Kadcyla®
ROCHE
Trastuzumab emtansina.
Agente antineoplásico. Anticuerpo monoclonal.
Composición.
Cada vial de uso único contiene 100 mg de trastuzumab emtansina, en un excipiente compuesto por sacarosa 300 mg, ácido succínico 5,90 mg, hidróxido de sodio 2,25 mg y polisorbato 20: 1,00 mg. Kadcyla 100 mg es un polvo liofilizado para preparar una solución concentrada administrable en infusión de color blanco o blanquecino. Luego de la reconstitución con 5 ml de agua para inyectables, el concentrado reconstituido de Kadcyla contiene 20 mg/ml de trastuzumab emtansina. Cada vial de uso único contiene 160 mg de trastuzumab emtansina, en un excipiente compuesto por sacarosa 480 mg, ácido succínico 9,44 mg, hidróxido de sodio 3,60 mg y polisorbato 20: 1,60 mg. Kadcyla 160 mg es un polvo liofilizado para preparar una solución concentrada administrable en infusión de color blanco o blanquecino. Luego de la reconstitución con 8 ml de agua para inyectables, el concentrado reconstituido de Kadcyla contiene 20 mg/ml de trastuzumab emtansina.
Farmacología.
Código ATC: L01XC14. Grupo farmacoterapéutico: Agente antineoplásico. Anticuerpo monoclonal. Propiedades farmacodinámicas: Mecanismo de acción: Kadcyla, trastuzumab emtansina, es un conjugado anticuerpo-fármaco dirigido a HER2 que contiene el IgG1 humanizado anti-HER2, trastuzumab, enlazado covalentemente con el medicamento inhibidor de microtúbulos DM1 (un derivado de maitansina) mediante el enlace tioéter estable MCC (4-[N-maleimidometil] ciclohexano-1-carboxilato). Emtansina se refiere al complejo MCC-DM1. Un promedio de 3,5 moléculas de DM1 se conjugan a cada molécula de trastuzumab. La conjugación del DM1 con trastuzumab confiere selectividad del agente citotóxico para células tumorales que sobreexpresan HER2, incrementando, de este modo, la entrega intracelular del DM1 directamente hacia el interior de las células malignas. Una vez unido a HER2, trastuzumab emtansina experimenta internalización mediada por receptor y la subsiguiente degradación lisosomal, lo que produce la liberación de catabolitos citotóxicos que contienen DM1 (principalmente lisina-MCC-DM1). Kadcyla posee los mecanismos de acción de trastuzumab y del DM1. Trastuzumab emtansina, como trastuzumab, se une al subdominio IV del dominio extracelular de HER2 (DEC), como también a los receptores Fcc y al complemento C1q. Además, Kadcyla, al igual que trastuzumab, inhibe el desprendimiento del DEC de HER2, inhibe la transmisión de señales a través de la vía fosfatidilinositol 3-kinasa (PI3-K) y actúa como mediador de la citotoxicidad mediada por células dependiente de anticuerpos (CMCDA) en células humanas de cáncer de mama que sobreexpresan HER2. DM1, el componente citotóxico de Kadcyla, se une a la tubulina. Al inhibir la polimerización de la tubulina, el DM1 y Kadcyla hacen que las células se detengan en la fase G2/M del ciclo celular, provocando finalmente muerte celular apoptótica. Los resultados de los estudios de citotoxicidad in vitro demuestran que el DM1 es entre 20 y 200 veces más potente que los taxanos y los alcaloides de vinca. El enlace MCC está diseñado para limitar la liberación sistémica e incrementar la entrega específica de DM1, como se demuestra mediante la detección de los niveles muy bajos de DM1 libre en el plasma. Seguridad y eficacia clínica: Estudio TDM4370g/BO21977: Se realizó un estudio clínico de Fase III, aleatorizado, multicéntrico, internacional, abierto, en pacientes con cáncer de mama localmente avanzado no resecable (CMLA) o cáncer de mama metastásico (CMM) HER2 positivo que habían sido tratados previamente con un taxano y trastuzumab, incluidos los pacientes con tratamiento anterior con trastuzumab y un taxano en el ámbito adyuvante y que habían recaído durante la terapia adyuvante o en los seis meses siguientes a su terminación. Antes de la incorporación en el estudio, se solicitaron muestras de tumores mamarios para confirmar principalmente el estado de HER2 positivo definido como puntaje de 3+ por técnicas inmunohistoquímicas (IHQ) o hibridación in situ por fluorescencia (FISH). Las características iniciales del paciente y del tumor estaban equilibradas entre los grupos del tratamiento. Los pacientes con metástasis cerebrales tratadas eran aptos para participar en el ensayo si no requerían tratamiento para el control de los síntomas. En aquéllos aleatorizados a Kadcyla, la edad promedio era de 53 años, con predominio de mujeres (99,8%); la mayoría eran caucásicos (72%) y el 57% padecía enfermedad con receptor de estrógeno y/o receptor de progesterona positiva. El estudio comparaba la seguridad y eficacia de Kadcyla con la de lapatinib más capecitabina. Un total de 991 pacientes fueron distribuidos al azar a los grupos de tratamiento con Kadcyla o con lapatinib en combinación con capecitabina del siguiente modo: Grupo Kadcyla: 3,6 mg/kg de Kadcyla, administrados por vía intravenosa durante 30-90 minutos, en el Día 1 del ciclo de tratamiento de 21 días. Grupo control (lapatinib en combinación con capecitabina): 1.250 mg por día de lapatinib, administrados por vía oral una vez por día, durante un ciclo de 21 días, más 1.000 mg/m2 de capecitabina, por vía oral dos veces por día, del Día 1 al 14 durante un ciclo de tratamiento de 21 días. Los criterios de valoración principales de eficacia del estudio fueron la sobrevida libre de progresión (SLP), según lo evaluado por el Comité de Revisión Independiente (CRI), y los índices de sobrevida global (SG) y sobrevida a 1 año y 2 años (véanse Tabla 1 y Figuras 1 y 2). El tiempo hasta la progresión de los síntomas, definido por una reducción de 5 puntos en el puntaje derivado de la subescala del Índice de los Resultados de los Ensayos Mamarios (TOI-B) del Cuestionario de Evaluación funcional de la terapia del cáncer de mama y de la calidad de vida (FACT-B QoL), también fue evaluado durante el estudio clínico. Un cambio de 5 puntos en el TOI-B se considera clínicamente significativo. Kadcyla retrasó el tiempo hasta la progresión de síntomas reportado por el paciente 7,1 meses comparado con 4,6 meses para el grupo control (proporción de riesgo [HR] 0,796 [0,667, 0,951); valor de p 0,0121. Los datos son de un estudio abierto y no se pueden extraer conclusiones firmes.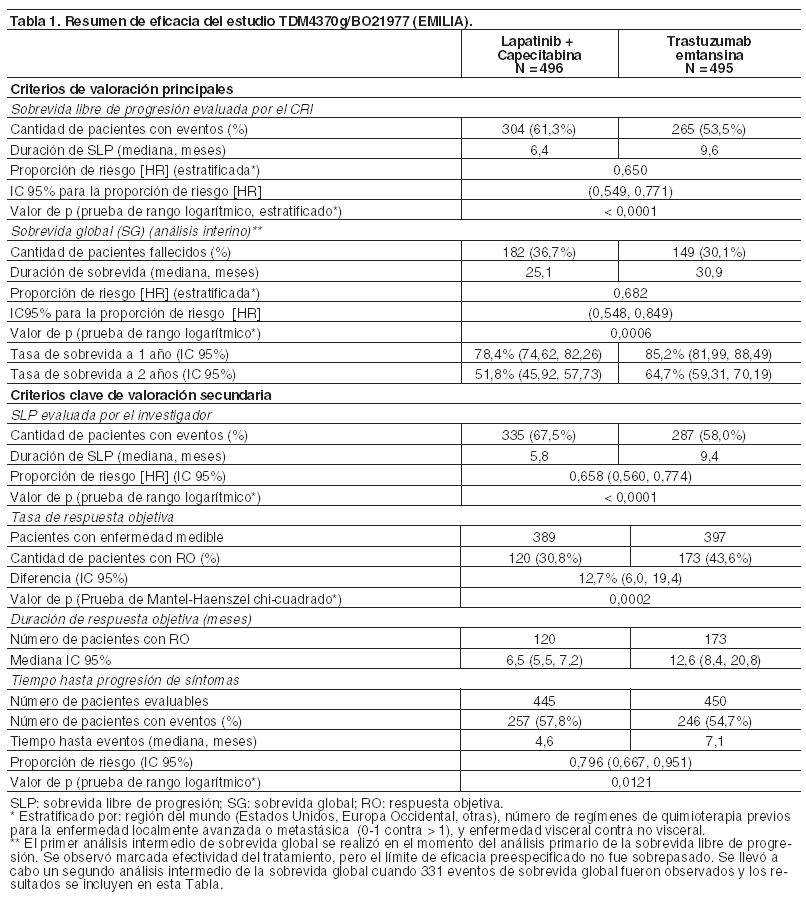
Se verificó beneficio terapéutico en el subgrupo de pacientes que habían recaído en los 6 meses siguientes a la terminación del tratamiento adyuvante y no habían recibido previamente terapia anticancerígena sistémica en el entorno metastásico (n=118); las proporciones de riesgo de la sobrevida libre de progresión y de la sobrevida global fueron de 0,51 (IC 95%: 0,30, 0,85) y 0,61 (IC 95%: 0,32, 1,16), respectivamente. La mediana de sobrevida libre de progresión fue de 10,8 meses y la de sobrevida global no se alcanzó en el grupo con trastuzumab emtansina, en comparación con los valores de 5,7 meses y 27,9 meses, respectivamente, que se lograron en el grupo tratado con lapatinib más capecitabina.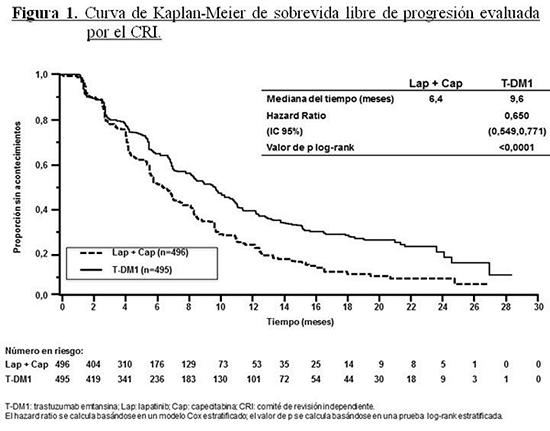
En el estudio TDM4370g/BO21977, se observó un beneficio consistente del tratamiento con trastuzumab emtansina en la mayoría de los subgrupos preespecificados de pacientes evaluados, lo que respalda la solidez del resultado global. En el subgrupo de los que tenían enfermedad con receptor hormonal negativo (n=426), la proporción de riesgo [HR] para la sobrevida libre de progresión y la sobrevida global fue de 0,56 (IC 95%: 0,44, 0,72) y 0,75 (IC 95%: 0,54, 1,03), respectivamente. En el subgrupo de aquéllos con enfermedad con receptor hormonal positivo (n=545), la proporción de riesgo [HR] para la sobrevida libre de progresión y la sobrevida global fue de 0,72 (IC 95%: 0,58, 0,91) y 0,62 (IC 95%: 0,46, 0,85), respectivamente. En el subgrupo de pacientes con enfermedad no medible (n=205), la proporción de riesgo [HR] para la sobrevida libre de progresión y la sobrevida global, basada en las evaluaciones del CRI, fue de 0,91 (IC 95%: 0,59, 1,42) y 0,96 (IC 95%: 0,54, 1,68), respectivamente. En pacientes ≥ 65 años de edad (n=138 entre ambos grupos de tratamiento) la proporción de riesgo [HR] para la sobrevida libre de progresión y la sobrevida global fue de 1,06 (IC 95%: 0,68, 1,66) y 1,05 (IC 95%: 0,58, 1,91), respectivamente. En pacientes de 65 a 74 años de edad (n=113), la proporción de riesgo [HR] para la sobrevida libre de progresión y la sobrevida global fue de 0,88 (IC 95%: 0,53, 1,45) y 0,74 (IC 95%: 0,37, 1,47), respectivamente, basándose en las evaluaciones del CRI. En los de 75 años de edad o más, la proporción de riesgo [HR] para la sobrevida libre de progresión y la sobrevida global, basada en las evaluaciones del CRI, fue de 3,51 (IC 95%: 1,22, 10,13) y 3,45 (IC 95%: 0,94, 12,65), respectivamente. En el subgrupo de pacientes de 75 años de edad o más no se demostró un beneficio en la sobrevida libre de progresión o la sobrevida global, pero la casuística era demasiado reducida (n=25) para extraer conclusiones definitivas. Estudio TDM4450g: En este estudio de Fase II, aleatorizado, multicéntrico, abierto, se evaluaron los efectos de Kadcyla en comparación con trastuzumab más docetaxel en pacientes con CMM HER2 positivo que no habían recibido previamente quimioterapia para la enfermedad metastásica. Los pacientes fueron asignados aleatoriamente para recibir 3,6 mg/kg de Kadcyla por vía intravenosa cada 3 semanas (n= 67) o una dosis inicial de 8 mg/kg de trastuzumab por vía intravenosa, seguida de 6 mg/kg por vía intravenosa cada tres semanas, más 75-100 mg/m2 de docetaxel por vía intravenosa cada 3 semanas (n=70). El criterio de valoración primario del estudio fue la sobrevida libre de progresión (SLP), evaluada por el investigador. La mediana de la sobrevida libre de progresión fue de 9,2 meses en el grupo de trastuzumab más docetaxel y de 14,2 meses en el de Kadcyla (proporción de riesgo [HR], 0,59; p = 0,035), después de una mediana de seguimiento de aproximadamente 14 meses en ambos grupos. La tasa de respuesta objetiva (TRO) fue del 58,0% con trastuzumab más docetaxel y del 64,2% con Kadcyla. La mediana de duración de respuesta no se alcanzó con Kadcyla, mientras que en el grupo control fue de 9,5 meses. Estudio TDM4374g: En este estudio de Fase II, abierto, con un solo grupo de tratamiento, se evaluaron los efectos de Kadcyla en pacientes con CMLA o CMM HER2 positivo incurable. Todos recibieron previamente tratamientos dirigidos contra HER2 (trastuzumab y lapatinib) y quimioterapia (antraciclinas, taxanos y capecitabina) en el entorno neoadyuvante, adyuvante, o de enfermedad localmente avanzada o metastásica. La mediana del número de agentes anticancerígenos suministrados a los pacientes en cualquiera de estos ámbitos fue de 8,5 (rango, 5-19) y, en el de la enfermedad metastásica de 7,0 (rango, 3-17), incluidos todos los medicamentos destinados para el tratamiento del cáncer de mama. Los pacientes (n=110) fueron tratados con 3,6 mg/kg de Kadcyla por vía intravenosa cada 3 semanas, hasta que se observara progresión de la enfermedad o toxicidad inaceptable. Los análisis principales de eficacia fueron TRO basadas en un examen radiológico independiente y la duración de la respuesta objetiva. La TRO fue de 32,7% (IC 95%: 24,1; 42,), n=36 pacientes con respuesta, basándose tanto en la evaluación del CRI como en la realizada por el investigador. No se alcanzó la mediana de duración de la respuesta según el CRI (IC 95%; 4,6 meses a no estimable). Inmunogenicidad: Al igual que sucede con todas las proteínas terapéuticas, existe la posibilidad de una respuesta inmune a Kadcyla. Un total de 836 pacientes de seis estudios clínicos fueron evaluados en diferentes puntos temporales para detectar la presencia de respuestas de anticuerpos antiterapéuticos (ATT) a Kadcyla. Luego de las dosis de Kadcyla, el 5,3% (44/836) de los pacientes obtuvieron resultados positivos de anticuerpos anti-Kadcyla en uno o más puntos temporales posteriores a la dosis; de éstos, 28 tenían muestras basales negativas de referencia. El significado clínico de los anticuerpos anti-trastuzumab emtansina es aún desconocido. Los resultados de los estudios de inmunogenicidad son altamente dependientes de varios factores, incluyendo especificidad y sensibilidad de análisis, metodología de determinación, manipulación de muestras, tiempo de recolección de muestras, medicación concomitante y enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos a Kadcyla con la de anticuerpos a otros productos puede inducir a error. Población pediátrica: La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar resultados de estudios realizados con Kadcyla en los diferentes grupos de población pediátrica con cáncer de mama (véase Dosificación). Propiedades farmacocinéticas: Absorción: Kadcyla se administra por vía intravenosa. No se han realizado estudios con otras vías de administración. Distribución: Los pacientes del estudio TDM4370g/BO21977 que recibieron 3,6 mg/kg de Kadcyla por vía intravenosa cada 3 semanas presentaron un promedio de concentración sérica máxima (Cmáx) de trastuzumab emtansina de 83,4 (± 16,5) mg/ml. Según el análisis farmacocinético poblacional, luego de la administración por vía intravenosa, el volumen central de distribución de trastuzumab emtansina fue de 3,13 litros y se aproximó al del volumen plasmático. Biotransformación (trastuzumab emtansina y DM1): Es previsible que trastuzumab emtansina experimente un proceso de desconjugación y catabolismo por medio de proteólisis en lisosomas celulares. Los estudios de metabolismo in vitro en microsomas hepáticos humanos sugieren que DM1, una pequeña molécula componente de trastuzumab emtansina, es metabolizado principalmente por CYP3A4 y en menor medida por CYP3A5. DM1 no inhibió las principales enzimas del sistema CYP450 in vitro. En plasma humano, se detectaron niveles bajos de los catabolitos de trastuzumab emtansina MCC-DM1, Lys-MCC-DM1 y DM1. In vitro, DM1 fue un sustrato de la p-glicoproteína (P-gp). Eliminación: Basados en el análisis farmacocinético poblacional (PK), luego de la administración de Kadcyla por vía intravenosa en pacientes con cáncer de mama metastásico HER2 positivo, el clearance de trastuzumab emtansina fue de 0,68 l/día y la vida media de eliminación (t1/2) de aproximadamente 4 días. No se observó acumulación de trastuzumab emtansina luego de dosis repetidas infundidas por vía intravenosa cada 3 semanas. Según un análisis farmacocinético poblacional (n=671), el peso corporal, la albúmina, la suma del diámetro más largo de lesiones blanco por los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST), el dominio extracelular (DEC) de HER2, las concentraciones iniciales de trastuzumab, y aspartato aminotransferasa (AST) fueron identificados como covariables estadísticamente significativas para los parámetros farmacocinéticos de trastuzumab emtansina. Sin embargo, la magnitud del efecto de estas covariables sobre la exposición a trastuzumab emtansina, sugiere que es poco probable que dichas covariables tengan efectos clínicamente significativos sobre la exposición a Kadcyla. Además, un análisis exploratorio mostró que la influencia de determinadas covariables (es decir, función renal, raza y edad) sobre la farmacocinética de trastuzumab total y DM1 era limitada y clínicamente irrelevante. En estudios preclínicos, los catabolitos de trastuzumab emtansina, incluidos DM1, Lis-MCC-DM1 y MCC-DM1, son principalmente excretados en la bilis, con eliminación mínima en la orina. Linealidad/no linealidad: Al administrar Kadcyla por vía intravenosa cada 3 semanas, exhibió farmacocinética lineal a lo largo de dosis de entre 2,4 y 4,8 mg/kg; los pacientes que recibieron dosis menores o iguales a 1,2 mg/kg tuvieron un clearance más rápido. Poblaciones especiales: Pacientes de edad avanzada: El análisis farmacocinético poblacional de Kadcyla demostró que la edad no afectó la farmacocinética de trastuzumab emtansina. No se observaron diferencias significativas en el análisis farmacocinético poblacional de trastuzumab emtansina en pacientes menores de 65 años (n=577), en los que tenían entre 65 y 75 años (n=78) y en aquellos mayores de 75 años (n=16). Pacientes con insuficiencia renal: No se han llevado a cabo estudios farmacocinéticos específicos en pacientes con insuficiencia renal. El análisis farmacocinético poblacional de Kadcyla demostró que el clearance de creatinina no altera la farmacocinética de trastuzumab emtansina, la cual, en pacientes con insuficiencia renal leve (clearance de creatinina [ClCr] 60 a 89 ml/min, n=254) o moderada (Clcr 30 a 59 ml/min, n=53) fue similar a la de aquéllos con función renal normal (Clcr ≥ 90 ml/min, n=361). Los datos de farmacocinética en pacientes con insuficiencia renal grave (Clcr 15 a 29 ml/min) son escasos (n=1); por lo tanto, no pueden realizarse recomendaciones posológicas. Pacientes con insuficiencia hepática: El hígado es el órgano principal para la eliminación de DM1 y de catabolitos que contienen DM1. La farmacocinética de trastuzumab emtansina y de catabolitos que contienen DM1 se evaluó después de la administración de 3,6 mg/kg de Kadcyla a los pacientes con cáncer de mama metastásico HER2 positivo con función hepática normal (n = 10), insuficiencia hepática leve (Child-Pugh A, n = 10) y moderada (Child-Pugh B, n = 8). Las concentraciones plasmáticas de DM1 y de catabolitos que contienen DM1 (Lis-MCC-DM1 y MCC-DM1) fueron bajas y comparables entre los pacientes con insuficiencia hepática o sin ella. Las exposiciones sistémicas (ABC) de trastuzumab emtansina en el ciclo 1 en pacientes con insuficiencia hepática leve y moderada fueron aproximadamente 38% y 67% menor que la de aquéllos con función hepática normal, respectivamente. La exposición de trastuzumab emtansina (ABC) en el ciclo 3 después de la administración reiterada en pacientes con disfunción hepática leve o moderada estuvo dentro del rango observado en los que tenían función hepática normal. Kadcyla no se ha estudiado en pacientes con insuficiencia hepática grave (Child-Pugh clase C). Otras poblaciones especiales: El análisis de farmacocinética poblacional de Kadcyla demostró que la raza no parecía influir sobre la farmacocinética de trastuzumab emtansina. Debido a que la mayoría de los pacientes de los estudios clínicos con Kadcyla fueron mujeres, el efecto del sexo en el análisis de farmacocinética poblacional de trastuzumab emtansina no se evaluó específicamente. Datos preclínicos sobre seguridad: Toxicología y/o farmacología en animales: La administración de trastuzumab emtansina fue bien tolerada en ratas y monos con dosis de hasta 20 y 10 mg/kg, respectivamente, que corresponden a 2.040 mg/m2 de DM1 en ambas especies, que es aproximadamente equivalente a la dosis clínica de trastuzumab emtansina en pacientes. A excepción de la neuropatía axonal periférica irreversible (observada sólo en monos tratados con dosis ≥ 10 mg/kg) y la toxicidad sobre los órganos reproductores (registrada únicamente en ratas que recibieron dosis de 60 mg/kg), las toxicidades dependientes de dosis identificadas en los estudios de toxicidad GLP fueron parcial o totalmente reversibles en ambos modelos animales. Las toxicidades principales incluyeron hepatotoxicidad (elevaciones de las enzimas hepáticas) con dosis de ≥ 20 mg/kg y ≥ 10 mg/kg, mielotoxicidad (disminución de los recuentos de plaquetas y leucocitos)/toxicidad hematológica, con dosis de ≥ 20 mg/kg y ≥ 10 mg/kg, y toxicidad en órganos linfoides con dosis de ≥ 20 mg/kg y ≥ 3 mg/kg, en ratas y monos, respectivamente. Mutagenicidad: DM1 fue aneugénico o clastogénico en un estudio de micronúcleos de médula ósea de rata in vivo utilizando una dosis única, a exposiciones que fueron comparables a las concentraciones máximas medias de DM1 determinadas en seres humanos tratados con trastuzumab emtansina. DM1 no fue mutagénico en un ensayo de mutación bacteriana inversa in vitro (Ames). Deterioro de la fertilidad y teratogenicidad: No se han llevado a cabo estudios específicos sobre la fertilidad con trastuzumab emtansina. Sin embargo, basados en los resultados obtenidos en los estudios generales de toxicidad en animales, es previsible que trastuzumab emtansina tenga efectos adversos sobre la fertilidad. No se han efectuado estudios específicos sobre el desarrollo embriofetal en animales con trastuzumab emtansina. Se ha identificado toxicidad sobre el desarrollo con trastuzumab en el entorno clínico, aunque no se preveía en el programa de estudios preclínicos. Además, en éstos se ha observado toxicidad sobre el desarrollo con maitansina, lo cual sugiere que DM1, el componente maitansinoide citotóxico de trastuzumab emtansina que inhibe el microtúbulo, será igualmente teratogénico y potencialmente embriotóxico.
Indicaciones.
Kadcyla, como agente único, se indica para el tratamiento de pacientes con cáncer de mama localmente avanzado no resecable, o metastásico HER2 positivo, que han recibido tratamiento previo con trastuzumab y un taxano por separado o en combinación. Los pacientes deben reunir los siguientes requisitos: haber recibido tratamiento previo para la enfermedad localmente avanzada o metastásica, o haber manifestado recurrencia de la enfermedad durante el tratamiento adyuvante o en los seis meses siguientes a su terminación.
Dosificación.
Kadcyla debe ser prescrito únicamente por un médico y administrado bajo la supervisión de un profesional de la salud con experiencia en el tratamiento de pacientes oncológicos. Los pacientes tratados con trastuzumab emtansina deben tener tumores HER2 positivos, que se definen por un puntaje 3+ mediante inmunohistoquímica (IHQ) o un cociente ≥ 2,0 mediante hibridación in situ (FISH), determinado por medio de un dispositivo médico para diagnóstico in vitro (IVD) con el marcado CE. Si no se dispone de un dispositivo IVD con el marcado CE, el estado de HER2 se debe evaluar utilizando una técnica alternativa validada. Para evitar errores de medicación, es importante controlar las etiquetas de los viales para asegurar que el medicamento que se está preparando y administrando es Kadcyla (trastuzumab emtansina) y no Herceptin (trastuzumab). Posología: La dosis recomendada de Kadcyla es de 3,6 mg/kg de peso corporal, administrada en infusión intravenosa cada 3 semanas (ciclos de 21 días). Los pacientes deben ser tratados hasta que se produzca progresión de la enfermedad o toxicidad inaceptable. La dosis inicial se debe administrar en infusión intravenosa de 90 minutos. Se debe vigilar a los pacientes durante su transcurso y por lo menos hasta 90 minutos después de la primera infusión por si se produjeran fiebre, escalofríos u otras reacciones relacionadas con la infusión. El lugar donde se realiza la infusión debe supervisarse cuidadosamente para la detección de posibles infiltraciones subcutáneas durante la administración (véase Precauciones). Si la infusión anterior fue bien tolerada, las dosis siguientes de Kadcyla pueden administrarse en infusiones de 30 minutos. Los pacientes deben ser controlados durante la infusión y por lo menos en los 30 minutos posteriores. Si se manifiestan síntomas relacionados con la infusión, se debe reducir la velocidad de infusión o interrumpir la administración de trastuzumab emtansina (véanse Precauciones y Reacciones adversas). Suspenda la administración de Kadcyla si observa reacciones a la infusión que pongan en peligro la vida del paciente. Tanto los medicamentos para el tratamiento de reacciones alérgicas/anafilácticas a la infusión, como el equipo de emergencia deben estar disponibles para su uso inmediato (véase Precauciones). Retrasos u omisiones de dosis: Si se omite una dosis programada, ésta se debe administrar lo antes posible, sin esperar hasta el siguiente ciclo programado. El esquema de administración debe ajustarse para mantener un intervalo de 3 semanas entre cada dosis. La siguiente dosis se debe administrar de acuerdo con las recomendaciones mencionadas. Modificación de la dosis: Es posible que el tratamiento de las reacciones adversas sintomáticas requiera interrupción temporaria, reducción de la dosis o suspensión de Kadcyla según lo establecido en las pautas proporcionadas en las Tablas 2 a 6. La dosis de Kadcyla no puede volver a aumentarse luego de haberse reducido.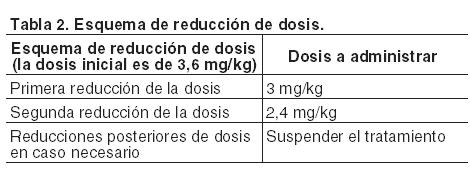



Neuropatía periférica: El tratamiento con Kadcyla se debe interrumpir temporalmente en los pacientes que manifiesten neuropatía periférica de Grados 3 o 4, hasta que ésta remita a Grado ≤ 2. Cuando se reanude el tratamiento, se puede considerar la reducción de la dosis de acuerdo con el esquema indicado en la Tabla 2. Población pediátrica: La seguridad y eficacia de Kadcyla en niños y adolescentes menores de 18 años no han sido establecidas, ya que no existe una recomendación de uso específica en la población pediátrica para la indicación de cáncer de mama metastásico (CMM). Pacientes de edad avanzada: No se requiere un ajuste de la dosis de Kadcyla en pacientes de 65 años o más. No se dispone de datos suficientes para establecer la seguridad y eficacia en aquéllos de 75 años o más. El análisis de farmacocinética poblacional indica que la edad no tiene un efecto clínicamente relevante en la farmacocinética de trastuzumab emtansina (véanse Farmacología, Propiedades farmacocinéticas). Pacientes con insuficiencia renal: No es preciso un ajuste de la dosis inicial de Kadcyla en pacientes con insuficiencia renal leve o moderada (véase Farmacología, Propiedades farmacocinéticas). La posible necesidad de modificar la dosis en pacientes con insuficiencia renal severa no puede determinarse, debido a que no se cuenta con datos suficientes, y por consiguiente, estos pacientes deben ser controlados estrechamente. Pacientes con insuficiencia hepática: No es necesario ajustar la dosis inicial en pacientes con insuficiencia hepática leve o moderada (véase Farmacología, Propiedades farmacocinéticas). Kadcyla no se ha estudiado en pacientes con insuficiencia hepática grave. El tratamiento de los pacientes con insuficiencia hepática debe realizarse con precaución debido a la hepatotoxicidad conocida observada con Kadcyla (véase Precauciones). Formas de administración: Kadcyla debe ser reconstituido y diluido por un profesional de la salud y administrado por vía intravenosa mediante infusión. No se debe administrar en infusión intravenosa rápida ni en bolo. Para consultar las instrucciones para la reconstitución y la dilución del medicamento previas a la administración, véase Precauciones especiales de eliminación y otras manipulaciones.
Contraindicaciones.
Kadcyla está contraindicado en pacientes con hipersensibilidad conocida a trastuzumab emtansina o a cualquiera de sus excipientes.
Reacciones adversas.
Resumen del perfil de seguridad: La seguridad de Kadcyla se ha evaluado en 1.871 pacientes con cáncer de mama en estudios clínicos. En esta población de pacientes: Las reacciones adversas a medicamentos (RAM) serias más frecuentes fueron fiebre, trombocitopenia, vómitos, dolor abdominal, náuseas, estreñimiento, diarrea, disnea y neumonitis. Las RAM más frecuentes (≥ 25%) con Kadcyla fueron hemorragia (incluida epistaxis), transaminasas elevadas, fatiga, dolor musculosquelético y cefalea. La mayoría de las RAM informadas fue de Grados 1 o 2 de severidad. Las RAM de Grados 3 o 4, de acuerdo con los Criterios de Terminología Común para Eventos Adversos del National Cancer Institute (NCI-CTCAE), más frecuentes ( > 2%) fueron trombocitopenia, fatiga, transaminasas elevadas, anemia, hipopotasemia, dolor musculosquelético y neutropenia. Tabla de reacciones adversas: En la Tabla 7 se presentan las RAM observadas en 1.871 pacientes tratados con Kadcyla. Las RAM se enumeran a continuación de acuerdo con la clasificación por órganos y sistemas (SOC) de MedDRA y por categorías de frecuencia. Estas se definen como muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1.000 a < 1/100), raras (≥ 1/10.000 a < 1/1.000), muy raras ( < 1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada grupo de frecuencia y SOC, las reacciones adversas se presentan en orden decreciente de gravedad. Las RAM se informaron utilizando los criterios NCI-CTCAE para la evaluación de la toxicidad.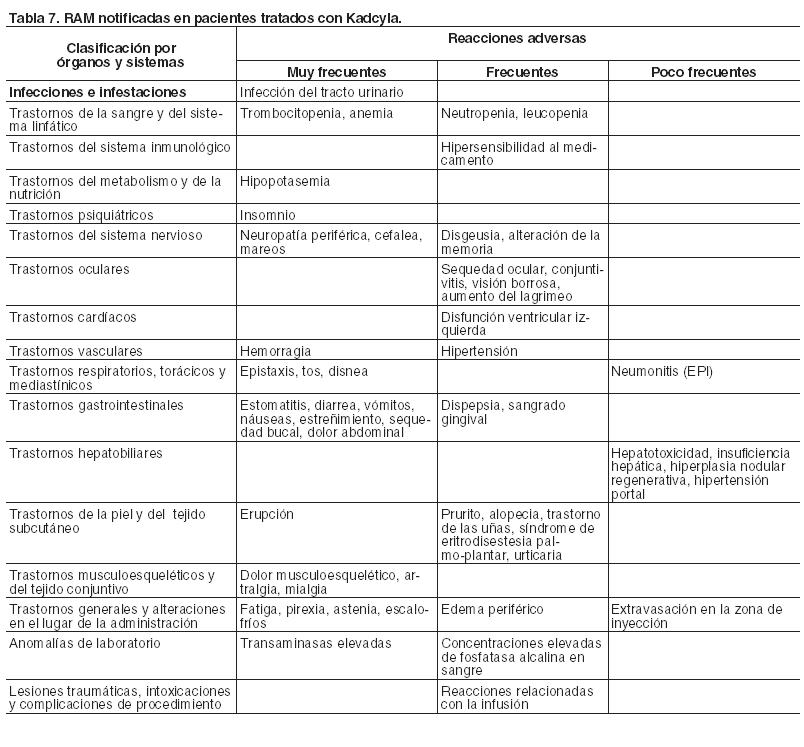
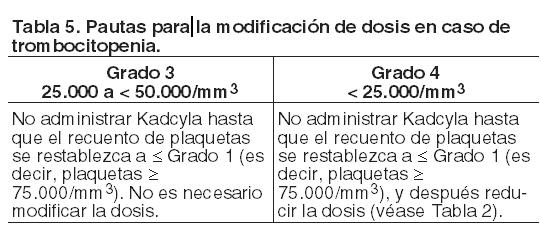
Descripción de reacciones adversas seleccionadas: Transaminasas elevadas (AST/ALT): Se han observado elevaciones (de Grados 1 - 4) de las transaminasas séricas durante el tratamiento con Kadcyla en estudios clínicos (véase Precauciones), que por lo general, fueron transitorias. Se ha observado un efecto acumulativo de Kadcyla en las transaminasas y los valores se restablecieron generalmente al interrumpir el tratamiento. Se notificaron aumentos de transaminasas en el 28% de los pacientes de los estudios clínicos. Se informaron incrementos de AST y ALT de Grados 3 o 4 en el 4,1% y el 2,8% de los pacientes respectivamente, y normalmente se produjeron en los primeros ciclos de tratamiento (1-6). En general, los eventos hepáticos de Grado ≥ 3 no se asociaron con evolución clínica desfavorable; los valores registrados posteriormente durante el seguimiento tendieron a mostrar una recuperación a rangos que permitieron al paciente permanecer en el estudio y continuar recibiendo el tratamiento del mismo con la idéntica dosis o con una dosis reducida. No se observó una relación entre la exposición a trastuzumab emtansina (ABC), la concentración sérica máxima (Cmáx) de trastuzumab emtansina, la exposición total a trastuzumab (ABC) o la Cmáx de DM1 y las elevaciones de las transaminasas. Para obtener información sobre la modificación de dosis en caso de transaminasas elevadas véase Dosificación y Precauciones. Disfunción ventricular izquierda: En estudios clínicos con Kadcyla, se detectó disfunción ventricular izquierda en el 2,0% de los pacientes. La mayoría de los eventos fueron disminuciones asintomáticas de Grados 1 o 2 en la FEVI. Se notificaron eventos de Grados 3 o 4 en el 0,3% de los pacientes. Estas reacciones poco frecuentes de Grados 3 o 4 se produjeron generalmente en los primeros ciclos de tratamiento (1-2). Se recomienda realizar controles adicionales de la FEVI en los pacientes que presenten valores de FEVI ≤ 45% (para consultar las pautas específicas para la modificación de la dosis, véase Dosificación; Tabla 6). Reacciones relacionadas con la infusión: Las reacciones relacionadas con la infusión (RRI) se caracterizan por la presencia de uno o varios de los siguientes síntomas: sofocos, escalofríos, fiebre, disnea, hipotensión, sibilancia, broncospasmo y taquicardia. Se notificaron RRI en el 4,5% de los pacientes de los estudios clínicos con trastuzumab emtansina, observándose un evento de Grado 3 y ninguno de Grado 4. Las RRI se resolvieron en el transcurso de varias horas a un día posterior a la finalización de la misma. No se observó una relación con la dosis en los estudios clínicos. Para consultar las pautas para la modificación de la dosis en caso de reacciones relacionadas con la infusión véanse Dosificación y Precauciones. Reacciones de hipersensibilidad: En los estudios clínicos con Kadcyla, se informaron reacciones de hipersensibilidad en el 2,6% de los pacientes y no se encontraron eventos de Grados 3 o 4. En general, la mayoría de estas reacciones fueron de intensidad leve o moderada y se resolvieron después del tratamiento. Para las modificaciones de la dosis en caso de reacciones de hipersensibilidad, véanse Dosificación y Precauciones. Trombocitopenia: Se notificó trombocitopenia, o disminución del recuento de plaquetas, en el 31,4% de los pacientes de los estudios clínicos de trastuzumab emtansina y fue la reacción adversa más frecuente que requirió la interrupción del tratamiento (1,4%). La trombocitopenia fue de Grados 1 o 2 (≥ 50.000/mm3) en la mayoría de los pacientes, el valor más bajo se alcanzó hacia el día 8 y los recuentos se restablecieron generalmente a Grados 0 o 1 (≥ 75.000/mm3) antes de administrar la siguiente dosis programada. En los estudios clínicos, la incidencia y la severidad de la trombocitopenia fueron mayores en los pacientes asiáticos. Independientemente de la raza, la incidencia de eventos de Grados 3 o 4 ( < 50.000/mm3) fue del 11,3% en los pacientes tratados con Kadcyla. La incidencia de eventos hemorrágicos graves (Grados ≥ 3) fue del 1,7% en todos los tratados con trastuzumab emtansina y del 1% en los participantes asiáticos a los que se administró trastuzumab emtansina. En algunos casos observados, los pacientes también estaban recibiendo anticoagulantes. Se han registrado casos de hemorragias con resultado mortal. Para obtener información sobre modificaciones de la dosis en caso de trombocitopenia, véanse Dosificación y Precauciones. Inmunogenicidad: Como sucede con todas las proteínas terapéuticas, existe el potencial de que se produzca una respuesta inmune contra Kadcyla. Se realizaron análisis en un total de 836 pacientes de seis estudios clínicos en diferentes puntos temporales para detectar la presencia de respuestas de anticuerpos antiterapéuticos (ATT) contra Kadcyla. El 5,3% (44/836) de los pacientes resultaron positivos para anticuerpos anti-Kadcyla en uno o más puntos temporales después de la administración de la dosis. Todavía no se conoce la importancia clínica de los anticuerpos anti-trastuzumab emtansina. Extravasación: En los estudios clínicos con Kadcyla, se han observado reacciones debidas a extravasación que fueron generalmente leves e incluyeron eritema, sensibilidad, irritación de la piel, dolor o inflamación en la zona de administración de la infusión. Estas reacciones se han observado con más frecuencia en las 24 horas siguientes a la infusión. Hasta el momento, no se conoce el tratamiento específico para la extravasación de Kadcyla. Anomalías de laboratorio: En la Tabla 8 se muestran las anomalías de laboratorio observadas en pacientes tratados con Kadcyla en el estudio clínico TDM4370g/BO21977.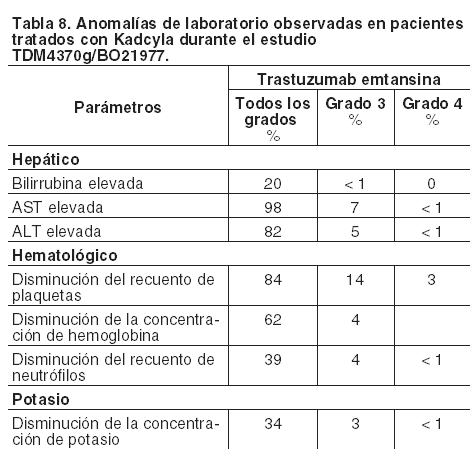
Comunicación de reportes de reacciones adversas: Es importante comunicar las presuntas reacciones adversas después de la autorización del medicamento. Esto permite la monitorización continua de la relación riesgo/beneficio. Se solicita a los profesionales de la salud informar de cualquier sospecha de eventos adversos asociados con el uso de Kadcyla® al Área de Farmacovigilancia de Roche al siguiente teléfono 0800-77-ROCHE (76243). En forma alternativa, esta información puede ser reportada ante ANMAT. Ante cualquier inconveniente con el producto, el paciente puede llenar la ficha que está en la Página Web de la ANMAT: http://www.anmat.gov.ar/farmacovigilancia/Notificar.asp o llamar a ANMAT responde al 0800-333-1234.
Precauciones.
Para mejorar la trazabilidad de los productos medicinales biológicos, el nombre comercial y el número de lote del producto administrado debe registrarse claramente (o mencionarse) en la historia clínica del paciente. La sustitución por cualquier otro medicamento biológico requiere el consentimiento del médico prescriptor. Para evitar errores de medicación, es importante controlar las etiquetas de los viales para asegurar que el medicamento que se está preparando y administrando es Kadcyla (trastuzumab emtansina) y no Herceptin (trastuzumab). Toxicidad pulmonar: En estudios clínicos con Kadcyla se informaron casos de enfermedad pulmonar intersticial (EPI), incluida neumonitis, algunos de los cuales condujeron a síndrome de dificultad respiratoria aguda o con consecuencias fatales (véase Reacciones adversas). Los signos y síntomas incluyen disnea, tos, fatiga e infiltrados pulmonares. Se recomienda interrumpir permanentemente el tratamiento con Kadcyla en pacientes con diagnóstico de EPI o neumonitis. Es posible que los pacientes con disnea en reposo causada por complicaciones de la enfermedad maligna avanzada y por comorbilidades puedan tener mayor riesgo de padecer disfunciones pulmonares. Hepatotoxicidad: Durante el tratamiento con Kadcyla en estudios clínicos, se observó hepatotoxicidad, principalmente en la forma de incrementos asintomáticos en las concentraciones de transaminasas séricas (transaminitis Grados 1 - 4) (véase Reacciones adversas). Estas elevaciones fueron generalmente transitorias, alcanzando valores máximos el día 8 después de la terapia y restableciéndose posteriormente a Grado 1 o menor antes del siguiente ciclo. También se verificó un efecto acumulativo de Kadcyla en las transaminasas (la proporción de pacientes con anomalías de ALT/AST de Grados 1 - 2 aumenta con los ciclos sucesivos). En la mayoría de los casos, las transaminasas se restablecieron a Grado 1 o a valores normales dentro de los 30 días siguientes a la administración de la última dosis de Kadcyla (véase Reacciones adversas). En pacientes tratados con trastuzumab emtansina se han detectado trastornos hepatobiliares graves, incluyendo hiperplasia nodular regenerativa (HNR) del hígado, que en algunas ocasiones tuvieron un desenlace fatal debido a lesión hepática inducida por fármacos. Los casos observados pudieron haber sido confundidos por comorbilidades y/o medicación concomitante con potencial hepatotóxico conocido. La función del hígado debe ser controlada antes de iniciar el tratamiento y antes de cada dosis de Kadcyla. Los pacientes con un incremento basal de ALT (por ejemplo, debido a metástasis hepáticas) pueden estar predispuestos a daño hepático con un riesgo mayor de evento hepático Grados 3 - 5 o de aumento de las pruebas de función hepática. Las pautas para la reducción de la dosis o la interrupción del tratamiento en caso de elevación de las transaminasas séricas y bilirrubina total se especifican en Dosificación.Se han identificado casos de hiperplasia nodular regenerativa (HNR) del hígado en biopsias hepáticas en pacientes tratados con Kadcyla. La HNR es un trastorno raro que se caracteriza por una transformación benigna generalizada del parénquima hepático en pequeños nódulos regenerativos; la HNR puede producir hipertensión portal no cirrótica. Su diagnóstico solamente puede confirmarse mediante un estudio histopatológico. Se debe considerar la existencia de HNR en todos los pacientes con síntomas clínicos de hipertensión portal y/o patrones cirróticos observados en tomografía computarizada (TC) del hígado, aunque las concentraciones de transaminasas sean normales y no presenten otras manifestaciones de cirrosis. Después del diagnóstico de HNR, el tratamiento con Kadcyla debe interrumpirse en forma permanente. Trastuzumab emtansina no se ha estudiado en pacientes con concentraciones de transaminasas séricas > 2
