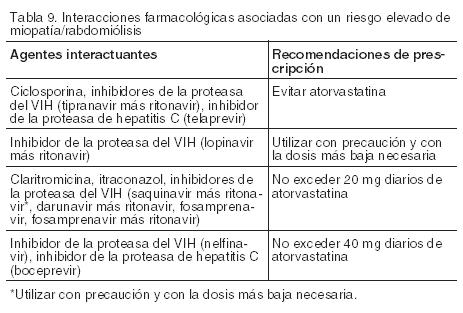
Hiperlipidemia y Dislipidemia Mixta: La dosis inicial recomendada de LIPITOR es 10 o 20 mg una vez al día. Los pacientes que requieren una reducción más grande en el C-LDL (más de 45%) pueden comenzar el tratamiento con 40 mg una vez al día. El rango de dosificación de LIPITOR es de 10 a 80 mg una vez al día. LIPITOR puede administrarse como una dosis única en cualquier momento del día, con o sin alimentos. La dosis inicial y la dosis de mantenimiento de LIPITOR deben individualizarse de acuerdo con las características del paciente tales como los objetivos de la terapia y las respuestas. Después del inicio y/o titulación de LIPITOR, los niveles de lípidos deben analizarse dentro de las 2 a 4 semanas y se debe ajustar la dosis. Hipercolesterolemia Heterocigota Familiar en Pacientes Pediátricos (10-17 años de edad). La dosis inicial de LIPITOR recomendada es de 10 mg/día; el rango usual de dosis es 10 a 20 mg/día oral, 1 vez al día (ver Estudios clinicos). Las dosis deberán ser individualizadas de acuerdo al objetivo de terapia recomendado (Propiedades farmacológicas e Indicaciones). Los ajustes deberán ser hechos a intervalos de 4 semanas o más. Hipercolesterolemia Homocigota Familiar: La dosis de LIPITOR en pacientes con hipercolesterolemia familiar (HF) homocigota es de 10 a 80 mg una vez al día. LIPITOR puede ser administrado como un complemento a otros tratamientos para reducir el colesterol (por ej. aféresis de LDL) en estos pacientes o si tales tratamientos no estuvieran disponibles. Terapia Concomitante de reducción de lípidos: Atorvastatina cálcica puede usarse en combinación con una resina captadora de ácidos biliares. La combinación de los inhibidores HMG-CoA reductasa con fibratos, generalmente debe ser evitada (ver Advertencias y precauciones e Interacción con otras drogas). Dosis en Pacientes con Insuficiencia Renal: La enfermedad renal no afecta las concentraciones plasmáticas ni la reducción de colesterol LDL por atorvastatina cálcica; por lo que el ajuste de dosis en pacientes con disfunción renal no es necesario (ver Propiedades farmacologicas, Farmacocinética). Dosis en pacientes que toman ciclosporina, claritromicina, itraconazol o ciertos inhibidores de la proteasa: Se debe evitar el tratamiento con atorvastatina cálcica en pacientes que toman ciclosporina o inhibidores de la proteasa del VIH (tipranavir más ritonavir) o el inhibidor de la proteasa de la hepatitis C (telaprevir). En pacientes con VIH que toman lopinavir más ritonavir se debe tener cuidado al recetar atorvastatina cálcica y se debe emplear la dosis más baja necesaria. En pacientes que toman claritromicina, itraconazol o en los pacientes con VIH que toman una combinación de saquinavir más ritonavir, darunavir más ritonavir, fos