Hemlibra®
ROCHE
Antihemorrágico. Hemostático sistémico.
Composición.
Cada vial de 1 ml contiene 30 mg de emicizumab en una concentración de 30 mg/ml, en un excipiente compuesto por: L-histidina 3,1 mg, ácido L-aspártico c.s.p., L-arginina 26,1 mg, Poloxámero 188: 0,5 mg y agua para inyectables c.s.p. 1,0 ml. Cada vial de 0,4 ml contiene 60 mg de emicizumab en una concentración de 150 mg/ml, en un excipiente compuesto por: L-histidina 1,2 mg, ácido L-aspártico c.s.p., L-arginina 10,5 mg, Poloxámero 188: 0,2 mg y agua para inyectables c.s.p. 0,4 ml. Cada vial de 0,7 ml contiene 105 mg de emicizumab en una concentración de 150 mg/ml, en un excipiente compuesto por: L-histidina 2,2 mg, ácido L-aspártico c.s.p., L-arginina 18,3 mg, Poloxámero 188: 0,4 mg y agua para inyectables c.s.p. 0,7 ml. Cada vial de 1 ml contiene 150 mg de emicizumab en una concentración de 150 mg/ml, en un excipiente compuesto por: L-histidina 3,1 mg, ácido L-aspártico c.s.p., L-arginina 26,1 mg, Poloxámero 188: 0,5 mg y agua para inyectables c.s.p. 1,0 ml.
Farmacología.
Código ATC: B02BX06. Grupo farmacoterapéutico: Sangre y órganos hematopoyéticos; antihemorrágicos; otros hemostáticos sistémicos. Propiedades farmacodinámicas: Mecanismo de acción: Emicizumab es un anticuerpo monoclonal humanizado modificado inmunoglobulina G4 (IgG4) con una estructura de anticuerpo biespecífico. Emicizumab une el factor IX activado y el factor X para restaurar el funcionamiento del factor VIII activado deficiente, que es necesario para una hemostasia efectiva. Emicizumab no tiene relación estructural ni homología de secuencia con el factor VIII y, como tal, no induce ni potencia el desarrollo de inhibidores directos del factor VIII. Efectos farmacodinámicos: El tratamiento profiláctico con Hemlibra reduce el aPTT y aumenta la actividad del factor VIII observada (utilizando un ensayocromogénico con factores de coagulación humanos). Estos dos marcadores farmacodinámicos no reflejan el verdadero efecto hemostático de emicizumab in vivo (el aPTT está claramente reducido y la actividad del factor VIII observada podría estar sobrestimada), pero proporcionan una estimación relativa del efecto pro-coagulante de emicizumab. Eficacia clínica y seguridad: Se evaluó la eficacia de Hemlibra para la profilaxis de rutina en pacientes con hemofilia A con o sin inhibidores FVIII en cuatro estudios clínicos (tres estudios en adultos y adolescentes [HAVEN 3, HAVEN 1 y HAVEN 4] y un estudio pediátrico [HAVEN 2]). Estudios clínicos en adultos y adolescentes: Pacientes (≥ 12 años y > 40 kg) con hemofilia A sin inhibidores del FVIII (estudio BH30071 - HAVEN 3): El estudio HAVEN 3 era un estudio clínico fase III, aleatorizado, multicéntrico y abierto con 152 pacientes varones adultos y adolescentes (≥ 12 años y > 40 kg) con hemofilia A severa sin inhibidores del FVIII que previamente habían recibido tratamiento episódico ("a demanda") o profilácticoc on FVIII. Los pacientes recibieron Hemlibra por vía subcutánea, 3mg/kg una vez a la semana durante las cuatro primeras semanas, seguido de 1,5 mg/kg una vez a la semana (grupos A y D) o 3 mg/kg cada dos semanas (grupo B), o sin profilaxis (grupo C). Los pacientes del grupo C pudieron cambiar al tratamiento con Hemlibra (3 mg/kg cada dos semanas) después de completar al menos 24 semanas sin profilaxis. Para los grupos A y B, se permitió un incremento de la dosis hasta 3 mg/kg a la semana luego de 24 semanas para los pacientes que experimentaron dos o más sangrados considerados relevantes (esto es, sangrados espontáneos y clínicamente significativos que tengan lugar en equilibrio dinámico). Los pacientes del grupo D podían llegar a un incremento de la dosis después de la segunda hemorragia significativa. En el momento del análisis primario, cinco pacientes tuvieron un incremento de su dosis de mantenimiento. Se aleatorizaron con un ratio 2:2:1 ochenta y nueve pacientes, que previamente habían recibido tratamiento episódico ("a demanda") con FVIII, para recibir Hemlibra una vez por semana (grupo A; N = 36), cada dos semanas (grupo B; N = 35) o sin profilaxis (grupo C; N = 18), con estratificación por tasa de sangrado previa de 24 semanas ( < 9 o ≥ 9). Se incluyeron sesenta y tres pacientes en tratamiento profiláctico previo con FVIII en el grupo D para recibir Hemlibra (1,5 mg/kgunavezpor semana). El objetivo principal del estudio era evaluar, en pacientes tratados anteriormente con FVIII episódico, la eficacia de la profilaxis semanal con Hemlibra (grupoA) o cada dos semanas (grupo B) en comparación con la ausencia de profilaxis (grupo C) según el número de sangrados que requirieron tratamiento con factores de coagulación (véase Tabla 4). Otros objetivos del estudio incluyeron la evaluación de la comparación aleatorizada de los grupos A o B, y el grupo C para la eficacia de la profilaxis con Hemlibra en la reducción del número de todos los sangrados, los sangrados espontáneos, los sangrados articulares y los sangrados de las articulaciones diana (véase Tabla 4), además de evaluar la preferencia de tratamiento del paciente mediante una encuesta. La eficacia de la profilaxis con Hemlibra también se comparó con el tratamiento profiláctico previo con FVIII (grupo D) en pacientes que habían participado en un estudio no intervencional (NIS por sus siglas en inglés) antes de su incorporación (véase Tabla 5). En esta comparación, solo se incluyeron pacientes del NIS ya que se recogieron datos de sangrado y tratamiento con el mismo nivel de detalle que en HAVEN 3. El NIS es un estudio observacional con el objetivo principal de recoger datos clínicos detallados acerca de los episodios de sangrado y el uso de medicación para hemofilia en pacientes con hemofilia A, fuera de un estudio intervencional. Pacientes (≥ 12 años) con hemofilia A con inhibidores del factor VIII (estudio BH29884 - HAVEN 1): El estudio HAVEN 1, era un estudio clínico, aleatorizado y multicéntrico, en 109 varones adultos y adolescentes (≥ 12 años) con hemofilia A con inhibidores del factor VIII que habían recibido anteriormente tratamiento episódico o profiláctico con agentes bypaseantes (CCPa y rFVIII). En el estudio, los pacientes recibieron semanalmente profilaxis con Hemlibra (grupos A, C y D), 3 mg/kg una vez por semana durante cuatro semanas seguidos de 1,5 mg/kg por semana en adelante o ninguna profilaxis (grupo B). Los pacientes randomizados del grupo B pudieron cambiar a profilaxis con Hemlibra después de completar al menos 24 semanas sin profilaxis. Se permitió un incremento de la dosis hasta 3 mg/kg una vez por semana después de 24 semanas de profilaxis con Hemlibra para los pacientes que experimentaron dos o más sangrados considerados relevantes (esto es, sangrados espontáneos y clínicamente significativos verificados que tengan lugar en equilibrio dinámico). En el momento del análisis interino, dos pacientes incrementaron la dosis de mantenimiento hasta 3 mg/kg una vez por semana. Cincuenta y tres pacientes previamente tratados con agentes bypaseantes episódicos ("a demanda") fueron aleatorizados en una proporción de 2:1 a recibir profilaxis con Hemlibra (grupo A) o ninguna profilaxis (grupo B), con estratificación por tasa de sangrados previa en 24 semanas ( < 9 o ≥ 9). Cuarenta y nueve pacientes tratados anteriormente con agentes bypaseantes profilácticos fueron incluidos en el grupo C para recibir profilaxis con Hemlibra. Siete pacientes previamente tratados con agentes bypaseantes episódicos ("a demanda") que habían participado en el estudio NIS previo a la incorporación, pero que no pudieron ser incluidos en HAVEN 1 antes del cierre de los grupos A y B fueron incluidos en el grupo D para recibir profilaxis con Hemlibra. El objetivo principal del estudio era evaluar, en pacientes previamente tratados con agentes bypaseantes episódicos ("a demanda"), el efecto del tratamiento de profilaxis con Hemlibra semanal en comparación con la ausencia de profilaxis (grupo A frente a grupo B) en el número de sangrados que requirieron tratamiento con factores de coagulación a lo largo del tiempo (mínimo de 24 semanas o fecha de la suspensión) (véase Tabla 6). Otros objetivos secundarios de la comparación aleatorizada de los grupos A y B fueron la eficacia de la profilaxis con Hemlibra semanal en la reducción del número de todos los sangrados, los sangrados espontáneos, los sangrados articulares y los sangrados de las articulaciones diana (véase Tabla 6), además de evaluar la calidad de vida relacionada con la salud (HRQoL por sus siglas en inglés) y el estado de salud de los pacientes (véanse Tablas 9 y 10). El tiempo medio de exposición (+DE) en todos los pacientes del estudio fue de 21,38 semanas (12,01). En cada grupo de tratamiento, el tiempo medio de exposición (+DE) fue de 28,86 semanas (8,37) en el grupo A, 8,79 (3,62) en el grupo B, 21,56 (11,85) en el grupo C y 7,08 (3,89) en el grupo D. Un paciente del grupo A se retiró del estudio antes del inicio de Hemlibra. El estudio también evaluó la eficacia de la profilaxis con Hemlibra semanal en comparación con los agentes bypaseantes episódicos ("a demanda") y profilácticos anteriores (comparaciones separadas) en pacientes que habían participado en el NIS antes de la incorporación (grupos A y C, respectivamente) (véase Tabla 7). Pacientes (≥ 12años) con hemofilia A con o sin inhibidores del factor VIII (estudio BO39182 - HAVEN 4): Se evaluó Hemlibra en un estudio clínico fase III de un único grupo, multicéntrico con 41 pacientes varones adultos y adolescentes (≥ 12 años y > 40 kg) que tienen hemofilia A con inhibidores de FVIII o hemofilia A severa sin inhibidores del FVIII que previamente habían recibido tratamiento episódico ("a demanda") o profiláctico con agentes bypaseantes o FVIII. Los pacientes recibieron profilaxis con Hemlibra - 3 mg/kg una vez a la semana durante las cuatro primeras semanas, seguido de 6 mg/kg cada cuatro semanas. El objetivo principal del estudio era evaluar la eficacia de la profilaxis con Hemlibra administrado cada cuatro semanas para mantener un control del sangrado adecuado, según el número de sangrados tratados. Otros objetivos fueron evaluar la eficacia clínica de la profilaxis con Hemlibra en todos los sangrados, los sangrados espontáneos tratados, los sangrados articulares tratados y los sangrados en las articulaciones diana tratados (véase Tabla 8). Además de evaluar la preferencia de tratamiento del paciente mediante una encuesta. Resultados de eficacia en adultos y adolescentes: HAVEN 3: Los resultados de eficacia de la profilaxis con Hemlibra, comparados con la no profilaxis respecto a la tasa de sangrados tratados, todos los sangrados, los sangrados espontáneos tratados, los sangrados articulares tratados y los sangrados en las articulaciones diana tratados, se muestran en la Tabla 4.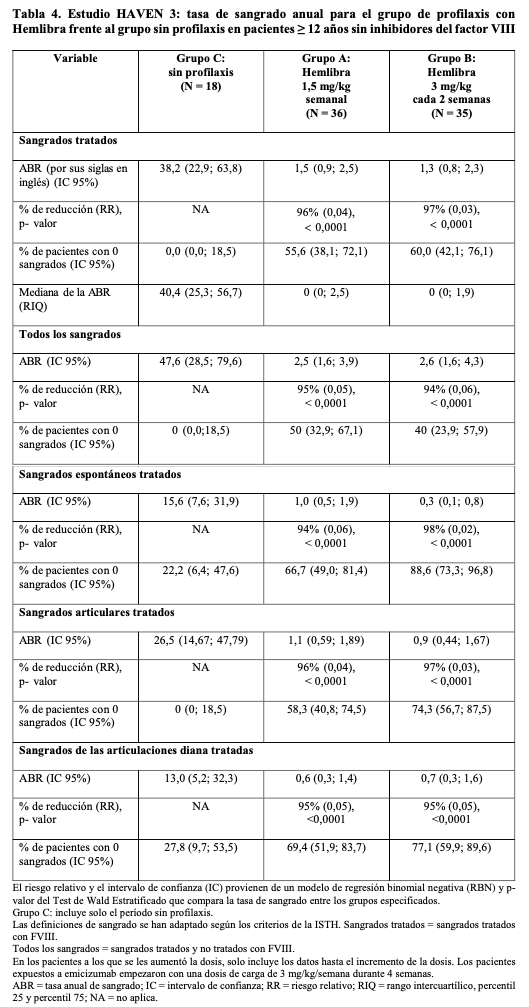
En el análisis intrapaciente del estudio clínico HAVEN 3, la profilaxis con Hemlibra dió lugar a una reducción (68%) estadísticamente significativa (p < 0,0001) de la tasa de sangrados para los episodios tratados en comparación con la profilaxis anterior con FVIII recogida en el NIS antes de la incorporación (véase Tabla 5).
HAVEN 1: Los resultados de eficacia de la profilaxis con Hemlibra, comparados con la no profilaxis respecto a la tasa de sangrados tratados, todos los sangrados, los sangrados espontáneos tratados, los sangrados articulares tratados y los sangrados en las articulaciones diana tratados, se muestran en la Tabla 6.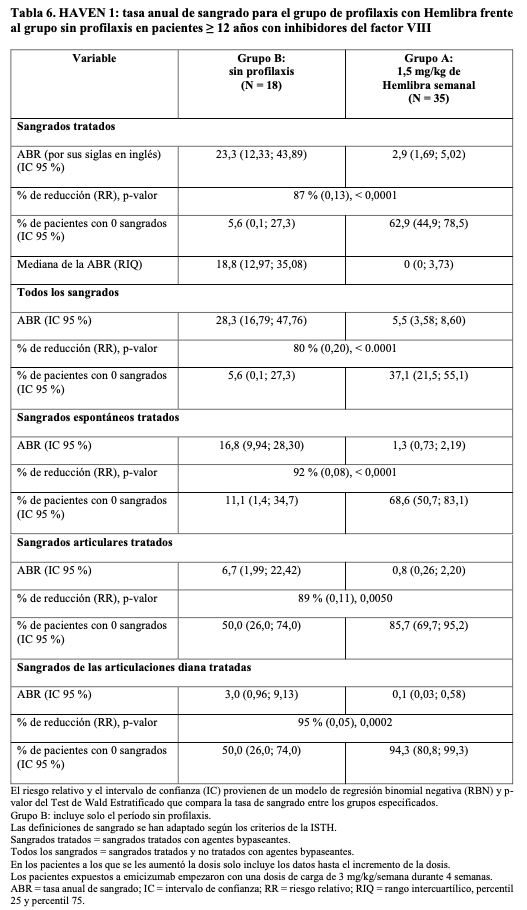
En el análisis intrapaciente de HAVEN 1, la profilaxis con Hemlibra dio lugar a una reducción estadística (p = 0,0003) y clínicamente significativa (79%) de la tasa de sangrados para los episodios tratados en comparación con la profilaxis anterior con agentes bypaseantes recogida en el NIS antes de la inclusión (véase Tabla 5).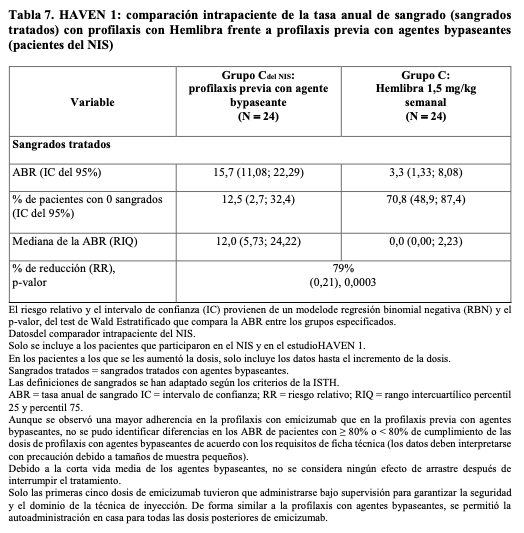
HAVEN 4: Los resultados de eficacia del análisis primario de la profilaxis con Hemlibra cada cuatro semanas respecto a la tasa de sangrados tratados, todos los sangrados, los sangrados espontáneos tratados, los sangrados articulares tratados y los sangrados en las articulaciones diana tratados, se muestran en la Tabla 8. Se evaluó la eficacia en cuarenta y un pacientes ≥ 12 años con una mediana del tiempo de observación de 25 - 6 semanas (rango 24,1 - 29,4).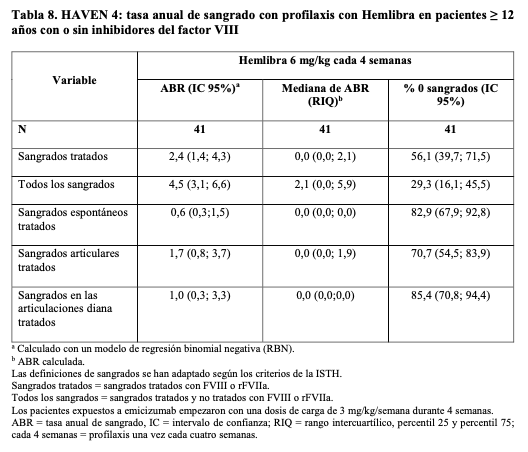
Resultados de las medidas relacionadas con la salud de adultos y adolescentes: Los estudios clínicos HAVEN en adultos y adolescentes, evaluaron los resultados de calidad de vida relacionados con la hemofilia, notificados por los pacientes mediante el cuestionario de Calidad de Vida Específico para Hemofilia (Haem-A-QoL) para adultos ( > 18 años) y su versión adolescente (Haemo-QoL-SF, de 8 a < 18 años), la Puntuación de Salud Física (es decir, hinchazón dolorosa, presencia de dolor en las articulaciones, dolor al moverse, dificultad para caminar lejos y necesitar más tiempo para prepararse) y la puntuación total (resumen de todas las puntuaciones) fueron puntos finales de interés definidos en el protocolo. Para medir el cambio en el estado de salud, se examinó la Puntuación del Índice de Utilidad (IUS por sus siglas en inglés) y la escala visual analógica del Cuestionario EuroQoL Five-Dimension-Five Levels (EQ-5D-5L). HAVEN 1 resultados relacionados con la salud: En este estudio, las Puntuaciones Totales basales (media = 41,14 y 44,58 respectivamente y las puntuaciones en la escala de la Salud Física (media = 52,41 y 57,19 respectivamente) fueron similares con profilaxis con Hemlibra y sin profilaxis. En la Tabla 9 se proporciona un resumen de la comparación entre el grupo de profilaxis con Hemlibra (grupo A) y el grupo sin profilaxis (grupo B) en la Puntuación Total en el Haem-A-QoL y en la escala de Salud Física después de 24 semanas de tratamiento ajustando por los valores basales. La profilaxis con Hemlibra semanal reveló una mejora estadística y clínicamente significativa en comparación con la ausencia de profilaxis en las variables pre-especificadas en el Haem-A-QoL y en la escala de Salud Física en la evaluación de la semana 25.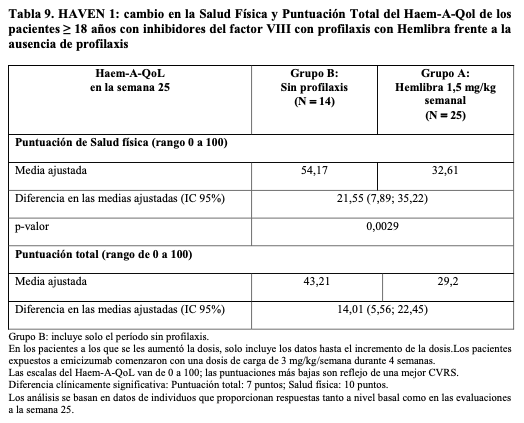
HAVEN 1 resultados en el estado de salud: En la Tabla 10 se proporciona un resumen de la comparación entre el grupo de profilaxis con Hemlibra (grupo A) y el grupo sin profilaxis (grupo B) en la escala del índice de utilidad del EQ- 5D-5L y la escala visual analógica después de 24 semanas de tratamiento ajustando por los valores basales.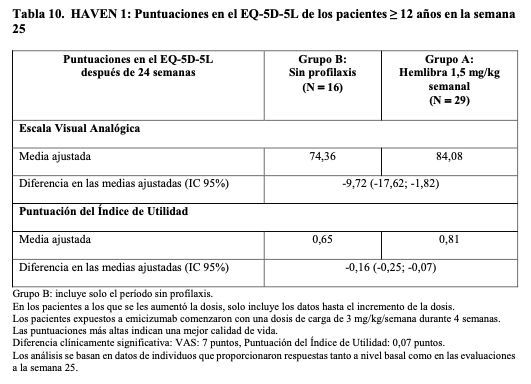
Estudio clínico en pacientes pediátricos: Pacientes pediátricos (edad < 12 años o 12 - 17 años y < 40 kg de peso) con hemofilia A con inhibidores del factor VIII (estudio BH29992 - HAVEN 2): La profilaxis con Hemlibra se evaluó en un estudio clínico abierto y multicéntrico de un solo grupo en pacientes pediátricos (edad < 12 años o 12 a 17 años y < 40 kg de peso) con hemofilia A con inhibidores del factor VIII. Los pacientes recibieron profilaxis con Hemlibra a 3 mg/kg una vez por semana durante las primeras 4 semanas, seguidos de 1,5 mg/kg una vez por semana en adelante. El estudio evaluó la farmacocinética, seguridad y eficacia, incluida la eficacia de la profilaxis con Hemlibra semanal en comparación con el tratamiento episódico y profiláctico previo con agentes bypaseantes en pacientes que habían participado en el NIS antes de la incorporación (comparación intrapaciente). HAVEN 2 resultados pediátricos de eficacia (análisis interino) En el momento del análisis interino, se evaluó la eficacia en 59 pacientes < 12 años y que habían recibido profilaxis semanal con Hemlibra durante al menos 12 semanas, incluidos cuatro pacientes < 2 años, 17 pacientes de 2 a < 6 años y 38 pacientes de 6 a < 12 años. Se calcularon la tasa anual de sangrado y el porcentaje de pacientes con 0 sangrados (véase Tabla 11). La mediana del tiempo de observación para estos pacientes fue de 29,6 semanas (rango: 18,4 a 63,0 semanas).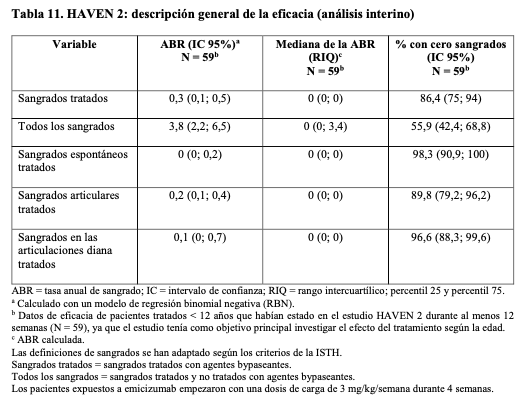
En el análisis intrapaciente, la profilaxis con Hemlibra dio lugar una reducción clínicamente significativa (98%) de la tasa de sangrados tratados en 18 pacientes pediátricos que recibieron al menos 12 semanas de profilaxis con Hemlibra en comparación con su tasa de sangrados recogida en el NIS antes de la incorporación.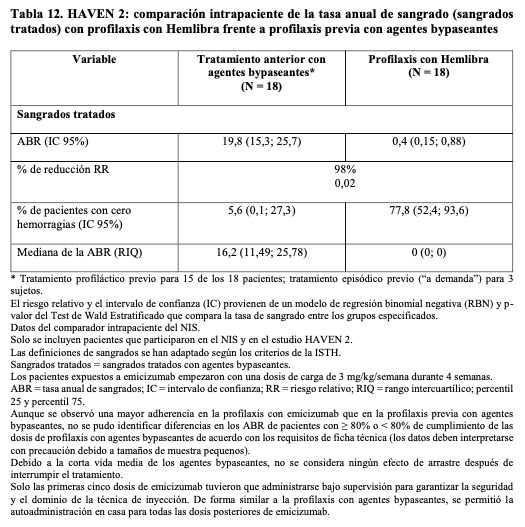
Resultados pediátricos relacionados con la salud: HAVEN 2 resultados relacionados con la salud: En HAVEN 2, se evaluó la HRQoL en la semana 25 en pacientes con edades comprendidas entre ≥ 8 y < 12 años, según el cuestionario Haemo-QoL-SF para niños (véase Tabla 13). El Haemo- QoL-SF es una medida válida y confiable de la HRQoL. También se evaluó la HRQoL de los pacientes < 12 años en la semana 25 según el cuestionario InhibQoL adaptado con Aspectos de la Carga del Cuidador completado por los cuidadores (véase Tabla 13). El InhibQoL adaptado es una medida válida y confiable de la HRQoL.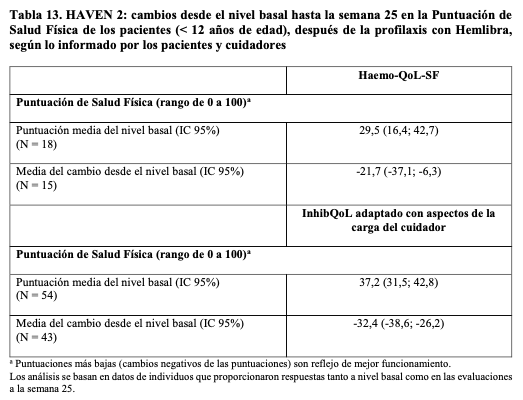
Existe experiencia limitada sobre el uso de agentes bypaseantes o FVIII durante cirugías y procedimientos. El uso de agentes bypaseantes o FVIII durante las cirugías y los procedimientos lo determinó el investigador. En caso de sangrado intercurrente, los pacientes que reciben profilaxis con emicizumab deben tratarse con las terapias disponibles. Para guía sobre los agentes bypaseantes, consultar Precauciones y advertencias. Inmunogenicidad: Como con todas las proteínas terapéuticas, existe la posibilidad de una respuesta inmune en los pacientes tratados con emicizumab. Se sometió a un total de 398 pacientes a pruebas de detección de anticuerpos anti-emicizumab en los ensayos clínicos HAVEN 1-4. Menos del 5% de los pacientes obtuvieron un resultado positivo de anticuerpos anti-emicizumab y < 1% de los pacientes desarrollaron anticuerpos anti-emicizumab con potencial neutralizante (basado en la disminución de farmacocinética). La pérdida de eficacia fue reportada en 1 de 398 pacientes. En el caso de signos clínicos de pérdida de eficacia, debe considerarse un cambio de tratamiento. Pacientes de edad avanzada: El uso de Hemlibra en pacientes de 65 años en adelante con hemofilia A, está respaldado por estudios en adultos y adolescentes HAVEN 1, HAVEN 3 y HAVEN 4. De acuerdo con los datos limitados, no hay evidencia que sugiera una diferencia en eficacia o seguridad en pacientes de 65 años o más. Población pediátrica: La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los estudios realizados con Hemlibra en uno o más grupos de la población pediátrica en el tratamiento del déficit hereditario del factor VIII (véase Posología y formas de administración para consultar la información sobre el uso en la población pediátrica). Propiedades farmacocinéticas: La farmacocinética de emicizumab se determinó mediante un análisis no compartimental en sujetos sanos, utilizando un análisis farmacocinético poblacional de una base de datos compuesta por 389 pacientes con hemofilia A. Absorción: Después de la administración subcutánea en pacientes con hemofilia A, la vida media de absorción fue de 1,6 días. Luego de la administración de múltiples dosis subcutáneas de 3 mg/kg una vez por semana durante las primeras 4 semanas en pacientes con hemofilia A, la media de las concentraciones plasmáticas valle (DE) alcanzó 52,6 ± 13,6 mg/ml en la semana 5. Las medias previstas (DE) de Cvalle, Cmáx y el ratio de Cmáx/Cvalle en equilibrio dinámico para las dosis de mantenimiento recomendadas de 1,5 mg/kg semanal, 3 mg/kg cada dos semanas o 6 mg/kg cada cuatro semanas, se muestran en la Tabla 14.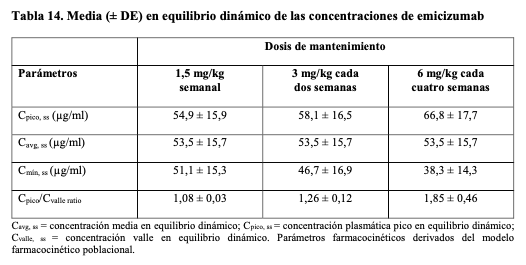
Se observaron perfiles farmacocinéticos similares después de la administración semanal (3 mg/kg/semana durante 4 semanas seguido de 1,5 mg/kg/semana) en adultos/adolescentes (≥ 12 años) y niños ( < 12 años) (véase Figura 1).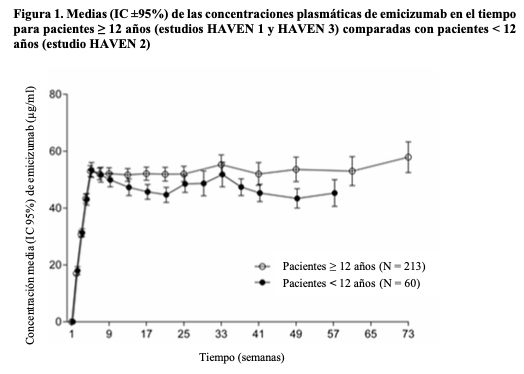
En los sujetos sanos, la biodisponibilidad absoluta después de la administración subcutánea de 1 mg/kg se situó entre el 80,4% y el 93,1%, según el lugar de inyección. Los perfiles farmacocinéticos observados después de la administración subcutánea en el abdomen, la parte superior del brazo y el muslo fueron parecidos. Emicizumab puede administrarse indistintamente en estos puntos anatómicos (véase Posología y formas de administración). Distribución: Después de una dosis intravenosa única de 0,25 mg/kg de emicizumab en sujetos sanos, el volumen de distribución en equilibrio dinámico fue de 106 ml/kg (es decir, 7,4 litros para un adulto de 70 kg). El volumen de distribución aparente (V/F), estimado a partir del análisis farmacocinético poblacional, en pacientes con hemofilia A después de múltiples dosis subcutáneas de emicizumab fue de10,4 litros. Biotransformación: No se ha estudiado el metabolismo de emicizumab. Los anticuerpos de IgG son catabolizados principalmente mediante proteólisis lisosómica y luego eliminados o reutilizados por el organismo. Eliminación: Después de la administración intravenosa de 0,25 mg/kg en sujetos sanos, el clearance total de emicizumab fue de 3,26 ml/kg/día (es decir, 0,228 litro/día para un adulto de 70 kg) y la vida media terminal promedio fue de 26,7 días. Luego de una única inyección subcutánea en sujetos sanos, la vida media de eliminación fue de aproximadamente 4 a 5 semanas. Después de múltiples inyecciones subcutáneas en pacientes con hemofilia A, el clearance aparente fue de 0,272 litro/día y la vida media de eliminación aparente de 26,8 días. Linealidad de la dosis Emicizumab mostró una farmacocinética proporcional a la dosis en pacientes con hemofilia A después de la primera dosis de Hemlibra en el intervalo de dosis de entre 0,3 y 6 mg/kg. La exposición (Cavg, ss) de dosis múltiples, es comparable entre 1,5 mg/kg cada 3 semanas, 3 mg/kg cada 2 semanas y 6 mg/kg cada 4 semanas. Poblaciones especiales: Pacientes pediátricos: El efecto de la edad en la farmacocinética de emicizumab se evaluó en un análisis farmacocinético poblacional que incluyó 5 lactantes (≥ 1 mes a < 2 años), 55 niños (menores de 12 años) y 50 adolescentes (de 12 a < 18 años) con hemofilia A. La edad no afectó a la farmacocinética de emicizumab en los pacientes pediátricos. Pacientes de edad avanzada: El efecto de la edad en la farmacocinética de emicizumab se evaluó en un análisis farmacocinético de población que incluyó a trece sujetos de 65 años o más (ninguno de los sujetos tenía más de 77 años). La biodisponibilidad relativa disminuía con la edad, pero no se observaron diferencias clínicamente relevantes en la farmacocinética de emicizumab entre los sujetos < 65 años y los sujetos ≥ 65 años. Etnia El análisis de farmacocinética poblacional en pacientes con hemofilia A demostraron que la etnia no afectaba a la farmacocinética de emicizumab. No es necesario ajustar la dosis para este factor demográfico. Insuficiencia renal: No se han llevado a cabo estudios especiales del efecto de la insuficiencia renal en la farmacocinética de emicizumab. En el análisis farmacocinético poblacional, la mayoría de los pacientes con hemofilia A tenían función renal normal (N = 332, clearance de creatinina [ACr] ≥ 90 ml/min) o insuficiencia renal leve (N = 27; ACr de 60 - 89 ml/min). La insuficiencia renal leve no afectó a la farmacocinética de emicizumab. Existen datos limitados disponibles sobre el uso de Hemlibra en pacientes con insuficiencia renal moderada (solo 2 pacientes con ACr de 30 - 59 ml/min) y ningún dato disponible en pacientes con insuficiencia renal grave. No se puede inferir el impacto de la insuficiencia renal moderada y grave en la farmacocinética de emicizumab. Emicizumab es un anticuerpo monoclonal y se elimina mediante catabolismo y no excreción renal, por lo que no se prevé que sea necesario modificar la dosis en los pacientes con insuficiencia renal. Insuficiencia hepática: No se han llevado a cabo estudios especiales del efecto de la insuficiencia hepática en la farmacocinética de emicizumab. La mayoría de los pacientes con hemofilia A en el análisis farmacocinético poblacional tenía función hepática normal (bilirrubina y AST ≤ LSN, N = 300) o insuficiencia hepática leve (bilirrubina ≤ LSN y AST > LSN o bilirrubina desde 1,0 a 1,5 × LSN y cualquier AST, N = 51). Solo 6 pacientes tenían insuficiencia hepática moderada (1,5 × LSN < bilirrubina ≤ 3 × LSN y cualquier AST). La insuficiencia hepática leve no afectó la farmacocinética de emicizumab (véase Posología y formas de administración). La seguridad y eficacia de emicizumab no se han estudiado específicamente en pacientes con insuficiencia hepática. En los ensayos clínicos se incluyó a pacientes con insuficiencia hepática leve y moderada. No se dispone de datos sobre el uso de Hemlibra en pacientes con insuficiencia hepática grave. Emicizumab es un anticuerpo monoclonal y se elimina mediante catabolismo y no a través del metabolismo hepático, por lo que no se prevé que sea necesario modificar la dosis en los pacientes con insuficiencia hepática. Otras poblaciones especiales: El modelado muestra que, en pacientes con hipoalbuminemia y bajo peso corporal para su edad, la dosificación menos frecuente da lugar a una menor exposición al emicizumab; las similaciones indican que estos pacientes todavía se beneficiarían de un control de sangrado clínicamente significativo. Ningún paciente con tales características fue incluido en los ensayos clínicos. Datos preclínicos sobre seguridad: Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios de toxicidad aguda y a dosis repetidas incluyendo variables de farmacología de seguridad y variables de la toxicidad reproductiva. Fertilidad: Emicizumab no provocó alteraciones en los órganos reproductivos de monos cynomolgus machos o hembras hasta la dosis más alta estudiada de 30 mg/kg/semana (equivalente a 11 veces la exposición humana a la dosis más alta de 3 mg/kg/semana, según el ABC). Teratogenicidad: No se dispone de datos sobre las posibles reacciones adversas de emicizumab en el desarrollo embriofetal. Reacciones en el lugar de inyección: Se ha observado hemorragia reversible, infiltración perivascular de células mononucleares, degeneración/necrosis de la hipodermis e hinchazón del endolelio en la hipodermis en animales después de una inyección subcutánea.
Indicaciones.
Hemlibra está indicado para la profilaxis de rutina de los episodios de sangrado en pacientes con: hemofilia A (deficiencia congénita del factor VIII) con inhibidores del factor VIII, hemofilia A severa (deficiencia congénita del factor VIII, FVIII < 1%) sin inhibidores del factor VIII. Hemlibra puede ser usado en todos los grupos etarios.
Dosificación.
General: El reemplazo por cualquier otro agente biológico requiere el consentimiento del médico prescriptor. El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y/o trastornos hemorrágicos. Posología: El tratamiento (incluida la profilaxis de rutina) con agentes bypaseantes (por ejemplo, CCPa y rFVIIa) debe suspenderse el día anterior al comienzo de la terapia con Hemlibra (véase Precauciones y advertencias). La profilaxis con el factor VIII (FVIII) puede prolongarse durante los 7 primeros días de tratamiento con Hemlibra. La dosis recomendada es de 3 mg/kg una vez por semana durante las primeras 4 semanas (dosis de carga), seguida de una dosis de mantenimiento de 1,5 mg/kg una vez por semana 3 mg/kg cada dos semanas, o 6 mg/kg cada cuatro semanas, todas las dosis administradas en forma de inyección subcutánea. El régimen de dosis de carga es el mismo, independientemente del régimen de dosis de mantenimiento. El régimen de dosis de mantenimiento debe seleccionarse basándose en la preferencia del médico y el paciente/cuidador para su cumplimiento. La dosis del paciente (en mg) y el volumen (en ml) deben calcularse de la siguiente manera: Dosis de carga (3 mg/kg) una vez por semana durante las 4 primeras semanas: Peso corporal del paciente (kg) x dosis (3 mg/kg) = cantidad total (mg) de emicizumab que debe administrarse. Seguida de una dosis de mantenimiento de 1,5 mg/kg una vez por semana, 3 mg/kg cada dos semanas o 6 mg/kg cada cuatro semanas, a partir de la semana 5: Peso corporal del paciente (kg) x dosis (1,5; 3 o 6 mg/kg) = cantidad total (mg) de emicizumab que debe administrarse. El volumen total de Hemlibra que debe administrarse por vía subcutánea se calcula de la manera siguiente: Cantidad total (mg) de emicizumab que debe administrarse ÷ concentración del vial (mg/ml) = volumen total de Hemlibra (ml) que debe inyectarse. No se deben combinar concentraciones diferentes de Hemlibra (30 mg/ml y 150 mg/ml) en la misma jeringa para llegar al volumen total a administrar. No se debe administrar un volumen superior a 2 ml por inyección. Ejemplos: Paciente con peso corporal de 16 kg, con un régimen de dosis de mantenimiento de 1,5 mg/kg una vez a la semana: Ejemplo de dosis de carga (4 primeras semanas): se necesitan 16 kg x 3 mg/kg = 48 mg de emicizumab para la dosis de carga una vez a la semana. Para calcular el volumen que debe administrarse, divida la dosis calculada de 48 mg por 150 mg/ml: se deben inyectar 48 mg de emicizumab ÷ 150 mg/ml = 0,32 ml de Hemlibra a una concentración de 150 mg/ml. Escoja la posología apropiada y el volumen de las concentraciones de los viales disponibles. Ejemplo de dosis de mantenimiento (a partir de la semana 5): se necesitan 16 kg x 1,5 mg/kg = 24 mg de emicizumab para la dosis de mantenimiento. Para calcular el volumen que debe administrarse, divida la dosis calculada de 24 mg por 30 mg/ml: se deben inyectar 24 mg de emicizumab ÷ 30 mg/ml = 0,8 ml de Hemlibra a una concentración de 30 mg/ml una vez a la semana. Escoja la posología apropiada y el volumen de las concentraciones de los viales disponibles. Paciente con un peso corporal de 40 kg, con un régimen de dosis de mantenimiento de 3 mg/kg cada dos semanas: Ejemplo de dosis de carga (4 primeras semanas): se necesitan 40 kg x 3 mg/kg = 120 mg de emicizumab para la dosis de carga una vez a la semana. Para calcular el volumen que debe administrarse, divida la dosis calculada de 120 mg por 150 mg/ml: se deben inyectar 120 mg de emicizumab ÷ 150 mg/ml = 0,8 ml de Hemlibra a una concentración de 150 mg/ml. Escoja la posología apropiada y el volumen de las concentraciones de los viales disponibles. Ejemplo de dosis de mantenimiento (a partir de la semana 5): se necesitan 40 kg x 3 mg/kg = 120 mg de emicizumab para la dosis de mantenimiento. Para calcular el volumen que debe administrarse, divida la dosis calculada de 120 mg por 150 mg/ml: se deben inyectar 120 mg de emicizumab ÷ 150 mg/ml = 0,8 ml de Hemlibra a una concentración de 150 mg/ml cada dos semanas. Escoja la posología apropiada y el volumen de las concentraciones de los viales disponibles. Paciente con un peso corporal de 60 kg, con un régimen de dosis de mantenimiento de 6 mg/kg cada cuatro semanas: Ejemplo de dosis de carga (4 primeras semanas): se necesitan 60 kg x 3 mg/kg = 180 mg de emicizumab para la dosis de carga una vez a la semana. Para calcular el volumen que debe administrarse, divida la dosis calculada de 180 mg por 150 mg/ml: se deben inyectar 180 mg de emicizumab ÷ 150 mg/ml = 1,20 ml de Hemlibra a una concentración de 150 mg/ml. Escoja la posología apropiada y el volumen de las concentraciones de los viales disponibles. Ejemplo de dosis de mantenimiento (a partir de la semana 5): se necesitan 60 kg x 6 mg/kg = 360 mg de emicizumab para la dosis de mantenimiento. Para calcular el volumen que debe administrarse, divida la dosis calculada de 360 mg por 150 mg/ml: se deben inyectar 360 mg de emicizumab ÷ 150 mg/ml = 2,4 ml de Hemlibra a una concentración de 150 mg/ml cada cuatro semanas. Escoja la posología apropiada y el volumen de las concentraciones de los viales disponibles. Duración del tratamiento: Hemlibra está destinado para el tratamiento profiláctico a largo plazo. Ajustes de dosis durante el tratamiento: No se recomiendan ajustes de dosis durante el tratamiento con Hemlibra. Retraso u omisión de dosis: Si un paciente olvida una inyección subcutánea de Hemlibra, se debe indicar al paciente que deberá administrarse la dosis olvidada tan pronto como sea posible, hasta un día antes del día de la siguiente dosis programada. A continuación, el paciente debe recibir la dosis siguiente el día previsto habitual. El paciente no debe recibir dos dosis el mismo día para compensar la dosis olvidada. Poblaciones especiales: Pacientes pediátricos: No se recomiendan ajustes de dosis en pacientes pediátricos (véase Características farmacológicas - Propiedades, Propiedades farmacocinéticas). No se dispone de datos en pacientes menores de 1 año de edad. Pacientes de edad avanzada: No se recomiendan ajustes de dosis en pacientes ≥ 65 años de edad (véanse Características farmacológicas - Propiedades, Propiedades farmacodinámicas y Propiedades farmacocinéticas). No se dispone de datos en pacientes mayores de 77 años de edad. Pacientes con insuficiencia renal y hepática No se recomienda ajustar la dosis en pacientes con insuficiencia renal o hepática leve (véase Características farmacológicas - Propiedades, Propiedades farmacocinéticas). Los datos disponibles sobre el uso de Hemlibra en pacientes con insuficiencia renal o hepática moderada son limitados. No se ha estudiado emicizumab en pacientes con insuficiencia renal o hepática grave. Manejo en el marco perioperatorio La seguridad y eficacia de emicizumab no se ha evaluado formalmente en el marco quirúrgico. En los ensayos clínicos, algunos pacientes han tenido cirugías sin interrumpir profilaxis con emicizumab. Si se necesitan agentes bypaseantes (por ejemplo, CCPa y rFVIIa) en el período perioperatorio, consulte la guía de administración sobre el uso de agentes bypaseantes en Precauciones y advertencias. Si se requiere FVIII en el marco perioperatorio, consultar Interacciones. Cuando se monitorice la actividad hemostática subyacente de un paciente, consultar Precauciones y advertencias sobre pruebas de laboratorio no afectadas por emicizumab. Inducción de inmunotolerancia (ITI, por sus siglas en inglés). Todavía no se ha determinado la seguridad y eficacia de emicizumab en los pacientes que están recibiendo inducción de inmunotolerancia. No se dispone de datos. Formas de administración: Hemlibra es para administración por vía subcutánea exclusivamente y debe administrarse utilizando una técnica aséptica apropiada (véase Observaciones particulares). La inyección debe limitarse a los lugares de inyección recomendados: el abdomen, el área superior externa de los brazos y los muslos (véase Características farmacológicas - Propiedades, Propiedades farmacocinéticas). La administración de la inyección subcutánea de Hemlibra en el área superior externa del brazo debe ser realizada por un cuidador o profesional de la salud. Alternar el sitio de la inyección puede ayudar a prevenir o reducir las reacciones en el lugar de la inyección (véase Reacciones adversas). La inyección subcutánea de Hemlibra no se debe administrar en lunares, cicatrices o áreas donde la piel esté sensible, con hematoma, enrojecida, dura o no intacta. Durante el tratamiento con Hemlibra es preferible que otros medicamentos de administración subcutánea se inyecten en lugares anatómicos diferentes. Administración por el paciente y/o el cuidador Un profesional de la salud guiará al paciente y/o al cuidador sobre la administración de Hemlibra. Después de un entrenamiento adecuado en técnicas de inyección subcutánea, el paciente podrá autoinyectarse o bien su cuidador podrá administrarle Hemlibra, si su médico determina que es apropiado. El médico y el cuidador deben determinar la idoneidad del niño que se autoinyecta Hemlibra. Sin embargo, la autoadministración no se recomienda para niños menores de 7 años de edad. Para obtener instrucciones completas sobre la administración de Hemlibra, consulte Información para el Paciente e Instrucciones de uso.
Contraindicaciones.
Hemlibra está contraindicado en pacientes con hipersensibilidad conocida a emicizumab o a cualquiera de los excipientes.
Reacciones adversas.
Resumen del perfil de seguridad: Las reacciones adversas al medicamento (RAMs) más graves informadas en los ensayos clínicos con Hemlibra fueron microangiopatía trombótica (MAT) y eventos trombóticos, incluyendo trombosis del seno cavernoso (TSC) y trombosis venosa superficial concomitantemente con necrosis cutánea (véanse Precauciones y advertencias y la información detallada a continuación). Las RAMs más frecuentes notificadas en ≥ 10% de los pacientes tratados con al menos una dosis de Hemlibra fueron: reacciones en el lugar de inyección (20%), artralgia (15%) y cefalea (14%). En total, tres pacientes (0,8%) de los ensayos clínicos que recibían profilaxis con Hemlibra suspendieron el tratamiento debido a las RAMs, las cuales fueron MAT, necrosis cutánea simultánea con tromboflebitis superficial y cefalea. Tabla de reacciones adversas: Las siguientes RAMs se basan en los datos agrupados de cuatro ensayos clínicos de fase III (estudios en adultos y adolescentes [BH29884 - HAVEN 1, BH30071 - HAVEN 3, y BO39182 - HAVEN 4] y un estudio pediátrico [BH29992 - HAVEN 2]), en los cuales un total de 373 pacientes de sexo masculino con hemofilia A recibieron por lo menos una dosis de Hemlibra como profilaxis de rutina. Doscientos setenta y seis (71%) eran adultos, 47 (13%) eran adolescentes (≥ 12 a < 18 años), 55 (15%) eran niños (≥ 2 a < 12 años) y cinco (1%) eran lactantes y niños pequeños (de 1 mes a < 2 años). La mediana de la duración de la exposición en los estudios fue de 33 semanas (intervalo: de 0,1 a 94,3semanas). Las RAMs de los ensayos clínicos fase III en pacientes que recibieron Hemlibra se enumeran dentro de la clasificación de órganos del sistema MedDRA en la Tabla 2 Las categorías de frecuencia correspondientes para cada RAM se basan en la siguiente definición: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1.000 a < 1/100), raras (≥ 1/10.000 a < 1/1.000), muy raras ( < 1/10.000) y no conocida (no puede estimarse a partir de los datos disponibles).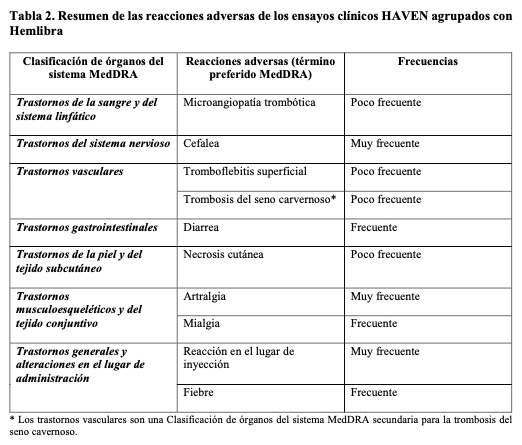
Descripción de algunas reacciones adversas: Microangiopatía trombótica (MAT): En los ensayos clínicos agrupados fase III, se reportaron eventos de MAT en menos del 1% de los pacientes (3/373) y en el 9,7% de los pacientes (3/31) que recibieron por lo menos una dosis de CCPa mientras estaban tratados con emicizumab. Las 3 MAT se dieron cuando se administró una cantidad acumulada media > 100 U/kg/24 horas de CCPa durante 24 horas o más durante un evento de tratamiento (véase Precauciones y advertencias). Los pacientes presentaron trombocitopenia, anemia hemolítica microangiopática y lesión renal aguda, sin deficiencias graves en la actividad de ADAMTS13. Un paciente reanudó el tratamiento con Hemlibra después de la resolución de la MAT sin recurrencia. Eventos trombóticos: En los ensayos clínicos agrupados fase III, se informaron eventos trombóticos graves en menos del 1% de los pacientes (2/373) y en el 6,5% de los pacientes (2/31) que recibieron por lo menos una dosis de CCPa mientras estaban tratados con emicizumab. Ambos episodios trombóticos graves se dieron cuando se administró una cantidad acumulada media > 100 U/kg/24 ho