GLIVEC®
NOVARTIS
Inhibidor de la proteína tirosina kinasa.
Composición.
Cada comprimido recubierto de 100 mg contiene: Imatinib (como mesilato) 100 mg. Excipientes: celulosa microcristalina, crospovidona, hipromelosa, estearato de magnesio, sílice coloidal anhidra, óxido de hierro rojo, óxido de hierro amarillo c. s. Cada comprimido recubierto de 400 mg contiene: Imatinib (como mesilato) 400 mg. Excipientes: celulosa microcristalina, crospovidona, hipromelosa, estearato de magnesio, sílice coloidal anhidra, óxido de hierro rojo, óxido de hierro amarillo c. s.
Farmacología.
Modo de acción: El imatinib es una pequeña molécula inhibidora de proteína tirosina-quinasas que inhibe con gran potencia la Bcr-Abl tirosina-cinasa, así como varias tirosina-quinasas de receptores: Kit, el receptor del factor de células precursoras (SCF) codificado por el protooncogén c-Kit, los receptores del dominio de discoidina (DDR1 y DDR2), el receptor del factor estimulante de colonias (CSF-1R) y los receptores a y b de factor de crecimiento derivado de plaquetas (PDGFR-a y PDGFR-b). El imatinib también puede inhibir procesos celulares mediados por la activación de estas cinasas de receptores. Acción farmacológica: Imatinib es un inhibidor de la proteína tirosina kinasa, que inhibe de forma potente la tirosina-quinasa Bcr-Abl in vitro, a nivel celular e in vivo. El compuesto inhibe selectivamente la proliferación e induce la apoptosis de líneas celulares Bcr-Abl positivas y de células leucémicas frescas de pacientes con LMC o leucemia linfoblástica aguda (LLA) cromosoma Filadelfia positivo. En ensayos de cultivo de colonias utilizando sangre periférica ex vivo y muestras de médula ósea, imatinib muestra una inhibición selectiva en las colonias Bcr-Abl positivas de pacientes con LMC. In vivo el compuesto muestra tener actividad antitumoral como agente único en modelos animales, utilizando células tumorales Bcr-Abl positivas. Además, imatinib es un potente inhibidor del receptor tirosina-quinasa para el factor de crecimiento derivado de plaquetas (PDGF) y factor de células stem (SCF), c-Kit, e inhibe los procesos celulares mediados por el PDGF y SCF. In vitro, imatinib inhibe la proliferación e induce la apoptosis de células neoplásicas de estroma gastrointestinal (TEGI/GIST), que expresa una mutación activadora de kit. Se ha propuesto que en la patogenia de SMD/TMP, SHE/LEC y DFSP podrían estar implicadas la activación constitutiva del PDGFR o de las tirosina-quinasas Abl a raíz de una fusión con diversas proteínas o de la producción constitutiva de PDGF. Por otro lado, la activación constitutiva de c-Kit o PDGFR podría estar implicada en la patogenia de la MS. Imatinib inhibe la señalización y la proliferación de células impulsada por la actividad desregulada de las cinasas PDGFR, Kit y Abl. Ensayos clínicos: Leucemia mieloide crónica: La efectividad de Glivec® está basada en las tasas de respuesta hematológica y citogenética y en la sobrevida sin progresión. Se llevaron a cabo tres ensayos internacionales grandes de Fase II, abiertos sin rama paralela, en pacientes con leucemia mieloide crónica (LMC) cromosoma Filadelfia positivo (Ph+), en fases avanzadas de la enfermedad (fase acelerada o crisis blástica), otras leucemias Ph+, o con LMC en fase crónica, en los que había fallado el tratamiento con interferón-a- (IFN). Se llevó a cabo un ensayo internacional a gran escala, multicéntrico, abierto, aleatorizado de Fase III, en pacientes con LMC Ph+ recientemente diagnosticada. Además, el tratamiento se administró a niños en dos estudios de Fase I y en un estudio de Fase II, multicéntrico, abierto y con un solo grupo de tratamiento. En todos los ensayos clínicos, el 38-40% de los pacientes tenían ≥60 años y el 10-12% de los pacientes tenía ≥70 años. Fase crónica recientemente diagnosticada: Este estudio de Fase III comparó el tratamiento de Glivec® como agente único con una combinación de interferón-a (IFN) más citarabina (Ara-C). Se permitió a los pacientes cambiar de rama si no lograban remisión hematológica completa a los 6 meses, o remisión citogenética significativa a los 24 meses, si perdían la remisión citogenética alcanzada, si aumentaba el recuento leucocitario, o si presentaban severa intolerancia al tratamiento. En la rama de Glivec®, los pacientes fueron tratados con 400 mg diarios. En la rama de IFN, los pacientes fueron tratados con una dosis de IFN de 5 MUI/m2/día vía subcutánea en combinación con Ara-C 20 mg /m2/día durante 10 días al mes. Se aleatorizaron un total de 1106 pacientes en 177 centros de 16 países, correspondiendo 553 pacientes a cada rama. Las características basales estuvieron bien balanceadas entre las dos ramas. La mediana de edad fue 51 años (rango 18-70 años), con 21,9% de pacientes con ≥60 años de edad. Participaron 59% de hombres y 41% de mujeres; el 89,9% era de raza blanca y el 4,7% eran pacientes de raza negra. Al cierre de este análisis (7 años después de la admisión del último paciente), la mediana de duración del tratamiento de primera línea era de 82 meses en el grupo de Glivec® y de 8 meses en el grupo del IFN. La mediana de duración del tratamiento de segunda línea con Glivec® era de 64 meses, y el 60% de los pacientes asignados al tratamiento con Glivec® siguen recibiendo el tratamiento de primera línea. En estos pacientes, la dosis promedio de Glivec® fue de 403 ± 57 mg. En total, en los pacientes que recibieron el tratamiento de primera línea con Glivec®, la dosis diaria promedio fue de 406 ± 76 mg. A consecuencia del mayor porcentaje de interrupciones del tratamiento y de cambios de grupo terapéutico, solamente el 2% de los pacientes asignados al grupo de IFN continúan recibiendo el tratamiento de primera línea. En el grupo del IFN, el motivo más frecuente de interrupción del tratamiento de primera línea fue el retiro del consentimiento (14%) y la razón más frecuente de cambio al grupo terapéutico de Glivec® fue una grave intolerancia al tratamiento (26%) y la progresión del cáncer (14%). El criterio principal de eficacia primaria del estudio es la sobrevida libre de progresión. La progresión fue definida como cualquiera de los siguientes eventos: progresión a fase acelerada o crisis blástica, muerte, pérdida de RHC o RCM o un aumento del recuento de glóbulos blancos (WBC) en pacientes que no alcanzan una RHC a pesar del manejo terapéutico apropiado. La respuesta citogenética mayor, la respuesta hematológica, la respuesta molecular (evaluación de la enfermedad residual mínima), el tiempo a la fase acelerada o crisis blástica y la sobrevida constituyen puntos finales secundarios mayores.
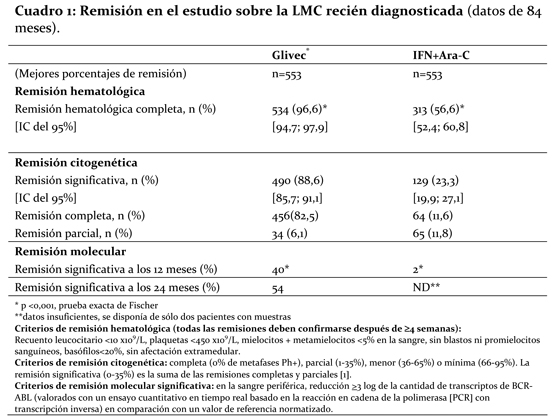
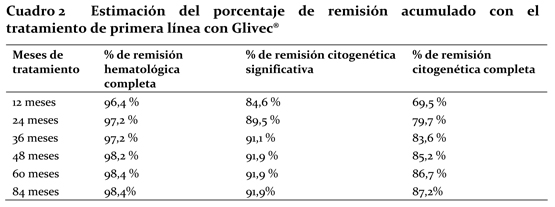
Los porcentajes de remisión hematológica completa, remisión citogenética significativa y remisión citogenética completa con el tratamiento de primera línea se estimaron por el método de Kaplan-Meier, suprimiendo del análisis los datos de los pacientes que no habían conseguido una remisión en la fecha del último examen. Así, este método arrojó los porcentajes acumulados de remisión con el tratamiento de primera línea con Glivec® que figuran en el Cuadro 2.
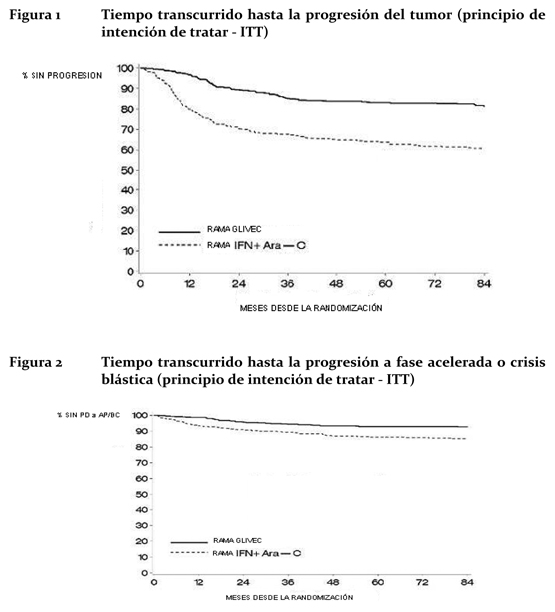
Para analizar los resultados a largo plazo, los pacientes asignados al tratamiento con Glivec® se compararon con los que recibieron IFN. Los datos de los pacientes que cambiaron de grupo terapéutico antes de la progresión no fueron suprimidos del análisis en el momento del cambio, y los acontecimientos sufridos por estos pacientes después del cambio se atribuyeron al tratamiento asignado originalmente. Durante el período de observación de 7 años se registraron 93 (16,8%) progresiones en el grupo de Glivec®: 37 (6,7%) que implicaron una progresión a fase acelerada o crisis blástica, 31 (5,6%) pérdida de remisión citogenética significativa, 15 (2,7%) pérdida de remisión hematológica completa o aumento del recuento leucocitario y 10 (1,8%) decesos sin relación con la LMC. En cambio, hubo 165 (29,8%) progresiones en el grupo tratado con la combinación de IFN+Ara-C, de los cuales 130 ocurrieron durante el tratamiento de primera línea con esta combinación. El porcentaje estimado de sobrevida libre de progresión a los 84 meses es del 81,2% con un IC del 95% (78; 85) en el grupo de Glivec® y del 60,6% (56; 5) en el grupo control (p < 0,001) (Figura 1). Los porcentajes anuales de progresión con Glivec® fueron del 3,3% en el primer año después del inicio del estudio, del 7,5% en el segundo año, y del 4,8%; 1,7%; 0,8%; 0,3% y 2,0% en los tercero, cuarto, quinto, sexto y séptimo años del estudio, respectivamente. El porcentaje estimado de pacientes sin progresión a fase acelerada o crisis blástica después de 84 meses fue significativamente mayor en el grupo de Glivec® que en el del IFN [del 92,5% (90, 95) frente al 85,1%, (82, 89) p < 0,001] (Figura 2). El porcentaje anual de progresión disminuyó al prolongarse el tratamiento: los porcentajes anuales de progresión a fase acelerada o crisis blástica fueron del 1,5%, 2,8%, 1,6%, 0,9%, 0,5%, 0% y 0,4% del primero al séptimo año, respectivamente.
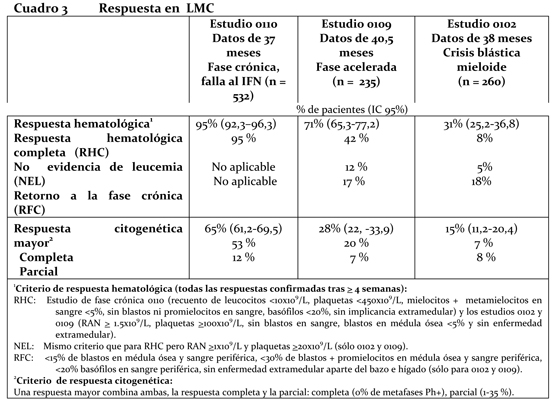
En total murieron 71 (12,8%) y 85 (15,4%) pacientes en los grupos tratados con Glivec® e IFN+Ara-C, respectivamente. A los 84 meses, la sobrevida global estimada es del 86,4% (83, 90) frente al 83,3% (80, 87) en los grupos aleatorizados a Glivec® e IFN+Ara-C, respectivamente (p=0,073, prueba log-posicional). El alto porcentaje de cambio de grupo terapéutico de IFN+Ara-C a Glivec® influyó considerablemente en el tiempo transcurrido hasta el evento. Además, el número de pacientes que recibió un trasplante de médula ósea después de suspender el tratamiento estudiado fue mayor en el grupo IFN+Ara-C (n=66, 38 después de cambiar a Glivec®) que en el grupo de Glivec® (n=50, 8 después de cambiar al IFN) en la actualización de 84 meses. Al suprimir del análisis las 48 muertes que ocurrieron después del trasplante de médula ósea, la tasa de sobrevida estimada a 84 meses fueron de 89,6% vs 88,1% (p=0,200 en el log rank test). En el grupo de Glivec®, sólo se atribuyeron a la LMC 31 muertes (5,6%) (anteriores al trasplante), contra 40 (7,2%) en el grupo de IFN+Ara-C. Al considerar únicamente estas muertes relacionadas con la LMC y al suprimir del análisis las muertes posteriores al trasplante de médula ósea o por algún otro motivo, la tasa de sobrevida estimada a 84 meses fueron del 93,6% frente al 91,1% (p=0,1, en el log rank test). En la LMC recién diagnosticada en fase crónica, el efecto del tratamiento con Glivec® en la sobrevida se examinó más a fondo mediante un análisis retrospectivo de los datos anteriores obtenidos con Glivec® y los datos básicos de otro estudio de Fase III que utilizó un régimen idéntico de IFN+Ara-C (n=325). En esta publicación se demostró la superioridad de Glivec® sobre IFN+Ara-C en cuanto a sus efectos respectivos en la sobrevida global (p < 0,001); después de 42 meses habían muerto 47 (8,5%) pacientes tratados con Glivec® contra 63 (19,4%) tratados con la combinación de IFN+Ara-C. El grado de remisión citogenética influyó claramente en los resultados a largo plazo en los pacientes tratados con Glivec®. Mientras que un 96% de los pacientes con remisión citogenética completa (93% con remisión citogenética parcial) a 12 meses no presentaba progresión a fase acelerada o crisis blástica a 84 meses, sólo el 81% de los pacientes sin remisión citogenética significativa a 12 meses no presentaba signos de progresión a LMC avanzada a los 84 meses (valor global: p < 0,001, p=0,25 entre remisión citogenética completa y parcial). Basándose en la referencia de 18 meses, las estimaciones fueron del 99%; 90% y 83% respectivamente, incluyendo ahora una diferencia estadísticamente significativa entre remisión citogenética completa y parcial (p < 0,001). La evaluación de respuesta molecular proporcionó información adicional importante para el pronóstico. En los pacientes con remisión citogenética completa y una reducción ≥3 log de los transcriptos Bcr-Abl a 12 meses, la probabilidad de mantenerse sin progresión a los 60 meses fue numéricamente mayor que en los pacientes con una remisión citogenética completa pero con una reducción < 3 log (95% frente al 89%, p=0,068), y significativamente mayor que la observada en pacientes sin remisión citogenética completa a 12 meses (70%; p < 0,001). Considerando únicamente la progresión a fase acelerada o crisis blástica, los porcentajes estimados de pacientes sin eventos fueron del 100%, 95% y 88% respectivamente (valor global: p < 0,001, p=0,007 entre la remisión citogenética completa con y sin remisión molecular significativa). Utilizando la referencia de 18 meses, los porcentajes estimados de pacientes que no habían evolucionado a fase acelerada o crisis blástica a 60 meses fueron del 100% en los pacientes con remisión citogenética completa y remisión molecular significativa, del 98% en los pacientes con remisión citogenética completa pero sin remisión molecular significativa y de sólo el 87% en los pacientes sin remisión citogenética completa (valor global: p < 0,001; p=0,105 entre la remisión citogenética completa con y sin remisión molecular significativa). En este estudio se permitió aumentar la dosis diaria de Glivec® de 400 mg a 600 mg, y luego de 600 mg a 800 mg. Al cabo de 42 meses de seguimiento, 11 pacientes que habían mostrado una remisión hematológica completa a los 3 meses y una remisión citogenética significativa a los 12 meses, mientras recibían dosis diarias de 400 mg, presentaron una pérdida confirmada (en un plazo de 4 semanas) de la remisión citogenética. Cuando se aumentó la dosis a 800 mg diarios en cuatro de estos once pacientes, dos volvieron a dar signos de remisión citogenética (uno parcial y el otro completa, en este último caso junto con remisión molecular). En los siete pacientes restantes no se aumentó la dosis y sólo uno de ellos volvió a conseguir una remisión citogenética completa. Los 40 pacientes en quienes se aumentó la dosis diaria a 800 mg presentaron algunas reacciones adversas con mayor frecuencia que la población total antes del aumento (n=551); estas reacciones consistieron en hemorragias gastrointestinales, conjuntivitis y elevaciones de las transaminasas o de la bilirrubina. Las otras reacciones adversas se manifestaron con igual o menor frecuencia. La calidad de vida se evaluó utilizando la escala validada FACT-BRM (Functional Assessment of Cancer Therapy - Biologic Response Modifiers). Se evaluaron todas las esferas y se obtuvieron puntuaciones significativamente mayores en el grupo de Glivec® que en el de IFN. Los datos de calidad de vida indicaron que los pacientes experimentaban un constante bienestar mientras recibían Glivec®. Fase crónica, falla al Interferón: 532 pacientes fueron tratados con una dosis inicial de 400 mg. Los pacientes fueron distribuidos en tres categorías principales: falla hematológica (29%), falla citogenética (35%) o intolerancia al interferón (36%). Los pacientes habían recibido tratamiento previo con IFN durante una mediana de 14 meses a una dosis de ≥25x106 U.I. / semana y todos estaban en una fase crónica tardía, con una mediana de tiempo desde el diagnóstico de 32 meses. La variable principal de eficacia del estudio fue la tasa de respuesta citogenética mayor (respuesta completa más parcial, 0-35% de metafases Ph+ en médula ósea). En este estudio, el 65% de los pacientes alcanzaron una respuesta citogenética mayor que fue completa en el 53% de los pacientes (Cuadro 2). En el 95% de los pacientes se consiguió una respuesta hematológica completa. Fase acelerada: Se incluyeron 235 pacientes con la enfermedad en fase acelerada. Los primeros 77 pacientes iniciaron el tratamiento con 400 mg, se modificó posteriormente el protocolo para permitir una dosis más alta y los restantes 158 pacientes iniciaron el tratamiento con 600 mg. La variable principal de eficacia, fue la tasa de respuesta hematológica, informada ya sea como respuesta hematológica completa, como no evidencia de leucemia (es decir, desaparición de blastos de médula ósea y sangre, pero sin una recuperación total de sangre periférica como en una respuesta completa) o como retorno a la fase crónica de la LMC. Se consiguió una respuesta hematológica, confirmada en el 71,5% de los pacientes (Cuadro 3). Igualmente importante es que el 27,7% de los pacientes también tuvo una respuesta citogenética mayor, que fue completa en el 20,4% de ellos. Para los pacientes tratados con 600 mg, las medianas de sobrevida libre de progresión y de sobrevida global estimadas fueron de 22,9 y 42,5 meses, respectivamente. En un análisis multivariado, una dosis de 600 mg se asoció con una mejora del tiempo hasta la progresión independientemente del recuento de plaquetas, blastos en sangre y hemoglobina ≥10 g/L. Crisis blástica mieloide: Se incluyeron 260 pacientes con crisis blástica mieloide. Noventa y cinco pacientes (37%) habían recibido anteriormente quimioterapia para el tratamiento de la fase acelerada o de crisis blástica (pacientes pretratados), mientras que 165 (63%) no habían recibido anteriormente tratamiento (pacientes no tratados). Los primeros 37 pacientes iniciaron el tratamiento con 400 mg, se modificó posteriormente el protocolo, para permitir la administración de una dosis más alta y los restantes 223 pacientes iniciaron el tratamiento con 600 mg. El criterio principal de eficacia fue la tasa de respuesta hematológica, informada tanto como respuesta hematológica completa, no evidencia de leucemia o regreso a la fase crónica de la LMC utilizando el mismo criterio que para el estudio en fase acelerada. En este estudio, el 31% de los pacientes alcanzó una respuesta hematológica (36% de los no tratados previamente y 22% de los previamente tratados). La tasa de respuesta también fue superior en los pacientes tratados con 600 mg (33%) en comparación con los tratados con 400 mg (16%, p=0,0220). La mediana estimada actual de sobrevida de los pacientes no tratados previamente y los tratados fue de 7,7 y 4,7 meses, respectivamente.
Pacientes pediátricos: Un total de 51 niños con LMC recién diagnosticada y no tratada en fase crónica participaron en un ensayo multicéntrico abierto de Fase II, de única rama. Los pacientes recibieron 340 mg/m2/día de Glivec®. El tratamiento con Glivec® produjo una remisión rápida en niños con LMC recién diagnosticada, registrándose un porcentaje de RHC del 78% al cabo de 8 semanas de tratamiento y una completa respuesta citogenética en el 65% de los pacientes, (cifra comparable con los resultados observados en adultos). Después de 3 a 10 meses de tratamiento. Se incorporó en un estudio de Fase I de escalamiento de dosis un total de 31 pacientes pediátricos de una población con múltiples tratamientos previos (45% con trasplante de médula ósea (BMT) y el 68% a poliquimioterapia). Con LMC de fase crónica (n=15) o LMC en crisis blástica o Ph+ ALL (n=16) fueron enrolados en un estudio de fase I de escalación de dosis. Los pacientes fueron tratados con dosis de Glivec® entre 260 mg/m2/día y 570 mg/m2/día. Entre los 13 pacientes con LMC y datos citogenéticos disponibles, 7 (54%) y 4 (31%) alcanzaron respuesta citogenética completa y parcial, respectivamente, para una tasa del 85% de RCM. Ensayos clínicos en LLA Ph+: Un total de 758 pacientes con LLA Ph+ recién diagnosticada o recaída/ resistente al tratamiento participaron en diez ensayos clínicos, de los cuales nueve tuvieron un diseño no comparativo y uno un diseño aleatorizado. LLA Ph+ recién diagnosticada: En un ensayo comparativo (ADE10) entre imatinib y una quimioterapia de inducción en 55 pacientes mayores de 55 años con diagnóstico reciente, la monoterapia con imatinib produjo un porcentaje significativamente mayor de remisión hematológica completa que la quimioterapia (96,3% frente al 50%; p=0,0001). Cuando se administró un tratamiento de último recurso con imatinib en pacientes que no habían conseguido una remisión o sólo una remisión insuficiente con la quimioterapia, 9 de 11 pacientes (81,8%) consiguieron una remisión hematológica completa. Este efecto clínico se acompañó de una mayor reducción de transcriptos Bcr-Abl en los pacientes tratados con imatinib que en el grupo de quimioterapia después de 2 semanas de tratamiento (p=0,02). Todos los pacientes recibieron imatinib y una quimioterapia de consolidación después de la inducción, y las cantidades de transcriptos Bcr-Abl fueron idénticos en los dos grupos después de 8 semanas. Tal como dejaba suponer el diseño del estudio, no se observó ninguna diferencia en la duración de la remisión, la sobrevida libre de enfermedad ni la sobrevida global, aunque los pacientes que presentaron remisión molecular completa y que permanecieron con enfermedad residual mínima obtuvieron mejores resultados en cuanto a la duración de la remisión (p=0,01) y la sobrevida libre de enfermedad (p=0,02). Los resultados de cuatro ensayos clínicos no comparativos (AAU02, ADE04, AJP01 y AUS01) observados en una población de 211 pacientes con LLA Ph+ recién diagnosticada concuerdan con los descriptos anteriormente, como muestra el cuadro 4. Imatinib en asociación con una quimioterapia de inducción produjo un porcentaje de remisión hematológica completa del 93% (147 de 158 pacientes evaluables) y un porcentaje de remisión citogenética significativa del 90% (19 de 21 pacientes evaluables). La tasa de remisión molecular completa fue del 48% (49 de 102 pacientes evaluables). Asimismo, en dos ensayos clínicos no comparativos (AFR09 y AIT04) en los que 49 pacientes mayores de 55 años con LLA Ph+ recién diagnosticada recibieron imatinib y corticoesteroides asociados o no a quimioterapia, se registró un porcentaje de remisión hematológica completa del 89% en la población total y un porcentaje de remisión molecular completa del 26% en 39 pacientes evaluables. En tres estudios (AJP01, AUS01 y AFR09), la sobrevida libre de enfermedad y la sobrevida total fueron sistemáticamente superiores a 1 año y a los valores históricos de comparación (sobrevida sin enfermedad: p < 0,001; sobrevida global: p < 0,01).
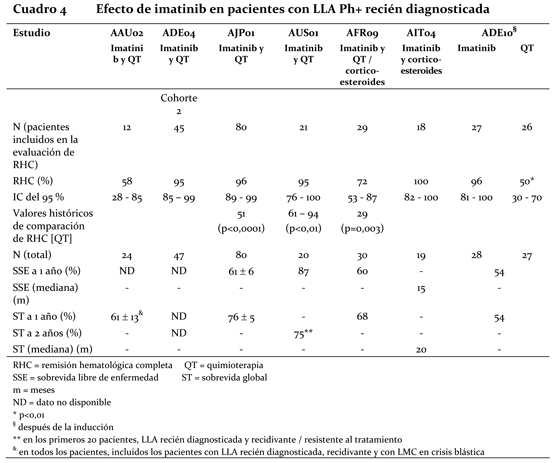
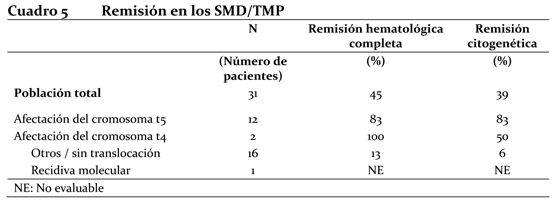
LLA Ph+ recaída / resistente al tratamiento: La monoterapia con imatinib en pacientes con LLA Ph+ recaída / resistente al tratamiento produjo, en 66 de 429 pacientes evaluables, un porcentaje de remisión hematológica del 33% (12% de remisión completa) y un porcentaje de remisión citogenética significativa del 23%. (Nótese que 353 de los 429 pacientes fueron tratados en el marco de un programa de acceso ampliado en el que no se recogieron datos sobre remisión primaria). La mediana del tiempo hasta progresión en los 429 pacientes varió de 1,9 a 3,1 meses, mientras que la mediana de la sobrevida total en los 409 pacientes evaluables varió de 5 a 9 meses. En 14 pacientes, imatinib combinado con una quimioterapia de inducción produjo un porcentaje de remisión hematológica completa del 92% en 12 pacientes evaluables y un porcentaje de remisión citogenética significativa del 100% en 8 pacientes evaluables. Se evaluó la remisión molecular en cuatro pacientes y dos de ellos mostraron una remisión completa. Una población de 146 pacientes mayores de 55 años con LLA recaída o resistente al tratamiento, que recibió monoterapia con imatinib, fue objeto de un análisis separado por la ausencia de un tratamiento curativo. En total, 14 de los 146 pacientes recibieron 600 mg de imatinib al día y se incluyeron en la evaluación de remisión; se observó una remisión hematológica completa en 5 pacientes (35%) y una remisión citogenética significativa en 7 pacientes (50%). Cabe notar que cuatro pacientes tratados con una dosis menor de imatinib (400 mg diarios) no consiguieron la remisión, lo cual indica que esta dosis es insuficiente. En la población total de 146 pacientes, la mediana de la sobrevida sin enfermedad varió de 2,8 a 3,1 meses, y la mediana de la sobrevida total de 7,4 a 8,9 meses. Ensayos clínicos en los Síndromes Mielodisplásicos / Trastornos Mieloproliferativos (SMD/TMP): Se efectuó un ensayo clínico multicéntrico abierto de Fase II (estudio B2225), para investigar el tratamiento con Glivec® en diversas poblaciones de pacientes con enfermedades mortales asociadas con las proteínas con actividad de tirosin-kinasas Abl, Kit o PDGFR. Dicho estudio incluyó 7 pacientes con SMD/TMP entre un total de 185 pacientes tratados, 45 de los cuales padecían enfermedades hematológicas y 140 diversos tumores sólidos. Estos pacientes recibieron 400 mg diarios de Glivec®. La edad de los pacientes admitidos en el ensayo era de 20 a 86 años. Los casos de otros 24 pacientes de 2 a 79 años con SMD/TMP se describieron en 12 informes publicados y en un ensayo clínico. Estos pacientes también recibieron una dosis de Glivec® de 400 mg diarios, salvo tres que fueron tratados con dosis más bajas. De la población total de 31 pacientes con SMD/TMP, 14 (45%) consiguieron una remisión hematológica completa y 9 (29%) una remisión citogenética completa (39% incluyendo remisiones completas y parciales). Nótese que, en 14 pacientes evaluables, la enfermedad presentaba una translocación que generalmente implicaba al cromosoma t5q33 o t4q12, provocando un reordenamiento del gen PDGFR. Todos estos pacientes consiguieron alguna remisión hematológica (12 una remisión completa). Se evaluó la respuesta citogenética en 11 de los 14 pacientes y todos mostraron alguna remisión (9 una remisión completa). Sólo 2 de los 16 pacientes (13%) sin translocación asociada con un reordenamiento del gen PDGFR consiguieron una remisión hematológica completa y uno (6%) una remisión citogenética significativa. Otro paciente con reordenamiento del gen PDGFR, en fase de recidiva molecular después de un trasplante de médula ósea, consiguió una remisión molecular. La mediana de duración del tratamiento fue de 12,9 meses (0,8 a 26,7) en los 7 pacientes tratados del estudio B2225, y varió entre 1 semana y más de 18 meses en los pacientes en remisión según los informes publicados en la literatura médica. El Cuadro 5 presenta un resumen de los resultados.
Ensayos clínicos en la MS: Se efectuó un ensayo clínico multicéntrico de Fase II, abierto (estudio B2225), para investigar el tratamiento con Glivec® en diversas poblaciones de pacientes con enfermedades mortales asociadas con las tirosin-kinasas Abl, Kit o PDGFR. Este estudio incluyó 5 pacientes con MS entre un total de 185 pacientes tratados, de los cuales 45 padecían enfermedades hematológicas y 140 distintos tumores sólidos. Los pacientes con MS recibieron de 100 a 400 mg diarios de Glivec®. La edad de estos pacientes variaba de 49 a 74 años. Los casos de otros 25 pacientes con MS de 26 a 85 años se describieron en 10 informes y casuísticas publicados. Dichos pacientes también recibieron de 100 a 400 mg diarios de Glivec®. De la población total de 30 pacientes con MS, 10 (33%) consiguieron una remisión hematológica completa y 9 (30%) una remisión hematológica parcial (porcentaje total de remisión=63%). Se evaluaron las anomalías citogenéticas en 21 de los 30 pacientes descriptos en informes publicados y en el estudio B2225. Ocho de estos 21 pacientes mostraron la presencia de la kinasa de fusión FIP1L1-PDGFR-a. Los pacientes portadores de esta anomalía citogenética tienen una gran probabilidad de ser de sexo masculino y de presentar eosinofilia asociada con su afección mastocítica sistémica. Dos pacientes mostraron una mutación de Kit en el área yuxtamembranosa (uno Phe522Cys y uno K509I). En 16 pacientes no se detectó ninguna anomalía citogenética o una anomalía desconocida. Cuatro pacientes mostraron una mutación D816V (el único paciente con remisión tenía simultáneamente LMC y MS). Se considera que la mayoría de los casos con la mutación D816V de c-Kit descriptos en la literatura médica analizada no son sensibles a Glivec®. La mediana de duración del tratamiento fue de 13 meses (rango: 1,4 a 22,3 meses) en los 5 pacientes tratados del estudio B2225 y de 1 mes a más de 30 meses en los pacientes en remisión descriptos en la literatura médica. El cuadro 6 presenta un resumen de los resultados.

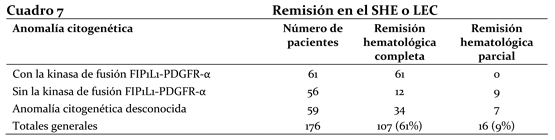
Ensayos clínicos en el SHE o la LEC: Se efectuó un ensayo clínico multicéntrico de Fase II, abierto (estudio B2225), para investigar el tratamiento con Glivec® en diversas poblaciones de pacientes con enfermedades mortales asociadas con las tirosina-kinasas Abl, Kit o PDGFR. Catorce pacientes con SHE o LEC entre un total de 185 pacientes tratados (de los cuales 45 padecían enfermedades hematológicas y 140 distintos tumores sólidos) recibieron de 100 a 1000 mg diarios de Glivec®. La edad de estos pacientes variaba de 16 a 64 años. Los casos de otros 162 pacientes de 11 a 78 años con SHE o LEC se describieron en 35 informes y casuísticas publicados. Dichos pacientes recibieron de 75 mg a 800 mg diarios de Glivec®. Entre la población total de 176 pacientes con SHE o LEC, 107 (61%) consiguieron una remisión hematológica completa y 16 (9%) una remisión hematológica parcial (porcentaje total de remisión = 70%). Se evaluaron las anomalías citogenéticas en 117 de los 176 pacientes descriptos en informes publicados y en el estudio B2225. De estos 117 pacientes, 61 mostraron la presencia de la kinasa de fusión FIP1L1-PDGFR-a y consiguieron una remisión hematológica completa. Un total de 115 no presentaba la kinasa de fusión FIP1L1-PDGFR-a o se desconocía este parámetro; de ellos, 62 (54%) consiguieron una remisión hematológica completa (n=46) o parcial (n=16). El cuadro 7 presenta un resumen de los resultados.
Por otro lado, en los informes de los casos, los investigadores señalaron mejoras en la sintomatología y en otras anomalías funcionales orgánicas. Se notificaron mejoras en el aparato cardivascular, sistema nervioso, piel y tejido subcutáneo, sistema respiratorio, torácico y mediastínico, sistema osteomuscular y tejido conjuntivo, y aparato digestivo. Ensayos clínicos en los TEGI/GIST de carácter irresecable o metastásico: Se realizaron dos ensayos clínicos multinacionales de Fase III, aleatorizados y sin ciego (SWOG, EORTC), en pacientes con tumores malignos del estroma gastrointestinal (TEGI/GIST) de carácter irresecable o metastásico. Por la similitud del diseño de estos dos ensayos, pudo efectuarse un análisis combinado predefinido de la seguridad y la eficacia. En estos dos ensayos participaron un total de 1640 pacientes aleatorizados para recibir, en una proporción de 1:1, 400 mg u 800 mg por vía oral una vez al día de manera continúa hasta que se observara una progresión de la enfermedad o una toxicidad inaceptable. En caso de progresión de la enfermedad, se permitió que los pacientes del grupo tratado con 400 mg una vez al día cambiaran a la dosis de 800 mg una vez al día. El diseño de los ensayos permitía la comparación de los porcentajes de remisión, de sobrevida libre de progresión del cáncer y de sobrevida global entre los dos grupos. La mediana de la edad de los pacientes al ingresar en el ensayo fue de 60 años (entre 17 y 94 años; percentiles 25-75: 50-69 años). Los varones representaron el 58% de los participantes. El diagnóstico de todos los pacientes era TEGI/GIST malignos de carácter irresecable o metastásico con CD117 positivo. En el ensayo EORTC, la evaluación de la sobrevida libre de progresión del cáncer constituyó el objetivo principal y la evaluación de la sobrevida global el objetivo secundario, mientras que en el ensayo SWOG, la evaluación de la sobrevida global fue el objetivo principal y la evaluación de la sobrevida libre de progresión del cáncer fue el objetivo secundario. El análisis de la sobrevida global y de la sobrevida libre de progresión del cáncer se realizó a partir de los conjuntos de datos combinados de los dos ensayos. Los resultados de dicho análisis figuran en el Cuadro 8.
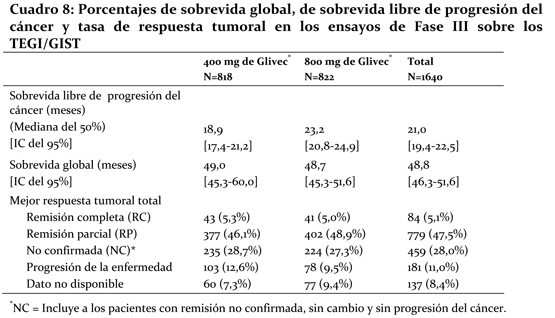
La mediana de la duración del seguimiento de los ensayos combinados fue de 37,5 meses (percentil 25-75 percentilo: 19-46 meses). Se observó una mejora estadísticamente significativa en la sobrevida libre de progresión del cáncer en el grupo tratado con 800 mg (23,2 meses [IC del 95%: 20,8-24,9 meses]) frente al grupo que recibió 400 mg (18,9 meses [IC del 95%: 17,4-21,2 meses]) (p=0,03). Sin embargo, no hubo diferencias entre los grupos en la sobrevida global (p=0,98). En los 1640 pacientes que participaron en estos ensayos de Fase III, la sobrevida libre de progresión estimada fue de 21 meses [IC del 95 %: 19,4-22,5 meses] y la sobrevida global de 48,8 meses [IC del 95%: 46,3-51,6 meses]. El 5,1% de los pacientes mostraron una remisión completa confirmada y el 47,5% una remisión parcial. El tratamiento con cualquiera de las dos dosis fue generalmente bien tolerado y, en total, el 5,4% de los pacientes se retiraron del ensayo debido a problemas de toxicidad. En los pacientes del grupo tratado con 400 mg al día que cambiaron a la dosis de 800 mg tras la progresión de la enfermedad (n=347), la duración de la exposición a Glivec® tras el cambio de la dosis fue de 3,4 meses (mediana) y de 7,7 meses (media). La sobrevida global después del cambio fue de 14,3 meses [IC del 95%: 12,2-16,7 meses] y el 19,3% de estos pacientes seguían vivos después de 48 meses. Se llevó a cabo un estudio clínico, multinacional, abierto, aleatorizado de Fase II, en pacientes de 18 años a 83 años, con TEGI/GIST para recibir Glivec® ya sea la dosis de 400 mg o 600 mg por vía oral una vez al día por hasta 36 meses. En este estudio participaron 147 pacientes con diagnóstico de TEGI/GIST no resecable y/o metastásico, con diagnóstico patológico c-Kit (CD117) positivo, con enfermedad medible en por lo menos un sitio. La evaluación primaria de la eficacia se basó en la tasa de respuestas objetivas (criterios SWOG South Western Oncology Group) que se detallan en el cuadro 9. En este estudio el 83% de los pacientes alcanzaron una remisión completa, remisión parcial o enfermedad estable.

No hubo diferencias entre las tasas de respuesta de los dos grupos de dosificación. Un número significativo de pacientes, que presentaba enfermedad estable en el análisis provisional, alcanzó una respuesta parcial al prolongar el tratamiento (mediana de seguimiento de 31 meses). La mediana del tiempo hasta la respuesta resultó de 13 semanas (IC del 95%: 12 a 23). La mediana del tiempo hasta el fracaso terapéutico alcanzó 122 semanas entre los respondedores (IC del 95%: 106 a 147), mientras que llegó a 84 semanas en la población general del estudio (IC del 95%: 71-109) (figura 3). La mediana de sobrevida global no se llegó a alcanzar. La sobrevida a los 36 meses del seguimiento es del 68%, según las estimaciones de Kaplan-Meier (figura 4). Por otro lado, no hubo ninguna diferencia de sobrevida entre los pacientes cuya enfermedad se estabilizó y los que consiguieron una remisión parcial.
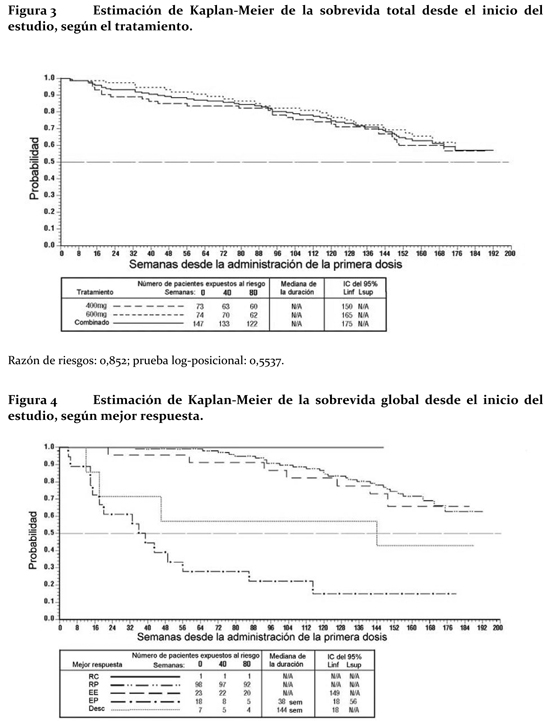
Ensayo clínico en el tratamiento adyuvante de TEGI / GIST: El tratamiento adyuvante con Glivec® se investigó en un ensayo de Fase III multicéntrico, de doble ciego, a largo plazo y controlado con placebo (Z9001) en el que participaron 773 pacientes de 18 a 91 años con diagnóstico histológico de TEGI / GIST primario que expresaba la proteína Kit según el ensayo inmunohistoquímico, y presentaban un tumor ≥3 cm de dimensión máxima, sometidos a resección completa del TEGI/ GIST primario de 14 a 70 días antes de su inclusión. Después de la resección del TEGI / GIST primario, los pacientes fueron aleatorizados a uno de dos grupos que recibirían 400 mg de Glivec® al día o un placebo equiparable durante un año. El criterio de valoración principal del ensayo fue la sobrevida libre de recurrencia, que se definió como el tiempo transcurrido desde la fecha de la aleatorización hasta la fecha de recidiva o el deceso por cualquier causa. Glivec® prolongó de manera significativa la sobrevida libre de recidiva ya que el 75% de los pacientes del grupo de Glivec® no habían presentado recidivas después de 38 meses, frente a 20 meses en el grupo placebo (IC del 95% [30 - valor imposible de estimar]; [14 - valor imposible de estimar], respectivamente); (razón de riesgos = 0,398 [0,259-0,610], p < 0,0001). Después de un año, la sobrevida libre de recidiva fue significativamente mayor con Glivec® (97,7%) que con el placebo (82,3%), (p < 0,0001), por lo que Glivec® redujo aproximadamente un 89% el riesgo de recidiva frente al placebo (razón de riesgos = 0,113 [0,049-0,264]). Un segundo estudio abierto de Fase III (SSG XVIII/AIO) comparó 12 meses de tratamiento con Glivec® 400 mg vs. 36 meses de tratamiento en pacientes después de resección quirúrgica de GIST y uno de los siguientes: diámetro de tumor > 5 cm y recuento mitótico > 5/50 campos de alto poder (HPF); o diámetro de tumor > 10 cm y cualquier recuento mitótico o tumor de cualquier tamaño con recuento mitótico > 10/50 HPF o tumores rotos en la cavidad peritoneal. Hay un total de 397 pacientes consentidos y aleatorizados en el estudio (199 pacientes en la rama de 12 meses y 198 pacientes en la rama de 18 meses), la mediana de edad fue de 61 años (rango 22 a 84 años de edad). La mediana de tiempo de seguimiento fue de 54 meses (desde la fecha de randomización a la fecha de corte), con un total de 83 meses entre el primer paciente randomizado y la fecha de corte. El objetivo primario del estudio fue la sobrevida libre de recurrencia (SLR) definida como el tiempo de la fecha de aleatorización a la fecha de recurrencia o muerte por cualquier causa. Los 36 meses de tratamiento con Glivec® prolongaron significativamente el SLR comparado con los 12 meses de tratamiento con Glivec® (con un Hazard Ratio general (HR)= 0.46 [0.32, 0.65], p < 0.0001 y un HR de 0.42 [0.28, 0.61] por encima del mes 12) (Cuadro 10, Figura 5). Hubo 84 (42%) y 50 (25%) eventos SLR totales para las ramas de 12 meses y 36 meses, respectivamente. Además, 36 meses de tratamiento con Glivec® prolongaron significativamente la sobrevida global (SG) comparado a los 12 meses de tratamiento con Glivec® (HR=0.45 [0.22, 0.89], p= 0.0187) (Cuadro 10, Figura 6). El número total de muertes fueron 25 para la rama de 12 meses de tratamiento y 12 para la rama de 36 meses de tratamiento.
Estudios clínicos en el DFSP: Se efectuó un ensayo clínico multicéntrico de Fase II, con un diseño abierto (estudio B2225), para investigar el tratamiento con Glivec® en diversas poblaciones de pacientes con enfermedades mortales asociadas con las tirosin-kinasas Abl, Kit o PDGFR. Este estudio incluyó a
12 pacientes con DFSP entre un total de 185 pacientes tratados, de los cuales 45 padecían enfermedades hematológicas y 140 distintos tumores sólidos. El criterio principal de eficacia en los pacientes del grupo de tumores sólidos se basó en los porcentajes de remisión objetiva. Esta población, cuya edad variaba de 23 a 75 años, recibió 800 mg diarios de Glivec®. En el momento de la admisión de los pacientes en el estudio, el DFSP era metastásico, localmente recaído después de una resección quirúrgica inicial y no se consideraba nuevamente resecable. Los casos de otros 6 pacientes con DFSP tratados con Glivec® (de 18 meses a 49 años de edad) se describen en 5 informes publicados. La población total que recibió tratamiento incluyó a 18 pacientes, 8 de ellos con enfermedad metastásica. Los adultos descriptos en la literatura médica publicada recibieron 400 mg diarios (4 casos) u 800 mg diarios (1 caso) de Glivec®. El niño recibió 400 mg/m2 diarios, dosis que se incrementó posteriormente hasta 520 mg/m2/día. El cuadro 11 presenta un resumen de las remisiones conseguidas con el tratamiento.

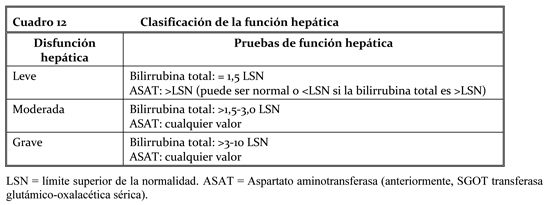
Doce de estos 18 pacientes consiguieron una remisión completa (7 pacientes) o se libraron de la enfermedad gracias a una intervención quirúrgica, tras haber conseguido una remisión parcial (5 pacientes, entre ellos un niño), lo que corresponde a un porcentaje de remisión completa del 67%. Otros 3 pacientes consiguieron una remisión parcial, lo que arroja un porcentaje total de remisión del 83%. De los 8 pacientes con enfermedad metastásica, cinco consiguieron una remisión (62%), la cual fue completa en tres de ellos (37%). La mediana de la duración del tratamiento en el estudio B2225 fue de 6,2 meses, y la máxima de 24,3 meses, mientras que en la literatura publicada esta duración varió de 4 semanas a más de 20 meses. Estudios clínicos en la insuficiencia hepática: En un estudio sobre pacientes con distintos grados de disfunción hepática (leve, moderada y grave; ver la clasificación de la función hepática en el cuadro 12 siguiente), la exposición media al imatinib (ABC normalizada para la dosis) no aumentó en comparación con los pacientes con una función hepática normal. En el citado estudio se administraron 500 mg al día, sin ningún problema de toxicidad, a pacientes con disfunción hepática leve y 300 mg al día, a los demás. Aunque los pacientes con disfunción hepática moderada o grave sólo recibieron una dosis diaria de 300 mg, el análisis farmacocinético prevé que la dosis de 400 mg se puede administrar de manera segura (ver Dosificación - modo de administración, Advertencias, Precauciones, Reacciones adversas y Propiedades farmacocinéticas).
Ensayos clínicos en pacientes con insuficiencia renal: En un estudio realizado en pacientes con distintos grados de insuficiencia renal (leve, moderada y grave; ver la clasificación de la función renal en el cuadro 13), la exposición media a imatinib (ABC normalizada en función de la dosis) aumentó de 1,5 a 2 veces con respecto a pacientes con función renal normal, que correspondió a una concentración plasmática elevada de glucoproteína ácida (AGP), una proteína a la que imatinib se une considerablemente. No se observó una correlación entre la exposición a imatinib y la gravedad de la insuficiencia renal. En este estudio, la dosis de 800 mg al día fue inocua en los pacientes con insuficiencia renal leve, y la dosis de 600 mg al día se empleó en la disfunción renal moderada. La dosis de 800 mg no se probó en los pacientes con disfunción renal moderada por el número limitado de pacientes admitidos. Asimismo, sólo 2 pacientes admitidos con insuficiencia renal grave recibieron una dosis baja de 100 mg, sin que se probaran dosis más altas. El estudio no incluyó pacientes hemodializados. Los datos publicados en la literatura mostraron que una dosis diaria de 400 mg fue bien tolerada por un paciente hemodializado con enfermedad renal terminal. La exposición plasmática de este paciente fue del mismo orden de magnitud que los valores de imatinib y de su metabolito CGP74588 observados en pacientes con función renal normal. La diálisis no alteró la cinética plasmática de imatinib. En vista de que la excreción renal representa una vía de eliminación menor de imatinib, los pacientes dializados con insuficiencia renal grave podrían recibir un tratamiento con una dosis inicial de 400 mg. Sin embargo, se recomienda cautela en estos pacientes. La dosis puede reducirse en caso de intolerancia o aumentarse en caso de ineficacia (ver Dosificación, Advertencias, Precauciones y Propiedades farmacocinéticas).

Propiedades farmacocinéticas: La farmacocinética de Glivec® (mesilato de imatinib) ha sido evaluada en un rango de dosis de 25 a 1000 mg. Los perfiles farmacocinéticos plasmáticos se analizaron en el día 1 y en el día 7 o día 28, cuando las concentraciones plasmáticas habían alcanzado el estado estacionario. Absorción: La media de la biodisponibilidad absoluta para imatinib es del 98%. El coeficiente de variación para el ABC plasmática de imatinib está en el rango del 40-60% después de una dosis oral. Cuando se administró junto con una comida rica en grasas, la tasa de absorción se redujo mínimamente (11% de reducción de la Cmáx y prolongación de tmáx de 1,5 h), con una pequeña reducción del ABC (7,4%) comparado con condiciones de ayuno. Distribución: En base a los experimentos in vitro, a concentraciones clínicamente relevantes de imatinib, el 95% aproximadamente está unido a proteínas plasmáticas, principalmente a la albúmina y a la alfa-ácido-glucoproteína, con una pequeña unión a lipoproteínas. Metabolismo: El principal metabolito circulante en humanos es el derivado piperazina N-desmetilado (CGP 71588), el cual muestra in vitro una potencia similar a la del compuesto madre. El área bajo la curva plasmática que se halló para el metabolito fue sólo 16% de ABC de imatinib. La unión a las proteínas plasmáticas del metabolito N-desmetilado es semejante a la del compuesto original. Eliminación: En base a la recuperación de compuesto(s) después de una dosis oral de imatinib marcado con 14C, aproximadamente el 81% de la dosis se recuperó en 7 días en las heces (68% de la dosis) y en orina (13% de la dosis). Imatinib inalterado alcanza el 25% de la dosis (5% orina, 20% heces), constituyendo el resto metabolitos. Farmacocinética plasmática: Tras la administración oral a voluntarios sanos, el t1/2 fue aproximadamente de 18 h, sugiriendo que una dosis única diaria es apropiada. El aumento en la media del ABC fue lineal con el incremento de dosis y proporcional a la dosis, en el rango de 25-1000 mg de imatinib. No hubo cambios en la cinética de imatinib a dosis repetidas y la acumulación fue de 1,5-2,5 veces en el estado estacionario, cuando se dosificó una vez al día. Farmacocinética de la población: En base al análisis farmacocinético de la población, hubo un pequeño efecto de la edad en el volumen de distribución (aumento del 12% en pacientes mayores de 65 años). No se cree que este cambio sea clínicamente significativo. El efecto del peso corporal en el clearance de imatinib es tal, que para un paciente que pesa 50 kg, el clearance medio esperado es de 8,5 L/h, mientras que, en un paciente que pese 100 kg, el clearance aumentará hasta 11,8 L/h. Estos cambios no son considerados suficientes para poder justificar un ajuste de dosis en base al peso corporal en kg. El sexo de los individuos no afecta la cinética de imatinib. Ulteriores análisis farmacocinéticos de la población en el estudio de Fase III en pacientes recientemente diagnosticados con LMC, mostraron que el efecto de covariables y tratamientos concomitantes tanto en el clearance como en el volumen, parecen ser pequeños y no son lo suficientemente pronunciados como para justificar un ajuste de dosis. Farmacocinética en niños: Al igual que en los pacientes adultos, imatinib fue rápidamente absorbido después de la administración oral en los pacientes pediátricos en estudios de Fase I y II. La dosificación en niños a 260 y 340 mg/m2 alcanzó la misma exposición que las dosis respectivas de 400 y 600 mg en los pacientes adultos. La comparación del ABC(0-24) en el día 8 y en día 1 de la dosis de 340 mg/m2 reveló una acumulación de 1,7 voz de droga, después de dosificaciones diarias únicas a repetición. Alteraciones de la función orgánica: Imatinib y sus metabolitos no se excretan en grado significativo por los riñones. En los pacientes con insuficiencia renal leve y moderada, la exposición plasmática parece ser mayor que en los pacientes con función renal normal. La exposición es aproximadamente 1,5 a 2 veces mayor, lo que corresponde a una elevación de 1,5 vez de la AGP plasmática, a la que imatinib se une considerablemente. La depuración del medicamento libre es probablemente similar entre los pacientes con insuficiencia renal y función renal normal ya que la excreción renal representa solamente una vía menor de eliminación de imatinib (ver Dosificación, Advertencias, Precauciones y Propiedades farmacocinéticas). Si bien los resultados del análisis farmacocinético revelaron una considerable variación interindividual, la exposición media a imatinib no aumentó entre los pacientes con diversos grados de disfunción hepática, en comparación con los que presentaban una función hepática normal (ver Dosificación, Advertencias, Precauciones, Reacciones adversas, Acción farmacológica y Propiedades farmacocinéticas). Datos sobre toxicidad preclínica: Imatinib ha sido objeto de estudios de farmacotoxicología, toxicidad de las dosis múltiples, genotoxicidad y toxicidad para la función reproductora. Los órganos diana asociados con la acción farmacológica de imatinib son la médula ósea, la sangre periférica, los tejidos linfáticos, las gónadas y el tubo digestivo. El hígado y los riñones se incluyen también entre ellos. Imatinib ha resultado embriotóxico y teratógeno para las ratas. La fecundidad no se vio afectada en el estudio preclínico sobre fecundidad y desarrollo embrionario inicial, aunque se observaron disminuciones del peso de los testículos y epidídimos, así como una reducción de la cantidad de espermatozoides móviles en los machos que recibieron la dosis alta. En el estudio preclínico sobre el desarrollo prenatal y posnatal en ratas, Glivec® tampoco afectó la fecundidad de las crías de la primera generación. En el estudio de carcinogenia de 2 años en ratas, la administración de 15, 30 y 60 mg/kg/día de imatinib produjo una reducción estadísticamente significativa de la longevidad de los machos con 60 mg/kg/día y de las hembras con dosis 30 mg/kg/día. El examen histopatológico de los animales que murieron reveló miocardiopatía (ambos sexos), nefropatía crónica progresiva (hembras) y papiloma de la glándula prepucial como causas principales de muerte o motivos de sacrificio. Los órganos efectores de las alteraciones neoplásicas fueron los riñones, la vejiga, la uretra, la glándula prepucial y la glándula clitorídea, el intestino delgado, las paratiroides, las suprarrenales y el estómago no glandular. Las mayores dosis examinadas que no produjeron efectos tóxicos en distintos órganos efectores con lesiones neoplásicas fueron las siguientes: 30 mg/kg/día para riñones, vejiga, uretra, intestino delgado, paratiroides, suprarrenales y estómago no glandular, y 15 mg/kg/día para la glándula prepucial y la glándula clitoridea. Los papilomas y carcinomas de las glándulas prepuciales y clitorideas ocurrieron con dosis de 30 y 60 mg/kg/día de imatinib aproximadamente de 0,5 a 4 veces o 0,3 a 2,4 veces la exposición diaria humana (basado en el ABC) a 400 mg/día u 800 mg diarios respectivamente, y de 0,4 a 3,0 veces la exposición diaria en niños (basada en el ABC) con dosis de 340 mg/m2. Los adenomas y carcinomas renales, así como los papilomas de la vejiga urinaria y uretra, adenocarcinomas del intestino delgado, adenomas de la paratiroides, tumores medulares benignos y malignos de las suprarrenales y papilomas/carcinomas del estómago no glandular sólo ocurrieron con dosis de 60 mg/kg/día. Se desconoce la relevancia que para la especie humana podrán tener estos datos del estudio de carcinogénesis con ratas. En un análisis sobre los datos de seguridad de los ensayos clínicos así como las notificaciones espontáneas de eventos adversos no se ha detectado que la incidencia general de neoplasias malignas aumente entre los pacientes tratados con imatinib, en comparación con la población general. Las lesiones no neoplásicas que no se habían identificado en estudios preclínicos anteriores implicaron el sistema cardiovascular, el páncreas, los órganos endocrinos y los dientes. Las alteraciones más importantes fueron la hipertrofia y dilatación cardíacas acompañadas de signos de insuficiencia cardíaca en algunos animales.
Indicaciones.
Glivec® está indicado para el tratamiento de: Pacientes adultos con diagnóstico reciente de leucemia mieloide crónica (LMC) con cromosoma Filadelfia positivo (Ph+) en fase crónica. Pacientes con LMC (Ph+) en fase crónica, acelerada o crisis blástica, cuando falla el tratamiento con interferón a. Pacientes pediátricos con LMC Ph+ en fase crónica recientemente diagnosticada o con enfermedad recidivada luego de transplante de médula ósea o resistentes al tratamiento con interferón a. No hay ensayos controlados en pacientes pediátricos que demuestren un beneficio clínico tales como mejoría de los síntomas relacionados con la enfermedad o mejoría de la sobrevida. Pacientes adultos con leucemia linfoblástica aguda con cromosoma Filadelfia positivo (LLA Ph+), recaída o refractaria. Pacientes adultos con síndrome mielodisplásico/síndrome mieloproliferativo (SMD/SMP) asociados con rearreglos genéticos del receptor del factor de crecimiento derivado de plaquetas (PDGFR). Pacientes adultos con mastocitosis sistémica agresiva (MSA/ASM) sin la mutación del D816V c-Kit o con el estado mutacional del c-Kit desconocido. Pacientes adultos con síndrome hipereosinofílico (SHE) y/o leucemia eosinofílica crónica (LEC) con sobreexpresión de la kinasa de fusión FIP1L1-PDGFR alfa (deleción de alelo CHIC2 demostrado por FISH o análisis mutacional) y para pacientes con SHE y/o LEC con kinasa de fusión FIP1L1-PDGFR a negativa desconocida. Pacientes adultos con dermatofibrosarcoma protuberans (DFSP) irresecable, recidivante y/o metastásico. Pacientes con tumor de estroma gastrointestinal (TEGI/GIST) metastásico maligno y/o Kit (CD117) positivo no resecable. Pacientes adultos después de la resección con criterio adyuvante de TEGI/GIST.
Dosificación.
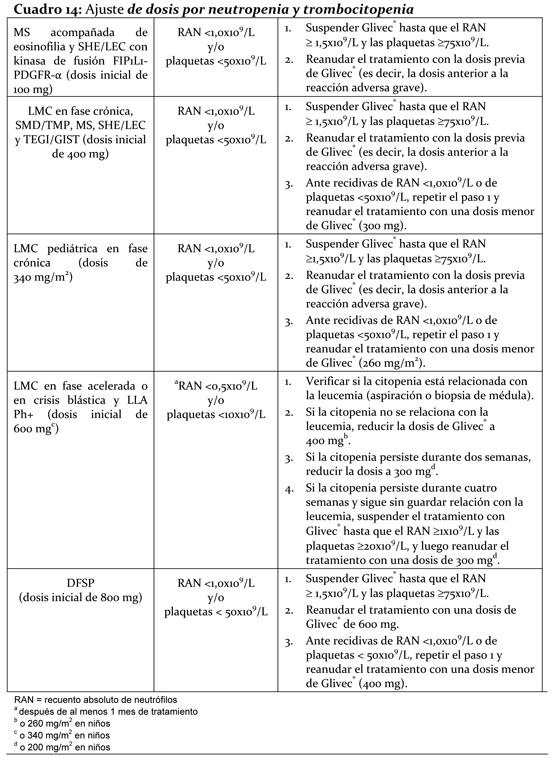
El tratamiento debe ser iniciado por un médico experimentado en el tratamiento de pacientes con hemopatías y sarcomas malignos según proceda. La dosis prescripta debe ser administrada por vía oral, con una comida y un vaso grande con agua. Las dosis de 400 o 600 mg deben administrarse una vez al día y la dosis diaria de 800 mg debe administrarse en dos tomas de 400 mg, una en la mañana y otra en la noche. Para los pacientes con dificultades para tragar los comprimidos recubiertos, se podrán dispersar los mismos en un vaso con agua no gasificada o con jugo de manzana. El número requerido de comprimidos será colocado en un vaso con el volumen apropiado de la bebida (aproximadamente 50 mL para un comprimido de 100 mg y 200 mL para el comprimido de 400 mg) revolviendo con una cuchara. La suspensión obtenida debe ser administrada inmediatamente después de haber obtenido la desintegración completa del/os comprimido/s. El tratamiento debe continuarse mientras el paciente se beneficie. Dosificación en LMC: La dosis recomendada de Glivec® es de 400 mg/día para pacientes adultos con LMC en fase crónica y de 600 mg/día para pacientes adultos en fase acelerada o crisis blástica. El aumento de la dosis de 400 a 600 u 800 mg en pacientes en la fase crónica de la enfermedad, o de 600 mg a un máximo de 800 mg (administrados en dosis de 400 mg dos veces al día), en pacientes en fase acelerada o crisis blástica, puede considerarse en ausencia de reacciones adversas severas y de neutropenia o de trombocitopenia severas no relacionadas con la leucemia en las siguientes circunstancias: progresión de la enfermedad (en cualquier momento); si no se consigue una respuesta hematológica satisfactoria después de por lo menos 3 meses de tratamiento; ausencia de remisión citogenética al cabo de 12 meses de tratamiento, o pérdida de la remisión hematológica o citogenética conseguida anteriormente. Posología en la LLA Ph+: La dosis recomendada de Glivec® en pacientes adultos con LLA Ph+ recaída o refractaria es de 600 mg al día. Síndrome mielodisplásico/ Enfermedad mieloproliferativa (SMD/EMP): La dosis recomendada de Glivec® para pacientes adultos es de 400 mg/día. Mastocitosis sistémica agresiva: La dosis recomendada de Glivec® es de 400 mg/día para pacientes adultos con MSA sin la mutación de D816V c-Kit o estado mutacional desconocido o que no responde a otras terapias. Para pacientes con MSA asociado a eosinofilia, una enfermedad hematológica clonal relacionada con la fusión de la kinasa FIP1L1-PDGFRa, se recomienda iniciar con una dosificación de 100 mg/día. El incremento de la dosis de 100 a 400 mg para estos pacientes puede ser considerado en ausencia de reacciones adversas a la droga y si las pruebas demuestran una respuesta insuficiente a la terapia. Síndrome eosinofílico / Leucemia eosinofílica: La dosis recomendada de Glivec® es de 400 mg/día. Pacientes con síndrome eosinofílico / leucemia eosinofílica con fusión de la kinasa FIP1L1-PDGRFa demostrada, se recomienda una dosis inicial de 100 mg diarios. El incremento de la dosis de 100 a 400 mg para estos pacientes puede ser considerado en ausencia de reacciones adversas a la droga y si las pruebas demuestran una respuesta insuficiente a la terapia. Dosificación en TEGI/GIST: La dosis recomendada de Glivec® es de 400 mg o para pacientes adultos con TEGI/GIST no resecable y/o metastásico. El aumento de la dosis de 400 a 600 u 800 mg debe ser considerado en ausencia de reacciones adversas de la droga cuando las evaluaciones demuestran una respuesta insuficiente al tratamiento. La dosis recomendada de Glivec® es de 400 mg al día para el tratamiento adyuvante de pacientes adultos después de la resección de TEGI/GIST. La duración mínima recomendada del tratamiento es de 36 meses. Se desconoce la duración óptima del tratamiento adyuvante con Glivec®. Posología en el DFSP: La dosis recomendada de Glivec® en pacientes con DFSP es de 800 mg al día. Ajuste de la dosis para reacciones adversas: Reacciones adversas no hematológicas: Si se desarrolla una reacción adversa severa no hematológica con el uso de Glivec®, el tratamiento se interrumpirá hasta que la reacción se haya resuelto. Después puede reanudarse el tratamiento, según corresponda, dependiendo de la severidad inicial de la reacción. Si se observara aumento de bilirrubina > 3 del límite superior normal (LSN) o de las transaminasas hepáticas en > 5xLSN, deberá suspenderse la administración de Glivec® hasta tanto los niveles de bilirrubina hayan retornado a < 1,5xLSN y los niveles de transaminasas a < 2,5xLSN. El tratamiento con Glivec® puede ser entonces continuado con una dosis diaria reducida. En adultos la dosis será reducida de 400 a 300 mg, de 600 a 400 mg o de 800 a 600 mg, y en los niños de 340 a 260 mg/m2/día. Reacciones adversas hematológicas: Se recomienda reducción de la dosis o interrupción del tratamiento si se produce neutropenia y/o trombocitopenia severas, tal como se indica en el siguiente cuadro.
Uso en pediatría: No hay experiencia en el uso de Glivec® en niños menores de 2 años de edad. Es muy limitada la experiencia con Glivec® en otras indicaciones en niños. La dosificación en niños debe realizarse en base al área de superficie corporal (mg/m2). En los niños con LMC en fases crónica o avanzada se recomienda administrar dosis diarias de 340 mg/m2 (sin superar un total de 600 mg al día). Se puede administrar una sola dosis diaria o se puede dividir la dosis diaria en dos tomas (una en la mañana y otra en la noche). La recomendación posológica se fundamenta en una pequeña población de pacientes pediátricos (ver Farmacología y Propiedades farmacocinéticas). Insuficiencia hepática: Imatinib se metaboliza principalmente en el hígado. Los pacientes con una disfunción hepática leve, moderada o grave recibirán la dosis mínima recomendada de 400 mg al día, que se podrá reducir en caso de toxicidad inadmisible (ver Advertencias, Precauciones, Reacciones adversas, Farmacología y Propiedades farmacocinéticas). Insuficiencia renal: Imatinib y sus metabolitos no se excretan significativamente por vía renal. Los pacientes con disfunción renal o en diálisis pueden empezar el tratamiento con la dosis mínima recomendada de 400 mg al día. Sin embargo, se recomienda cautela en estos casos. La dosis puede reducirse en caso de intolerancia. Si es tolerada la dosis puede aumentarse en caso de ineficacia (ver Advertencias y Precauciones). Pacientes de edad avanzada: No se observaron diferencias significativas farmacocinéticas relacionadas con la edad, en pacientes adultos incluidos en estudios clínicos donde más del 20% de los pacientes tenían 65 o más años de edad. No es necesaria una recomendación específica de ajuste de dosis en los pacientes de edad avanzada.
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes.
Reacciones adversas.
Resumen del perfil de seguridad: El perfil de toxicidad de Glivec® se ha caracterizado adecuadamente durante más de 12 años de utilización clínica. Durante el desarrollo clínico, la mayoría de los pacientes presentaron reacciones adversas en algún momento. Las más frecuentes ( > 10 %) consistieron en neutropenia, trombocitopenia, anemia, cefalea, dispepsia, edema, aumento de peso, náuseas, vómito, calambres musculares, dolor osteomuscular, diarrea, exantema, cansancio y dolor abdominal. Las reacciones fueron de intensidad leve a moderada y sólo del 2 al 5 % de los pacientes suspendieron el tratamiento definitivamente debido a un acontecimiento relacionado con el medicamento. El perfil de toxicidad muestra diferencias entre las leucemias Ph+ y los tumores sólidos: son mayores la incidencia y la intensidad de la mielodepresión en las leucemias Ph+ y las hemorragias gastrointestinales e intratumorales en pacientes con TEGI, probablemente por factores vinculados con la enfermedad. La mielodepresión, las reacciones adversas gastrointestinales, el edema y el exantema son frecuentes en estas dos poblaciones de pacientes. Otras afecciones gastrointestinales, como por ejemplo obstrucción, perforación y ulceración gastrointestinales, parecen ser más específicas de cada indicación. Se han observado las siguientes reacciones adversas importantes tras la exposición a Glivec®, que pueden tener una relación de causa y efecto con el medicamento: hepatotoxicidad, insuficiencia renal aguda, hipofosfatemia, reacciones adversas respiratorias graves, síndrome de lisis tumoral y retraso del crecimiento en niños. En función de la gravedad de las reacciones, puede ser necesario ajustar la dosis. En casos contados se tendrá que retirar el medicamento debido a reacciones adversas. Las reacciones adversas (Cuadros 15 y 16) se clasificaron en orden de frecuencia de la siguiente manera: muy frecuentes (≥1/10); frecuentes (≥1/100, < 1/10); infrecuentes (≥1/1000, < 1/100); raras (≥1/10000, < 1/1000); muy raras ( < 1/10000), incluidos los informes aislados. Las reacciones adversas y las frecuencias indicadas en el Cuadro 1 se derivan de los estudios que se efectuaron para justificar el registro del medicamento para el tratamiento de la LMC y los TEGI/GIST.

Los siguientes tipos de reacciones se han notificado durante la farmacovigilancia de Glivec® y algunos ensayos clínicos complementarios. Comprenden las notificaciones espontáneas y los acontecimientos adversos graves registrados durante ensayos clínicos más pequeños o en curso y durante programas de disponibilidad ampliada. Dado que estas reacciones se han notificado en una población de tamaño indeterminado, no siempre es posible dar una estimación fiable de su frecuencia o establecer una relación de causa y efecto con la exposición a imatinib.

Descripción de algunas reacciones adversas seleccionadas: Mielodepresión: La mielodepresión es muy frecuente en los pacientes cancerosos tratados con Glivec®. Las anomalías de laboratorio de grados 3 y 4 notificadas con mayor frecuencia mielosupresión, trombocitopenia, neutropenia y anemia. La mielosupresión de los pacientes con LMC tratados con Glivec® fue generalmente reversible y en la mayoría de los casos no obligó a interrumpir la administración ni a reducir la dosis. Pocos pacientes tuvieron que suspender definitivamente el tratamiento. También se han notificado otros acontecimientos adversos como pancitopenia, linfopenia y depresión medular. El mayor grado de depresión hematológica se observó con las dosis más altas y pareció depender del estadio de la LMC; la neutropenia y la trombocitopenia de grados 3 o 4 fueron entre 4 y 6 veces más frecuentes en los pacientes con LMC en crisis blástica o en fase acelerada (44 % y 63 %, respectivamente) que en los pacientes con LMC recién diagnosticada en fase crónica (16,7 % y 8,9 %, respectivamente). Normalmente, estas reacciones pueden tratarse reduciendo la dosis o interrumpiendo el tratamiento con Glivec®, y raramente requieren la retirada definitiva del tratamiento. La incidencia de toxicidad hematológica es menor en los pacientes con tumores sólidos (es decir, TEGI) que en los pacientes con leucemias Ph+, puesto que las frecuencias de neutropenia y trombocitopenia de grados 3 o 4 son de aproximadamente el 10 % y el 1 %, respectivamente. Hemorragia: Las hemorragias del SNC y gastrointestinales son frecuentes en los pacientes con LMC que presentan un deterioro de la función medular antes del tratamiento. Las hemorragias constituyen una de las complicaciones bien conocidas de la leucemia aguda y pueden ser una consecuencia de la trombocitopenia o, con menor frecuencia, de una disfunción plaquetaria. No obstante, no todos los pacientes que presentan hemorragias del SNC o gastrointestinales durante el tratamiento con el imatinib padecen trombocitopenia. Entre las hemorragias de importancia clínica, la hemorragia gastrointestinal fue la más frecuente, sobre todo en pacientes con LMC avanzada y en pacientes con TEGI metastásicos, en los que la hemorragia puede formar parte del cuadro subyacente debido a la hemorragia o necrosis tumoral. Las frecuencias más bajas de hemorragia gastrointestinal se observaron generalmente en el tratamiento de primera línea de la LMC y en el tratamiento adyuvante de los TEGI. Edema y retención de líquido: El edema es un efecto tóxico frecuente del imatinib que se produce en más del 50 % de los pacientes, combinando todas las indicaciones. El edema depende de la dosis, y al parecer existe una correlación entre su aparición y las concentraciones plasmáticas del fármaco. La manifestación más común es el edema periorbitario, mientras que el edema de las extremidades inferiores es un poco menos frecuente. No suele ser necesario administrar un tratamiento específico. Aunque los demás acontecimientos de retención de líquido son mucho menos frecuentes, pueden ser graves debido a la ubicación anatómica del sitio afectado. El más común fue el derrame pleural, sobre todo en los pacientes con LMC avanzada o TEGI metastásicos. La frecuencia de insuficiencia cardiaca fue generalmente baja entre los pacientes con edema y retención de líquido, pero fue mayor en los pacientes con LMC avanzada que en los demás grupos. Esto puede explicarse por el estado médico más deteriorado de los pacientes con LMC avanzada. Se observó la misma tendencia en la insuficiencia renal entre los pacientes con edema y retención de líquido. La mayoría de los pacientes que presentaron edema y retención de líquido eran ancianos (mayores de 65 años). En un ensayo clínico, la frecuencia de acontecimientos indicativos de insuficiencia cardiaca congestiva fue del 1,5 % con el imatinib frente al 1,1 % con IFN-a en pacientes con LMC de diagnóstico reciente. La frecuencia fue considerablemente más elevada en pacientes con LMC transformada (en fase acelerada o en crisis blástica), en pacientes de edad más avanzada o con un valor inicial de hemoglobina inferior a 8 g/dl. Combinando todas las indicaciones, la mayor frecuencia de insuficiencia cardiaca congestiva observada en pacientes con LMC que en pacientes con TEGI podría indicar diferencias de algunos factores de riesgo vinculados con la enfermedad. Además, un análisis especial de la toxicidad cardiaca publicado recientemente en el marco del estudio de la EORTC de 942 pacientes con TEGI de carácter irresecable o metastásico, concluyó que el imatinib no induce insuficiencia ventricular izquierda en pacientes con TEGI, en quienes la incidencia observada fue de alrededor del 0,2 %, mientras que puede alcanzar el 2 % en una población con cardiopatías preexistentes. Exantemas y reacciones adversas cutáneas graves: Se han notificado casos de exantema eritematoso, maculopapular, pruriginoso, que puede desaparecer sin interrumpir el tratamiento. Algunos pacientes pueden presentar prurito sin exantema asociado, y en ocasiones se observa un componente exfoliativo. La reexposición al medicamento ha provocado la reaparición del exantema en algunos pacientes, pero no en todos. Estas erupciones suelen responder al tratamiento con antihistamínicos y esteroides locales. A veces se necesitan corticoesteroides sistémicos. Se han observado exantemas hasta en un tercio de los pacientes tratados con el imatinib, combinando todas las indicaciones. Se trata frecuentemente de lesiones pruriginosas, generalmente eritematosas y maculopapulares en el antebrazo, el tronco o la cara. Las biopsias de piel han revelado una reacción tóxica al fármaco, con la presencia de un infiltrado celular combinado. Si bien la mayoría de los exantemas son leves y de curso limitado, los casos más graves pueden necesitar la interrupción temporal o definitiva del tratamiento. Como era de esperarse, las reacciones cutáneas fueron más frecuentes que con el placebo en el estudio sobre el tratamiento adyuvante de TEGI. Hepatotoxicidad: Puede producirse hepatotoxicidad, a veces grave pues se han observado casos durante los ensayos preclínicos y clínicos. Las anomalías de las pruebas de la función hepática generalmente consistieron en elevaciones leves de las transaminasas, aunque en una minoría de pacientes se observaron elevaciones de la bilirrubina. La hepatotoxicidad suele aparecer en los dos primeros meses de tratamiento, pero en ocasiones se ha presentado al cabo de un período de 6 a 12 meses de tratamiento. Los valores generalmente se normalizan después de interrumpir el tratamiento durante 1 a 4 semanas. Hipofosfatemia: Se han observado concentraciones bajas de fosfato e hipofosfatemia (hasta de grados 3 o 4) de manera relativamente frecuente en todas las indicaciones, pero no se ha dilucidado el origen ni la importancia clínica de este hallazgo. Se ha demostrado que el imatinib inhibe la diferenciación de los monocitos humanos en osteoclastos. Esta disminución se acompañó de una reducción de la capacidad de resorción de estas células. El imatinib provocó una disminución de RANK-L en los osteoclastos en función de la dosis. La inhibición constante de la actividad osteoclástica puede conducir a una contrarregulación que da lugar a un aumento de las concentraciones de PTH. Aún no se ha esclarecido la importancia clínica de los hallazgos preclínicos y no se ha demostrado una asociación con las reacciones adversas óseas (por ejemplo, fracturas). Durante el programa de desarrollo clínico no se midió sistemáticamente la concentración sérica de fosfato en todos los ensayos. Si bien se planteó inicialmente la hipótesis de que la hipofosfatemia podría depender de la dosis, los resultados interpretables al cabo de 24 meses del ensayo TOPS de Fase III, diseñado para investigar la relación entre la dosis y algunas variables de seguridad en pacientes con LMC recién diagnosticada, mostraron que el 19,1 % y el 15,5 % de los pacientes tratados con 400 mg presentaron disminuciones de grados 3 o 4 de las concentraciones séricas de fosfato o calcio, respectivamente, frente a porcentajes del 5,1 % y del 0,9 %, respectivamente, en los pacientes tratados con 800 mg. Obstrucción, perforación y ulceración gastrointestinales: La ulceración gastrointestinal que, en casos extremos, puede ser una manifestación de irritación local causada por el imatinib, se ha observado en una proporción reducida de pacientes en todas las indicaciones. La hemorragia y la necrosis tumorales, así como la obstrucción y perforación gastrointestinales, parecen relacionarse con la enfermedad y se han producido exclusivamente o con mayor frecuencia en pacientes con TEGI. En el caso de TEGI metastásicos, puede ocurrir necrosis tumoral en el contexto de la respuesta del tumor, que rara vez conduce a una perforación. La máxima incidencia de obstrucción o íleo gastrointestinales se observó en la población con TEGI, posiblemente por la obstrucción tumoral causada por los TEGI metastásicos, así como en el marco del tratamiento adyuvante por las adherencias de una intervención quirúrgica previa. Síndrome de lisis tumoral: Se considera posible una relación causal entre el síndrome de lisis tumoral y el tratamiento con Glivec®, aunque algunos casos incluyeron factores de confusión consistentes en medicamentos concomitantes y otros factores de riesgo independientes (ver Advertencias y Precauciones). Retraso del crecimiento en niños: Glivec® parece afectar el crecimiento de los niños, especialmente de los niños prepúberes. Es imposible descartar una relación causal entre el retraso del crecimiento en niños y el tratamiento con Glivec®, aunque se dispone solamente de información limitada sobre algunos casos de retraso del crecimiento (Ver Advertencias y Precauciones). Reacciones adversas respiratorias graves: Se han observado con Glivec® reacciones respiratorias graves, a veces mortales, que han incluido insuficiencia respiratoria aguda, hipertensión pulmonar, enfermedad pulmonar intersticial y fibrosis pulmonar. En muchos de estos casos existían afecciones cardiacas o pulmonares preexistentes que podían asociarse con acontecimientos pulmonares graves. Anormalidades en los exámenes de laboratorio: Hematología: En LMC un hallazgo constante en todos los estudios fueron las citopenias, particularmente neutropenia y trombocitopenia, sugiriendo una mayor frecuencia a dosis elevadas (≥750 mg: estudio de Fase I). Sin embargo, la presencia de citopenias fue también claramente dependiente de la fase de la enfermedad. En pacientes con LMC recientemente diagnosticada, las citopenias fueron menos frecuentes que en los otros pacientes con LMC. La frecuencia de neutropenias de Grado 3 o 4 (RAN < 1,0x109/L) y trombocitopenias (recuento de plaquetas < 50x109/L), fue entre 4 y 6 veces mayor en crisis blástica y en fase acelerada (59-64% y 44-63% para neutropenia y trombocitopenia, respectivamente) comparado con pacientes en fase crónica de LMC recientemente diagnosticada (16,7% neutropenia y 8,9% trombocitopenia). Se observó neutropenia de Grado 4 (RAN < 0,5x109/L) y trombocitopenia (recuento de plaquetas < 10x109/L) en el 3,6% y en menos del 1% respectivamente, de los pacientes con LMC recientemente diagnosticada en fase crónica. La mediana de la duración de los episodios de neutropenia y trombocitopenia normalmente fue de 2 a 3 semanas y de 3 a 4 semanas respectivamente. Estos efectos pueden ser tratados con reducción de dosis o con interrupción del tratamiento con Glivec®, pero en casos raros puede llevar a la discontinuación definitiva del tratamiento. En niños con LMC, las toxicidades más frecuentes fueron citopenias de Grado 3 o 4 que consistieron en neutropenia, trombocitopenia y anemia. Estas reacciones se presentaron generalmente durante los primeros meses de tratamiento. En pacientes con TEGI/GIST irresecable o metastásico maligno (Estudio B2222) se observó anemia Grado 3 y 4 en el 5,4% y 0,7% de los pacientes, respectivamente, y pueden haber estado relacionados a sangrado gastrointestinal o intratumoral en al menos algunos de estos pacientes. Se observó neutropenia grado 3 y 4 en el 7,5% y 2,7% de los pacientes respectivamente y trombocitopenia Grado 3 en 0,7% de los pacientes. Ningún paciente desarrolló trombocitopenia Grado 4. La disminución del recuento de glóbulos blancos y el recuento de neutrófilos ocurrió en su mayoría durante las primeras seis semanas del tratamiento, con valores que se mantuvieron después relativamente estables. Bioquímica: La elevación intensa de las transaminasas ( < 5%) o de la bilirrubina ( < 1%) ocurrieron entre los pacientes con LMC y se controló casi siempre mediante una reducción posológica o interrupción del tratamiento (la duración mediana de estos episodios se aproximó a una semana). El tratamiento se suspendió de forma permanente, a causa de las anomalías hepáticas de laboratorio, en menos del 1% de los casos de LMC. El 6,8% de los pacientes con TEGI/GIST (estudio B2222), manifestó elevaciones de la alanina-aminotransferasa de Grado 3 o 4 y el 4,8%, elevaciones de la aspartato-aminotransferasa de Grado 3 o 4. La elevación de la bilirrubina se registró entre menos del 3%. Se produjeron casos de hepatitis citolítica y colestásica así como de insuficiencia hepática, en ocasiones, de carácter mortal.
Precauciones.
Controles de laboratorio: Durante el tratamiento con Glivec® deben realizarse regularmente recuentos sanguíneos completos. El tratamiento con Glivec® de pacientes con LMC, se ha asociado con neutropenia y/o trombocitopenia. Sin embargo, la presencia de estas citopenias depende de la fase de la enfermedad que se está tratando, siendo más frecuentes en pacientes en fase acelerada de LMC o crisis blástica, en comparación con pacientes en fase crónica de LMC. El tratamiento con Glivec® puede ser interrumpido o la dosis reducida, tal como se recomienda en Dosificación. La función hepática (transaminasas, bilirrubina, fosfatasa alcalina) debe ser controlada regularmente en pacientes que reciben Glivec®. Tal como se recomienda en Dosificación: Reacciones adversas no hematológicas, estas anormalidades de laboratorio deben ser tratadas interrumpiendo y/o reduciendo la dosis de Glivec®. Glivec® y sus metabolitos no se excretan por vía renal en forma significativa. Se sabe que
el clearance de creatinina se reduce con la edad y que ésta no afecta de forma significativa la cinética de Glivec®. En los pacientes con insuficiencia renal, la exposición plasmática a imatinib parece ser mayor que en los que tienen una función renal normal, probablemente porque tales pacientes presentan concentraciones plasmáticas elevadas de glucoproteína ácida a (AGP, proteína a la que se une imatinib). Según la clasificación basada en la depuración de creatinina entre los pacientes con insuficiencia renal leve (CrCl de 40-59 mL/min) e insuficiencia renal grave (CrCl < 20 mL/min), no existe una correlación entre la exposición a imatinib y el grado de insuficiencia renal. Sin embargo, como se recomienda en Posología, puede reducirse la dosis inicial de imatinib en caso de intolerancia. Niños y adolescentes: Ha habido informes de casos de un retraso del crecimiento en niños y preadolescentes que recibieron imatinib. Se desconocen los efectos a largo plazo del tratamiento prolongado con imatinib en el crecimiento de los niños. Por consiguiente, se recomienda la vigilancia estrecha del crecimiento en los niños que reciben imatinib (Ver Reacciones adversas). Interacciones con otros medicamentos y otras formas de interacción: Fármacos que pueden alterar las concentraciones plasmáticas del imatinib: Fármacos que pueden aumentar las concentraciones plasmáticas de imatinib: Las sustancias que inhiben la actividad del citocromo P450, isoenzima CYP3A4 (por ejemplo: ketoconazol, itraconazol, eritromicina, claritromicina) podrían reducir el metabolismo y aumentar las concentraciones de imatinib. Hubo un aumento significativo en la exposición a imatinib (la Cmáx y el área bajo la curva medias de imatinib aumentaron en un 26% y 40% respectivamente) en sujetos sanos cuando se administró conjuntamente con una dosis única de ketoconazol (un inhibidor de la CYP3A4). Deberá tenerse precaución cuando se administre Glivec® con inhibidores de la familia de la CYP3A4. Fármacos que pueden reducir las concentraciones plasmáticas de imatinib: Las sustancias que son inductoras de la actividad de CYP3A4 por ejemplo: dexametasona, fenitoína, carbamazepina, rifampicina, fenobarbital, o hierba de San Juan [Hypericum perforatum] pueden reducir significativamente la exposición a Glivec®. El pretratamiento de 14 voluntarios sanos con dosis múltiples de 600 mg diarios de rifampicina durante 8 días, seguido de una dosis única de 400 mg de Glivec®, incrementó el clearance de la dosis oral de Glivec® en 3,8 veces (intervalo de confianza 90%=3,5 a 4,3 veces), que representa descensos promedio de Cmáx, ABC(0-24) y ABC(0-inf) en 54%, 68% y 74% de los respectivos valores sin tratamiento de rifampicina. Similares resultados fueron observados en pacientes con gliomas tratados con Glivec® mientras se medicaban con drogas antiepilépticas inductoras de enzimas tales como carbamazepina, oxacarbazepina, fenitoína, fosfenitoína, fenobarbital y primidona. El ABC plasmático para imatinib decrece al 73% comparado con pacientes no tratados con drogas antiepilépticas inductoras de enzimas. En dos estudios publicados, la administración concomitante de imatinib y un producto que contiene hierba de San Juan derivó en una reducción del 30-32% del ABC de Glivec® En pacientes donde está indicado el uso de rifampicina u otros inductores de CYP3A4, deberá ser considerado el uso de agentes terapéuticos alternativos con menor potencial de inducción enzimática. Fármacos a los que Glivec® puede alterar su concentración plasmática: Imatinib aumenta la Cmáx y ABC media de simvastatina (sustrato de CYP3A4) 2 y 3,5 veces respectivamente, lo que indica una inhibición del CYP3A4 por imatinib. Por lo tanto, se recomienda precaución cuando se administre Glivec® con sustratos de la CYP3A4 con ventana terapéutica estrecha (por ejemplo: ciclosporina o pimozida). Glivec® puede incrementar la concentración plasmática de otras drogas metabolizadas por la CYP3A4 (por ejemplo: triazol-benzodiacepinas, bloqueantes dihidropiridínicos del canal cálcico, ciertos inhibidores de la HMG-CoA reductasa como las estatinas, etc.). Imatinib también inhibe la actividad in vitro de CYP2C9 y CYP2C19. Se observó una prolongación del tiempo de protrombina luego de la co-administración con warfarina. Cuando se administran cumarinas, se hace necesario el seguimiento controlado a tiempos cortos del tiempo de protrombina al comienzo y finalización del tratamiento con Glivec® y cuando se modifica la dosificación. Alternativamente, deberá ser considerado el uso de heparina de bajo peso molecular. Glivec® inhibe in vitro la actividad del citocromo P450, isoenzima CYP2D6 a concentraciones similares a las que afectan la actividad del CYP3A4. Imatinib a una dosis de 400 mg dos veces al día tuvo un efecto inhibitorio moderado sobre metabolismo del metoprolol mediado por el CYP2D6, aumentando la Cmáx y el ABC del metoprolol en aproximadamente 23%. La coadministración de imatinib con sustratos del CYP2D6 como el metoprolol, no parece ser un factor de riesgo para la interacción farmacológica por lo que no sería necesario ajustar la dosis. In vitro, Glivec® inhibe la O-glucuronidación del paracetamol o acetaminofeno (valor de Ki igual a 58,5 mmol/L con concentraciones terapéuticas). Embarazo y lactancia: Embarazo: No existen datos adecuados sobre la utilización de imatinib en mujeres embarazadas. Sin embargo, los estudios en animales han mostrado toxicidad reproductiva (ver Datos de toxicidad preclínica) y se desconoce el riesgo potencial para el feto. Glivec® no debe utilizarse durante el embarazo excepto si fuese estrictamente necesario. Si se utiliza durante el embarazo, la paciente ha de ser informada del riesgo potencial para el feto. Hubo reportes post-marketing de abortos espontáneos y anormalidades congénitas en infantes de mujeres que habían tomado Glivec®. Mujeres en edad de procrear: Debe aconsejarse a las mujeres en edad fértil para que utilicen un método anticonceptivo altamente eficaz durante el tratamiento. Un anticonceptivo altamente eficaz es un método el cual resulta en una tasa baja de falla (por ejemplo menos de 1% al año) cuando se utiliza consistentemente y correctamente. Lactancia: Tanto imatinib como su metabolito activo pueden distribuirse en la leche humana. El gradiente leche plasma fue determinado en 0,5 para imatinib y 0,9 para su metabolito, sugiriendo una mayor distribución del metabolito en la leche. Considerando la combinación de concentración de imatinib y su metabolito y el máximo de ingesta diaria de leche de un infante, es de esperar que la exposición total sea baja (~10% de la dosis terapéutica). Sin embargo, como los efectos de una exposición a dosis bajas en los infantes es desconocida, las mujeres que se encuentren bajo tratamiento con Glivec® no pueden amamantar. Fertilidad: No han sido realizados estudios humanos en pacientes masculinos que habían recibido Glivec® para determina su efecto en la fertilidad masculina y la espermatogénesis. Los pacientes preocupados acerca de su fertilidad durante el tratamiento con Glivec® deben consultar a su médico. Efectos sobre la capacidad para conducir y utilizar máquinas: Informes de accidentes con vehículos a motor han sido informados en pacientes tratados con Glivec®. Mientras que la mayoría de estos informes no son sospechados de ser producidos por Glivec®, los pacientes deberán ser informados de que pueden experimentar efectos indeseados tales como mareos, visión borrosa o somnolencia durante el tratamiento con Glivec®. Se recomienda precaución al conducir un automóvil u operar maquinarias.
Advertencias.
Cuando Glivec® es administrado con otras medicaciones, hay potencial de interacción medicamentosa. Se debe tener precaución cuando se administra Glivec® con rifampicina y otros inductores potentes de CYP3A4, ketoconazol u otros inhibidores potentes del CYP3A4, sustratos de CYP3A4 con una ventana terapéutica estrecha (por ejemplo ciclosporina, pimozida) o sustratos de CYP2C9 con una ventana terapéutica estrecha (por ejemplo warfarina y los derivados de cumarinas). Un paciente que estaba tomando paracetamol regularmente por fiebre, murió por insuficiencia hepática aguda. Si bien la etiología es desconocida actualmente, debe tenerse precaución, especialmente cuando se usa el paracetamol. Hipotiroidismo: Se han notificado casos clínicos de hipotiroidismo en pacientes tiroidectomizados que recibieron una terapia sustitutiva con levotiroxina durante el tratamiento con Glivec®. En estos pacientes deben vigilarse de cerca las concentraciones de TSH. Hepatotoxicidad: Si la función hepática está alterada, puede esperarse que la exposición a Glivec® esté aumentada. Glivec® deberá ser utilizado con precaución en pacientes con disfunción hepática. En estos casos, los recuentos de sangre periférica y las enzimas hepáticas deben ser cuidadosamente controladas. Al combinar Glivec® con regímenes de quimioterapia agresiva, se ha observado toxicidad hepática transitoria que se manifiesta por elevación de las transaminasas e hiperbilirrubinemia. Además, se han notificado casos infrecuentes de insuficiencia hepática aguda. Se recomienda vigilar la función hepática al combinar imatinib con regímenes de quimioterapia que pueden provocar disfunción hepática (ver Reacciones adversas). Retención de líquidos: Se han notificado ocasionalmente casos de sobrecarga hídrica severa (derrame pleural, edemas, edema pulmonar, ascitis, edema superficial) en aproximadamente 2,5% de los pacientes con LMC recientemente diagnosticada tratados con Glivec®, por lo que se recomienda pesar de forma regular a los pacientes. Deberá estudiarse cuidadosamente cualquier aumento rápido e inesperado de peso y si se considerara necesario, deberán llevarse a cabo medidas terapéuticas y de soporte adecuadas. En los estudios clínicos hubo un aumento de la incidencia de estos eventos en los pacientes de edad avanzada y en aquéllos con historia previa de enfermedad cardíaca. Pacientes con falla cardiaca o renal: Pacientes con enfermedades cardiacas o con factores de riesgo de falla cardíaca o historia de falla renal deberán ser monitoreados cuidadosamente, y cualquier paciente con signos o síntomas relacionados con falla cardíaca o renal debe ser evaluado y tratado. En los pacientes con síndrome hipereosinofílico (SHE) y afectación cardíaca, el inicio del tratamiento con imatinib se ha asociado con casos aislados de shock cardiogénico / disfunción ventricular izquierda. Se ha informado que esta reacción es reversible con la administración de corticoesteroides sistémicos, medidas complementarias para sostener la circulación y la interrupción temporal del tratamiento con imatinib. Los trastornos mielodisplásicos / mieloproliferativos y la mastocitosis sistémica podrían asociarse con concentraciones elevadas de eosinófilos. Por lo tanto, debe considerarse la posibilidad de efectuar un ecocardiograma y determinar las concentraciones séricas de troponina en los pacientes con SHE o LEC y en los pacientes con SMD/TMP o MS asociados con concentraciones elevadas de eosinófilos. Si alguno de estos análisis resulta anormal, debe considerarse el uso profiláctico de corticoesteroides sistémicos (1-2 mg/kg) durante un período de una a dos semanas al principio del tratamiento con imatinib. Hemorragia gastrointestinal: En los ensayos de Fase III sobre los TEGI/GIST malignos de carácter irresecable o metastásico, 211 pacientes (12,9%) presentaron hemorragia grado 3/4 en algún sitio. En el ensayo de Fase II sobre los TEGI/ GIST malignos de carácter irresecable o metastásico (ensayo B2222) se registraron hemorragias gastrointestinales en ocho pacientes (5,4%) y hemorragias en la zona de metástasis tumorales en cuatro de ellos (2,7%). Las hemorragias tumorales eran intraabdominales o intrahepáticas según la localización anatómica de las lesiones tumorales. La ubicación gastrointestinal del tumor pudo haber sido la causa de las hemorragias gastrointestinales en esta población de pacientes. Los pacientes deben deben ser monitoreados para controlar los síntomas gastrointestinales al comienzo de la terapia (ver Reacciones adversas). Síndrome de lisis tumoral: Se han reportado casos de síndrome de lisis tumoral (SLT) en pacientes tratados con Glivec®. Debido a la posible ocurrencia de SLT, la corrección de la deshidratación clínicamente significante y el tratamiento de niveles de ácido úrico elevados son recomendados antes de comenzar el tratamiento con Glivec®.
Conservación.
Conservar a menos de 30° C. Proteger de la humedad. No usar Glivec® después de la fecha de vencimiento que figura en el envase. Conservar Glivec® en el envase original. No utilizar ningún envase de Glivec® si se observara que estuviera dañado o mostrara signos de deterioro.
Sobredosificación.
Es limitada la experiencia con dosis mayores de Glivec®. Casos aislados de sobredosis de Glivec® fueron reportados espontáneamente y en la literatura. Generalmente el reporte de evolución fue mejoría o recuperación en estos casos. En el caso de sobredosis el paciente debe ser observado y se le administrará el tratamiento sintomático apropiado. Los eventos que han sido reportados en diferentes rangos de dosis fueron: Sobredosis en Adultos: 1200 a 1600 mg (la duración varía en 1 a 10 días): náuseas, vómitos, diarrea, rash, eritema, edema, tumefacción, fatiga, espasmos musculares, trombocitopenia, pancitopenia, dolor abdominal, cefalea, pérdida del apetito. 1800 a 3200 mg (hasta 3200 mg diarios por 6 días): debilidad, mialgias, incremento de CPK, incremento de bilirrubina, dolor gastrointestinal. 6400 mg (dosis única): un caso en la literatura reportado, un paciente que experimentó náuseas, vómitos, dolor abdominal, pirexia, tumefacción facial, reducción del recuento de neutrófilos, aumento de las transaminasas. 8 a 10 g (dosis única): se reportaron vómitos y dolor gastrointestinal. Sobredosis pediátrica: un individuo masculino de 3 años expuesto a una dosis única de 400 mg experimentó vómitos, diarrea y anorexia y otro varón de 3 años con una dosis única de 980 mg experimentó reducción del recuento de glóbulos blancos y diarrea. Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Ricardo Gutiérrez: (011) 4962 6666 / 2247; Hospital A. Posadas: (011) 4654 6648, (011) 4658 7777.
Información al paciente.
Lea todo el prospecto detenidamente antes de empezar a tomar Glivec®. Conserve este prospecto, puede tener que volver a leerlo. Si tiene alguna duda, consulte a su médico. Este medicamento ha sido recetado solo para usted. No se lo administre a otra persona ni lo utilice para tratar otra enfermedad. ¿QUE ES GLIVEC® Y PARA QUE SE UTILIZA?: Glivec® es un medicamento para tratar el cáncer. Contiene la sustancia activa imatinib. Glivec® está indicado para el: Tratamiento de pacientes adultos y niños con leucemia mieloide crónica (LMC): La leucemia es un tipo de cáncer de los glóbulos blancos. Los glóbulos blancos o leucocitos ayudan al control de las infecciones. La leucemia mieloide crónica es una forma de leucemia en las que ciertos leucocitos anormales (llamadas células mieloides) crecen sin control. Glivec® inactiva una enzima llamada Bcr - Abl tirosina kinasa, que es decisiva en la aparición y evolución de la LMC. Por lo tanto, Glivec® bloquea los procesos celulares que causan que la médula ósea normal pase a ser maligna e inhibe el crecimiento de las células leucémicas. Leucemia linfoblástica aguda con positividad del cromosoma Filadelfia (LLA Ph+ en pacientes adultos): La leucemia es un cáncer de los glóbulos blancos, en la leucemia linfoblástica aguda Ph+ Glivec® bloquea los procesos celulares desencadenados por la tirosina kinasa Bcr-Abl inhibiendo los glóbulos blancos anormales (denominados linfoblastos). Tratamiento de pacientes con neoplasia de estroma gastrointestinal (TEGI/GIST): Los TEGI/GISTs son neoplasias que afectan el estómago y los intestinos, que se originan a partir del crecimiento descontrolado de células de los tejidos de soporte de estos órganos. Glivec® inhibe el crecimiento de estas células. Dermatofibrosarcoma protuberans (DFSP): El DFSP es un cáncer del tejido subcutáneo en el que determinadas células empiezan a desarrollarse de manera descontrolada. Glivec® inhibe el crecimiento de estas células. Mastocitosis sitémica (MS): La mastocitosis sistémica es una enfermedad en la que unas células de la sangre llamadas mastocitos empiezan a proliferar y crecer fuera de los mecanismos de control normales. Síndrome Hipereosinofílico y/o Leucemia Eosinofílica crónica (SHE/ LEC): El síndrome hipereosinofílico y la leucemia eosinofílica crónica son enfermedades en las que unas células de la sangre llamadas eosinófilos empiezan a proliferar y crecer fuera de los mecanismos de control normales. Síndrome Mielodisplásico/Enfermedad mieloproliferativa (SMD/EMP): El síndrome mielodisplásico/enfermedad mieloproliferativa es una enfermedad en la que unas células de la sangre empiezan a proliferar y crecer fuera de los mecanismos de control normales. ¿CÓMO FUNCIONA GLIVEC®?: Glivec® detiene la producción de células anormales en las enfermedades listadas arriba. Consulte a su médico si tiene alguna duda de por qué se le ha recetado este medicamento. MONITOREO DURANTE EL TRATAMIENTO CON GLIVEC®: Su médico monitoreará regularmente su condición para verificar si Glivec® está teniendo el efecto deseado. También se le realizarán exámenes de sangre regulares para ver si Glivec® es tolerado (por ejemplo células de la sangre, función del hígado, función tiroidea). Usted será pesado regularmente mientras tome Glivec®. ANTES DE TOMAR GLIVEC®: Su tratamiento con Glivec® solo será prescripto por un doctor con experiencia en terapias contra el cáncer. Siga cuidadosamente todas las instrucciones que le haya dado su médico. Estas pueden ser diferentes de las contenidas en este prospecto. Lea las siguientes instrucciones antes de usar Glivec®. No debe tomar Glivec®: Si es alérgico (hipersensible) a imatinib o a cualquiera de los demás componentes de Glivec®, detallados al principio de este prospecto. Consulte a su médico si usted cree que puede ser alérgico. Tenga especial cuidado con Glivec®: Si usted tiene o ha tenido algún problema de hígado, riñón o cardíaco. Si usted está o cree que puede estar embarazada (ver a continuación). Si usted está amamantando (ver a continuación). Si está en tratamiento con levotiroxina secundario a extirpación de tiroides. Si usted esta recibiendo tratamiento con otros medicamentos, en particular los medicamentos listados en Toma de otros medicamentos. Informe a su médico inmediatamente si usted experimenta algunos de los siguientes efectos durante el tratamiento con Glivec®: Ganó peso rápidamente, hinchazón de las extremidades (muñecas, tobillos), hinchazón generalizado como hinchazón de la cara (signos de retención de fluidos). Debilidad, sangrado espontáneo o moretones, infecciones frecuentes con signos como fiebre, escalofríos, dolor de garganta o úlceras bucales (signos de nivel bajo de células de sangre). Dolor abdominal severo, vómito con sangre, materia fecal con sangre o materia fecal oscura (signos de desórdenes gastroinstestinales). Náuseas, falta de aire, ritmo cardíaco irregular, orina turbia, cansancio y/o disconfort de las articulaciones asociado con valores de laboratorio anormales (como niveles altos de potasio, ácido úrico y fósforo y niveles bajos de calcio en la sangre). Uso en personas de edad avanzada (65 años de edad y más): Glivec® puede ser utilizado en personas mayores de 65 años. Uso en niños y adolescentes (menos de 18 años de edad): Glivec® es un tratamiento adecuado para niños y adolescentes con LMC. Se carece de experiencia sobre el uso de Glivec® en niños menores de 2 años. Algunos niños y adolescentes que tomen Glivec® pueden tener retraso en el crecimiento. El doctor monitoreará el crecimiento en visitar regulares. Embarazo: Glivec® no está recomendado durante el embarazo ya que puede dañar al bebe. Si usted está embarazada o piensa que puede estarlo informe a su médico. Su médico discutirá con usted los riesgos potencial de utilizar Glivec® durante el embarazo. Lactancia: Informe a su médico si está amamantando. No debe amamantar durante el tratamiento con Glivec®. Mujeres con potencial de procreación: Las mujeres que pueden quedar embarazadas deben utilizar un método anticonceptivo efectivo mientras toman Glivec®. Conducción y uso de máquinas: En caso de que usted experimente eventos adversos, tales como mareos, somnolencia y visión borrosa, deberá tener especial cuidado en la conducción o uso de máquinas. Toma de otros medicamentos: Glivec® puede interferir con otros medicamentos. Informe a su médico o farmacéutico si está tomando o ha tomado recientemente otros medicamentos, incluso los que no le han sido recetados por un médico, incluidos: Algunos medicamentos indicados para control de ciertas infecciones como ketoconazol, itraconazol, eritromicina o claritromicina. Algunos medicamentes utilizados en epilepsia como carbamazepina, oxcarbazepina, fenobarbital, fenitoína, fofenitoína o primidona. Algunos medicamentos para reducir el colesterol como simvastatina. Algunos medicamentos utilizados para controlar desórdenes mentales como ser benzodiazepina o pimozida. Algunos medicamentes para control de la presión arterial elevada o desórdenes cardíacos como los bloqueantes de canales de calcio o metoprolol. Rifampicina, droga utilizada para tratar la tuberculosis. Hierba de San Juan, hierba medicinal utilizada en el tratamiento de depresión y otras condiciones. Dexametasona, medicina antiinflamatoria. Ciclosporina, medicina inmunosupresora. Paracetamol, medicina para control del dolor y de la fiebre. Warfarina, medicina para tratar trastornos de la coagulación (trombos y coágulos). Estos medicamentos deben evitarse durante el tratamiento con Glivec®. Si se encuentra en tratamiento con alguno de ellos antes de iniciar el tratamiento con Glivec®, su médico puede ofrecerle alguna alternativa. Avísele a su médico sobre cualquier nueva medicina que se le indique durante el tratamiento con Glivec®. ¿COMO DEBE SER ADMINISTRADO GLIVEC®?: Siga exactamente las instrucciones que le dé su médico. No sobrepase la dosis recomendada. ¿QUE CANTIDAD TOMAR?: Adultos: Su médico le indicará cuántos comprimidos de Glivec® debe tomar. Si Usted está siendo tratado por leucemia mieloide crónica, la dosis inicial habitual administrada una vez al día por boca es de: 1 comprimido de 400 mg (400 mg de Glivec®) o 1 comprimido de 400 mg más 2 comprimidos de 100 mg cada uno (600 mg de Glivec®). Si usted está siendo tratado por TEGI/GIST, la dosis inicial es de: 1 comprimido de 400 mg (400 mg de Glivec®), administrado una vez por día por boca. Para aquellos pacientes con LMC o TEGI/GIST, su doctor puede prescribirle una dosis mayor o menor dependiendo de cómo responda al tratamiento. Si su dosis diaria es de 800 mg debe tomar una dosis de 400 mg (1 comprimido) en la mañana y otra dosis en la tarde. Si padece LLA Ph+: la dosis inicial es de 600 mg en forma de un comprimido de 400 mg más 2 comprimidos de 100 mg una vez al día. Si padece DFSP: la dosis inicial es de 800 mg diarios, en forma de un comprimido de 400 mg en la mañana y otro comprimido de 400 mg en la noche. Si padece MS: la dosis inicial es de 400 mg, en una dosis diaria de un comprimido de 400 mg al día. Para algunos pacientes (con MS asociada a eosinofilia) la dosis inicial es de 100 mg diarios (1 comprimido de 100 mg/día). Su doctor puede decidir incrementar la dosis a 400 mg por día dependiendo de la respuesta al tratamiento. Si padece SHE / LEC: La dosis usual de inicio de tratamiento es de 400 mg por día, como una única dosis diaria. En pacientes con SHE / LEC con FIPL1-PDGFR a fusión kinasa la dosis inicial es de 100 mg diarios (1 comprimido de 100 mg/día). Su doctor puede decidir aumentar la dosis a 400 mg una vez al día, dependiendo de la respuesta terapéutica que usted presente. Si padece (SMD/EMP): La dosis inicial es de 400 mg en forma de un comprimido de 400 mg por boca. Niños: El médico le indicará cuántos comprimidos de Glivec® le administrará al niño. La cantidad de Glivec® a ser administrada dependerá del estado en que se encuentra el niño, de su tamaño corporal y de su altura. La dosis diaria total a ser administrada a un niño no podrá exceder a 1 comprimido de 400mg más 2 comprimidos de 100mg (600mg de Glivec®), administrados en una única dosis diaria. También se puede dividir la dosis diaria total en dos tomas, una de 400 mg en la mañana y otra de 200 mg (2 comprimidos de 100 mg) a la noche. El tratamiento puede ser administrado como una dosis diaria o alternativamente la dosis diaria puede ser dividida en dos administraciones (una por la mañana y otra por la tarde). ¿COMO TOMAR GLIVEC®?: Debe tomar Glivec® con alimentos, esto ayudará a proteger su estómago. Trague los comprimidos enteros con un vaso grande con agua. Para los casos en que los pacientes tuvieran dificultades para tragar, podrán dispersarse los comprimidos en un vaso grande con agua no gasificada o con jugo de manzana. Para ello: Coloque la cantidad requerida de comprimido/s en suficiente cantidad de líquido (aproximadamente 50 mL para un comprimido de 100 mg y de 200 mL para un comprimido de 400 mg). Revuelva con una cuchara hasta la completa desintegración del/de los comprimido/s. De inmediato ingiera el contenido total del vaso. Usted puede dejar trazas del/de los comprimido/s en el vaso después de la ingesta. ¿Cuándo y durante cuánto tiempo tomar Glivec®?: Su médico determinará durante cuánto tiempo deberá tomar Glivec®. Asegúrese de que toma Glivec® durante el tiempo que su médico le indicó. Si usted toma más Glivec® del que debiera: Si piensa que puede haber tomado accidentalmente más Glivec® del que debiera, hable de inmediato con su médico. Pudiera requerir atención médica. POSIBLES EFECTOS ADVERSOS: Al igual que todos los medicamentos, Glivec® puede tener efectos adversos. Estos son normalmente de leves a moderados. No se alarme por la lista de posibles efectos adversos: usted puede no experimentar ninguno. Los efectos adversos pueden ocurrir con ciertas frecuencias las cuales se definen a continuación: Muy frecuentes: afectan a más de 10 por cada 100 pacientes. Frecuentes: afectan entre 1 y 10 por cada 100 pacientes. Infrecuentes: afectan entre 1 y 10 cada 10000 pacientes. Raros: afectan entre 1 y 10 cada 10000 pacientes. Muy raros: afectan menos de 1 cada 10000 pacientes. Algunos efectos pueden ser serios: Informe a su médico inmediatamente si usted experimenta alguno de los eventos adversos listados a continuación: Los siguientes efectos adversos son comunes o muy comunes: Rápido aumento de peso, edema de extremidades, edema generalizado como inflamación de la cara (signo de retención de líquidos). Debilidad, sangrado espontáneo o aparición de moretones, infecciones frecuentes acompañadas de fiebre y escalofríos, dolor de garganta o úlceras en la boca (aftas) (signos de disminución del recuento de glóbulos blancos). Muy raros o infrecuentes: Palidez cutánea, cansancio, falta de aire, orina oscura (signos de glóbulos rojos bajos). Disminución de la visión, visión borrosa, sangre en el ojo. Dolor de pecho, temperatura, cansancio, ritmo irregular del corazón (signo de enfermedad cardiaca como ataque cardíaco, angina). Náuseas y vómitos, diarrea, dolor abdominal, fiebre (signo de enfermedad inflamatoria de los intestinos). Erupción severa, piel roja, ampollas en los labios, piel o boca, despellejamiento, fiebre, picazón, quemazón, aparición de manchas rojas o púrpuras, erupción pustular (signo de desorden cutáneo). Dolor de cadera o dificultad para caminar. Inflamación de la piel causada por una infección (celulitis). Dolor de cabeza severo, debilidad o parálisis de piernas, brazos o cara, dificultad para hablar, pérdida brusca de la conciencia (signos de enfermedad del sistema nervioso). Convulsiones. Sensación de cabeza liviana, mareo o desmayo (pueden ser signos de presión sanguínea baja). Dedos fríos o entumecidos (signo de síndrome de Raynaud). Dolor abdominal severo, vómito de sangre, materia fecal negra o con sangre. Náuseas, pérdida de apetito, orina oscura o color amarillo de la piel o los ojos (signo de enfermedad del hígado), hinchazón del abdomen/fluidos en el abdomen, constipación, dolor de estómago (signos de desórdenes gastrointestinales). Sed, pérdida de peso y disminución de la cantidad de orina. (Signos de poca ingestión de agua). Sangre en la orina. Edema y dolor en alguna parte del cuerpo. Tos, dificultad para respirar, respiración dolorosa, silibancias, dolor al respirar (signos de infecciones de pulmón/desórdenes). Debilidad muscular, espasmos musculares, ritmo cardiaco anormal (signos de cambio de los niveles de potasio en sangre). Espasmos musculares, fiebre, orina rojiza amarronada, desórdenes de riñón, debilidad y dolor muscular (signo de desórdenes musculares). Dolor pelviano, a veces acompañado de náuseas y vómitos, sangrado vaginal inesperado, mareos (signos de desórdenes ginecológicos). Reacciones alérgicas severas que pueden resultar en dificultad para respirar y mareos. Náuseas, falta de aire, ritmo cardíaco irregular, orina oscura, cansancio y/o disconfort en articulaciones asociado con resultados de laboratorio anormales (como niveles elevados de potasio, ácido úrico, fósforo y niveles bajos de calcio en sangre). Cefalea severa, mareos, visión borrosa (signos de presión aumentada en el cráneo). Si usted experimenta alguno de estos efectos adversos, avise a su médico: Algunos otros efectos adversos son muy comunes: Dolor de cabeza. Náuseas, vómitos, diarrea, indigestión, dolor abdominal. Urticaria con picazón, enrojecimiento y quemazón. Calambres musculares, dolor muscular y de los huesos. Hinchazón de los párpados o alrededor de los ojos. Fatiga. Aumento de peso. Algunos otros efectos adversos son comunes: Insomnio. Mareo. Cosquilleo, dolor o entumecimiento de las manos, pies, piernas o alrededor de la cadera. Problemas del gusto. Disminución de la sensibilidad de la piel. Lagrimeo con picazón, enrojecimiento e hinchazón (conjuntivitis), aumento de la producción de lágrimas, ojos secos. Oleadas de calor. Nariz sangrante. Boca seca. Hinchazón en el abdomen, flatulencia, diarrea, constipación, quemazón en el pecho, náuseas y dolor de estómago (signos de gastritis). Resultados anormales de análisis de función hepática. Piel seca. Picazón. Pérdida o adelgazamiento del cabello. Sudor nocturno. Aumento de la sensibilidad de la piel al sol (fotosensibilidad). Hinchazón de las articulaciones. Escalofríos. Pérdida de peso. Pérdida de apetito. Ulceras en la boca. Debilidad. Fiebre. Otros efectos adversos son infrecuentes: Enrojecimiento y/o inflamación en las palmas de las manos y plantas de los pies que pueden acompañarse de sensación de adormecimiento y dolor quemante. Infección respiratoria superior produciendo tos. Nódulos de linfas agrandados. Congestión nasal. Estornudos. Dolor de garganta. Cefalea. Presión facial o estornudos. Cefalea severa acompañada por náuseas. Vómitos y sensibilidad a la luz (signos de migraña). Síntomas de gripe. Infección del tracto urinario. Articulaciones dolorosas y hinchadas. Depresión. Ansiedad. Somnolencia. Temblor. Desórdenes de la memoria. Necesidad de mover una parte del cuerpo (comúnmente la pierna) para detener sensaciones inconfortables. Irritación del ojo. Dolor del ojo o enrojecimiento. Hinchazón o picazón de los parpados. Mareos. Dificultad para oír. Ruido en el oído. Ritmo cardíaco acelerado. Hipertensión. Frio periférico. Inflamación de los labios. Dificultad para tragar. Sudoración incrementada. Decoloración de la piel. Uñas decoloradas. Uñas rotas de la mano o el pie. Inflamación de los folículos del pelo. Enrojecimiento alrededor de los codos o rodillas. Oscurecimiento de la piel. Crecimiento de los senos en hombres y mujeres. Edema en los testículos. Problemas de erección. Períodos menstruales irregulares y pesados. Desórdenes sexuales. Deseo sexual disminuido. Dolor en los pezones. Dolor de pecho. Malestar. Infección viral como resfrío. Infección del tracto respiratorio superior que incluye la nariz (sinusitis). Dolor de garganta. Dedos fríos (síndrome de Raynaud). Dolor de espalda resultante de desórdenes de hígado. Orinar más frecuentemente. Aumento del apetito. Ulcera en el estómago. Rigidez de articulaciones y músculos. Resultados de laboratorio anormales. Confusión. Retraso en el crecimiento en niños y adolescentes. Consulte a su médico si experimenta cualquiera de los efectos anteriormente mencionados.
Presentación.
100 mg: Envases con 180 comprimidos recubiertos divisibles. 400 mg: Envases con 30 comprimidos recubiertos no divisibles.
Revisión.
14/07/2011.