GENVOYA®
GADOR
Grupo farmacoterapéutico: Antivirales para uso sistémico; antivirales para el tratamiento de infecciones por VIH y combinaciones. Código ATC: J05AR18.
Composición.
Cada comprimido recubierto de GENVOYA® contiene: Elvitegravir 150mg, Cobicistat (en dióxido de silicio) 150mg, Emtricitabina 200mg, Tenofovir alafenamida fumarato 11.2mg (equivalente a 10 mg de tenofovir alafenamida) Excipientes: Lactosa monohidrato, Celulosa microcristalina, Croscarmelosa sódica, Hidroxipropilcelulosa, Dióxido de silicio coloidal, Lauril sulfato de sodio, Estearato de magnesio, Alcohol polivinílico*, Dióxido de titanio*, Polietilenglicol 3350*, Talco*, Colorante FD&C azul N°2*, Óxido de hierro amarillo (CIN°77492)* c.s. * Se refiere a los componentes del Opadry II verde 85F110095. Excipientes con efecto conocido: GENVOYA® contiene lactosa monohidrato.
Farmacología.
Descripción: Los comprimidos de GENVOYA® contienen elvitegravir, cobicistat, emtricitabina y tenofovir alafenamida fumarato. Elvitegravir es un inhibidor de la transferencia de las cadenas de la integrasa del VIH-1. La integrasa es una enzima codificada por el VIH-1 necesaria para la replicación viral. Cobicistat es un inhibidor selectivo del mecanismo de las enzimas del citocromo P450 (CYP) de la subfamilia CYP3A. La inhibición del metabolismo mediado por CYP3A por parte de cobicistat potencia la exposición sistémica a los sustratos de CYP3A, como elvitegravir. Emtricitabina es un inhibidor análogo de nucleósidos de la transcriptasa inversa (INTI) y un análogo nucleósido de 2'-desoxicitidina. Tenofovir alafenamida es un inhibidor análogo de nucleótidos de la transcriptasa inversa (INtTI) y un profármaco fosfonoamidato de tenofovir (análogo de 2'-desoxiadenosina monofosfato). Los comprimidos de GENVOYA® son de color verde, en forma de cápsula, marcado en una de las caras del comprimido con "GSI" y en la otra cara del comprimido con "510". Los comprimidos de GENVOYA® se administran por vía oral. Cada comprimido contiene 150mg de elvitegravir, 150mg de cobicistat, 200mg de emtricitabina y tenofovir alafenamida fumarato equivalente a 10mg de tenofovir alafenamida. Los comprimidos también incluyen los siguientes excipientes: Lactosa monohidrato, Celulosa microcristalina, Croscarmelosa sódica, Hidroxipropilcelulosa, Dióxido de silicio, Lauril sulfato sódico, Estearato magnésico. Los comprimidos están recubiertos con Alcohol polivinílico, Dióxido de titanio, Polietilenglicol 3350, Talco, Colorante FD&C Blue N°2, Óxido de hierro amarillo. Farmacología clínica: Mecanismo de acción: Elvitegravir es un inhibidor de la transferencia de las cadenas de la integrasa del VIH-1. La integrasa es una enzima codificada por el VIH-1 necesaria para la replicación viral. La inhibición de la integrasa impide la integración del ácido desoxirribonucleico (ADN) del VIH-1 en el ADN genómico del huésped, con el consiguiente bloqueo de la formación de provirus del VIH-1 y de la propagación de la infección viral. Cobicistat es un inhibidor selectivo del mecanismo de las enzimas del citocromo P450 (CYP) de la subfamilia CYP3A. La inhibición del metabolismo mediado por CYP3A por parte de cobicistat potencia la exposición sistémica a los sustratos de CYP3A, como elvitegravir, cuya biodisponibilidad resulta limitada y su vida media acortada por el metabolismo dependiente de CYP3A. Emtricitabina es un inhibidor análogo de nucleósidos de la transcriptasa inversa (INTI) y un análogo nucleósido de 2'-desoxicitidina. Emtricitabina es fosforilada por enzimas celulares para formar emtricitabina trifosfato. Emtricitabina trifosfato inhibe la replicación del VIH a través de su incorporación en el ADN viral mediante la transcriptasa inversa (TI) del VIH, lo que produce la interrupción de la cadena de ADN. Emtricitabina muestra actividad frente al VIH-1, el VIH-2 y el VHB. Tenofovir alafenamida es un inhibidor análogo de nucleótidos de la transcriptasa inversa (INtTI) y un profármaco fosfonamidato de tenofovir (análogo de 2'-desoxiadenosina monofosfato). Tenofovir alafenamida es permeable en las células y, debido a su mayor estabilidad plasmática y activación intracelular mediante hidrólisis por la catepsina A, tenofovir alafenamida es más eficaz que tenofovir disoproxilo para concentrar tenofovir en las células mononucleares de sangre periférica (CMSP) (incluyendo linfocitos y otras células diana del VIH) y los macrófagos. Tenofovir intracelular es subsecuentemente fosforilado al metabolito farmacológicamente activo tenofovir difosfato. Tenofovir difosfato inhibe la replicación del VIH mediante su incorporación en el ADN viral por la TI del VIH, lo que produce la interrupción de la cadena de ADN. Tenofovir muestra actividad frente al VIH-1, el VIH-2 y el VHB. Propiedades farmacocinéticas: Absorción: Después de la administración oral con alimentos en pacientes infectados por el VIH-1, las concentraciones plasmáticas máximas se observaron aproximadamente 4 horas después de la dosis para elvitegravir, 3 horas después de la dosis para cobicistat, 3 horas después de la dosis para emtricitabina y 1 hora después de la dosis para tenofovir alafenamida. Los valores medios en estado estacionario de Cmáx, AUCtau y Cmín (media ± DE) en pacientes infectados por el VIH-1, respectivamente, fueron de 1,7 ± 0,39 mg/mL, 23 ± 7,5 mgh/mL y 0,45 ± 0,26 mg/mL para elvitegravir, lo que genera un cociente inhibitorio de aproximadamente 10 (relación Cmín: IC95 ajustado para unión a proteínas para el virus VIH-1 de tipo salvaje). Los valores medios correspondientes al estado estacionario de Cmáx, AUCtau, y Cmín (media ± DE) fueron de 1,1 ± 0,40 mg/mL, 8,3 ± 3,8 mgh/mL y 0,05 ± 0,13 mg/mL para cobicistat, 1,9 ± 0,5 mg/mL, 13 ± 4,5 mgh/mL y 0,14 ± 0,25 mg/mL para emtricitabina. Los valores medios correspondientes al estado estacionario de Cmáx y AUCtau para tenofovir alafenamida fueron de 0,16 ± 0,08 mg/mL y de 0,21 ± 0,15 mgh/mL, respectivamente. Para elvitegravir, la Cmáx y el AUC aumentaron en un 22% y un 36% con la comida ligera y en un 56% y un 91% con la comida de alto contenido graso, con respecto a las condiciones de ayuno. Los valores de exposición a cobicistat no resultaron afectados por la comida ligera y, aunque se produjo un escaso descenso del 24% y del 18% en la Cmáx y el AUC respectivamente con la comida de alto contenido graso, no se observaron diferencias en su efecto de potenciación farmacológica sobre elvitegravir. Los valores de exposición a emtricitabina no resultaron afectados por las comidas ligeras o de alto contenido graso. En cuanto a las condiciones de ayuno, la administración de GENVOYA® con una comida ligera (unas 400 kcal, 20% de grasa) o con una comida de alto contenido graso (unas 800 kcal, 50% de grasa) no afectó los valores globales de exposición a tenofovir alafenamida en un grado clínicamente relevante (aproximadamente un AUC un 15% y un 18% mayores con una comida ligera o de alto contenido graso, respectivamente, en comparación a las condiciones de ayuno). Distribución: Elvitegravir se une en un 98-99% a las proteínas plasmáticas humanas y la unión es independiente de la concentración del fármaco en el intervalo de 1 ng/mL a 1,6 mg/mL. La relación de concentración media del fármaco entre plasma y sangre fue de 1,37. Cobicistat se une en un 97-98% a las proteínas plasmáticas humanas y la relación de concentración media del fármaco entre plasma y sangre fue de 2. La unión in vitro de emtricitabina a proteínas plasmáticas fue < 4% y resultó independiente de la concentración en el intervalo de 0,02 a 200 mg/mL. A la concentración plasmática máxima, la relación de concentración media del fármaco entre plasma y sangre fue de aproximadamente 1,0 y la relación de concentración media del fármaco entre semen y plasma fue de aproximadamente 4,0. La unión in vitro de tenofovir a proteínas plasmáticas es < 0,7% y fue independiente de la concentración en el intervalo de 0,01-25 mg/ml. La unión ex vivo de tenofovir alafenamida a proteínas plasmáticas en las muestras recogidas durante los estudios clínicos fue de aproximadamente el 80%. Biotransformación: Elvitegravir sufre principalmente metabolismo oxidativo mediante CYP3A y secundariamente glucuronidación por las enzimas UGT1A1/3. Tras la administración oral de [14C]-elvitegravir potenciado, elvitegravir fue la sustancia predominante en el plasma, representando aproximadamente el 94% de la radiactividad circulante. Los metabolitos generados mediante hidroxilación aromática y alifática o glucuronidación están presentes en niveles muy bajos, demostrandouna actividad antiviral considerablemente menor contra el VIH-1, y no contribuyen a la actividad antiviral global de elvitegravir. Cobicistat se metaboliza mediante una oxidación mediada por CYP3A (mayor) y CYP2D6 (menor) y no sufre glucuronidación. Tras la administración oral de [14C]-cobicistat, el 99% de la radiactividad circulante en plasma correspondió a cobicistat en forma inalterada. Los estudios in vitro indican que emtricitabina no es un inhibidor de las enzimas CYP humanas. Tras la administración de [14C]-emtricitabina, se obtuvo una recuperación completa de la dosis de emtricitabina en la orina (aproximadamente el 86%) y las heces (aproximadamente el 14%). El 13% de la dosis se recuperó en la orina en forma de tres aparentes metabolitos. La biotransformación de emtricitabina comprende la oxidación del radical tiólico, para dar los diastereómeros 3'-sulfóxido (~ el 9% de la dosis), y la conjugación con el ácido glucurónico, para formar el 2'-O glucurónido (~ el 4% de la dosis). No hubo otros metabolitos identificables. El metabolismo es la ruta de eliminación principal de tenofovir alafenamida en los seres humanos, suponiendo > 80% de una dosis oral. Los estudios in vitro han mostrado que tenofovir alafenamida se metaboliza a tenofovir (metabolito principal) por medio de la catepsina A en las CMSP (incluyendo linfocitos y otras células diana del VIH) y los macrófagos y por medio de la carboxilesterasa 1 en los hepatocitos. In vivo, tenofovir alafenamida se hidroliza en las células para formar tenofovir (metabolito principal), que es fosforilado al metabolito activo tenofovir difosfato. En los estudios clínicos humanos, una dosis oral de 10mg de tenofovir alafenamida en GENVOYA® dio lugar a concentraciones de tenofovir difosfato más de 4 veces superiores en las CMSP y concentraciones de tenofovir en plasma con reducción mayor al 90% en comparación con una dosis oral de 245mg de tenofovir disoproxil (en forma de fumarato) en E/C/F/TDF. In vitro, tenofovir alafenamida no es metabolizado por CYP1A2, CYP2C8, CYP2C9, CYP2C19 o CYP2D6. Tenofovir alafenamida es metabolizado mínimamente por CYP3A4. Cuando se administra de forma concomitante con el conocido inductor moderado de CYP3A efivarenz, la exposición a tenofovir alafenamida no se ve afectada significativamente. Después de la administración de tenofovir alafenamida, la radiactividad [14C] en plasma mostró un perfil dependiente del tiempo, siendo tenofovir alafenamida la especie más abundante en las primeras horas iniciales y el ácido úrico en el periodo restante. Eliminación: Tras la administración oral de [14C]-elvitegravir/ritonavir, el 94,8% de la dosis se recuperó en las heces, lo que concuerda con la excreción hepatobiliar de elvitegravir; el 6,7% de la dosis administrada se recuperó en la orina. La mediana de la vida media plasmática terminal de elvitegravir tras la administración de E/C/F/TDF es de aproximadamente 12,9 horas. Tras la administración oral de [14C]-cobicistat, el 86% y el 8,2% de la dosis se recuperaron en las heces y en la orina, respectivamente. La mediana de la vida media plasmática terminal de cobicistat tras la administración de E/C/F/TDF es de aproximadamente 3,5 horas y los valores de exposición a cobicistat asociados generan una Cmín de elvitegravir aproximadamente 10 veces mayor que el IC95 ajustado para unión a proteínas del virus VIH-1 de tipo salvaje. Emtricitabina se excreta fundamentalmente por el riñón y la dosis se recupera por completo en la orina (aproximadamente 86%) y en las heces (aproximadamente 14%). El trece por ciento de la dosis de emtricitabina se recoge en la orina en forma de tres metabolitos. El aclaramiento sistémico de emtricitabina alcanza un promedio de 307 mL/min. Después de la administración oral, la vida media de eliminación de emtricitabina es de aproximadamente 10 horas. La excreción renal de tenofovir alafenamida intacto es una ruta menor, con < 1% de la dosis eliminada por la orina. Tenofovir alafenamida se elimina principalmente después de la metabolización a tenofovir. Tenofovir alafenamida y tenofovir tienen una mediana de vida media plasmática de 0,51 y 32,37 horas, respectivamente. Tenofovir se elimina del organismo a través de los riñones por filtración glomerular y secreción tubular activa. Farmacocinética en poblaciones especiales: Edad, sexo y raza: No se ha identificado ninguna diferencia farmacocinética clínicamente relevante en relación con el sexo o la raza para elvitegravir potenciado con cobicistat, cobicistat, emtricitabina o tenofovir alafenamida. Las exposiciones a elvitegravir, cobicistat, emtricitabina, tenofovir y tenofovir alafenamida alcanzadas en 24 pacientes adolescentes de 12 a < 18 años que recibieron GENVOYA® en el estudio GS-US-292-0106 fueron similares a las exposiciones alcanzadas en adultos que nunca habían recibido tratamiento después de la administración de GENVOYA® (Tabla 1).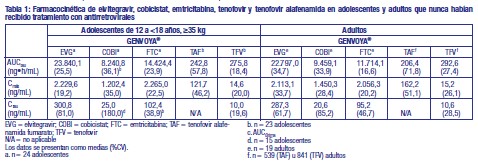
Las exposiciones medias a elvitegravir, cobicistat, emtricitabina, tenofovir y tenofovir alafenamida alcanzadas en niños de 8 a < 12 años ( > 25 kg; n = 23) que recibieron GENVOYA® en el estudio GS-US-292-0106 eran mayores (20 a 80%) que las exposiciones medias alcanzadas en adultos (Tabla 2).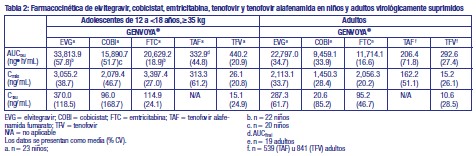
Insuficiencia renal: No se observaron diferencias clínicamente relevantes en la farmacocinética de elvitegravir, cobicistat, tenofovir alafenamida o tenofovir entre los individuos sanos y los pacientes con insuficiencia renal grave (ClCr estimado > 15 pero < 30 mL/min) en los estudios de elvitegravir potenciado con cobicistat o de tenofovir alafenamida, respectivamente. La exposición sistémica media a emtricitabina fue mayor en pacientes con insuficiencia renal grave (ClCr < 30 mL/ min) (33,7 mgh/mL) que en sujetos con función renal normal (11,8 mgh/mL). Insuficiencia hepática: Tanto elvitegravir como cobicistat se metabolizan y eliminan principalmente por vía hepática. Se realizó un estudio de la farmacocinética de elvitegravir potenciado con cobicistat en pacientes no infectados por el VIH-1 con insuficiencia hepática moderada (clase B de Child-Pugh). No se observaron diferencias clínicamente relevantes en la farmacocinética de elvitegravir o cobicistat entre los pacientes con insuficiencia hepáticamoderada y los individuos con función hepática normal. No se ha estudiado el efecto de la insuficiencia hepática grave (clase C de Child-Pugh) sobre la farmacocinética de elvitegravir o cobicistat. La farmacocinética de emtricitabina no se ha estudiado en pacientes con insuficiencia hepática; sin embargo, emtricitabina no sufre un metabolismo significativo a través de las enzimas hepáticas, por lo que la repercusión de la insuficiencia hepática debería ser escasa. No se observaron cambios clínicamente relevantes en la farmacocinética de tenofovir alafenamida o su metabolito tenofovir en los pacientes con insuficiencia hepática leve o moderada. En pacientes con insuficiencia hepática grave, las concentraciones plasmáticas totales de tenofovir alafenamida y tenofovir son más bajas que las observadas en sujetos con función hepática normal. Cuando se corrigen por la unión a proteínas, las concentraciones plasmáticas de tenofovir alafenamida no unido a proteínas (libre) son similares en pacientes con insuficiencia hepática grave y en sujetos con función hepática normal. Infección concomitante por el virus de la hepatitis B y/o de la hepatitis C: No se ha evaluado por completo la farmacocinética de emtricitabina y tenofovir alafenamida en los pacientes con infección concomitante por el virus de la hepatitis B y/o de la hepatitis C. Escasos datos procedentes deanálisis farmacocinéticos poblacionales (n = 24) indicaron que la infección concomitante por el virus de la hepatitis B y/o C no produce un efecto clínicamente relevante sobre la exposición a elvitegravir potenciado. Embarazo y posparto. Los resultados informados de un estudio prospectivo (IMPAACT P1026s) mostraron que el tratamiento con regímenes que contienen cobicistat y elvitegravir durante el embarazo da como resultado exposiciones más bajas de elvitegravir y cobicistat (Tabla 3).
Microbiología: Actividad antiviral in vitro: Elvitegravir, emtricitabina y tenofovir alafenamida demostraron actividad antiviral sinérgica en los cultivos celulares. La sinergia antiviral se mantuvo para elvitegravir, emtricitabina y tenofovir alafenamida cuando se analizaron en presencia de cobicistat. La actividad antiviral de elvitegravir frente a aislamientos clínicos y de laboratorio del VIH-1 se evaluó en células linfoblastoides, células monocíticas/macrofágicas y linfocitos de sangre periférica y los valores de la concentración efectiva al 50% (CE50) oscilaron entre 0,02 y 1,7 nM. Elvitegravir mostró actividad antiviral en cultivos celulares frente a los clados del VIH-1 A, B, C, D, E, F, G y O (con valores de CE50 de0,1 a 1,3 nM) y actividad frente al VIH-2 (CE50 de 0,53 nM). Cobicistat no presenta actividad antiviral detectable frente al VIH-1 y no antagoniza los efectos antivirales de elvitegravir, emtricitabina o tenofovir. La actividad antiviral de emtricitabina frente a aislamientos clínicos y de laboratorio del VIH-1 se evaluó en líneas celulares linfoblastoides, en la línea celular MAGI-CCR5 y en CMSP. Los valores de CE50 para emtricitabina oscilaron entre 0,0013 y 0,64 mM. Emtricitabina mostró actividad antiviral en cultivos celulares frente a los clados del VIH-1 A, B, C, D, E, F y G (con valores de CE50 de 0,007 a 0,075 mM) y presentó actividad específica de cepa frente al VIH-2 (con valores de CE50 de 0,007 a 1,5 mM). La actividad antiviral de tenofovir alafenamida frente a aislamientos clínicos y de laboratorio del subtipo B del VIH-1 se evaluó en líneas celulares linfoblastoides, CMSP, células monocíticas/macrofágicas primarias y linfocitosT CD4+. Los valores de la CE50 de tenofovir alafenamida oscilaron entre 2,0 y 14,7 nM. Tenofovir alafenamida mostró actividad antiviral en cultivos celulares frente a todos los grupos del VIH-1 (M, N y O), incluyendo los subtipos A, B, C, D, E, F y G (con valores de CE50 de 0,10 a 12,0 nM) y mostró actividad específica de cepa frente al VIH-2 (con valores de CE50 de 0,91 a 2,63 nM). Resistencia: In vitro: La sensibilidad reducida a elvitegravir se asocia con mayor frecuencia a las mutaciones primarias de la integrasa T66I, E92Q y Q148R. Las mutaciones adicionales de la integrasa observadas en la selección mediante cultivos celulares fueron H51Y, F121Y, S147G, S153Y, E157Q y R263K. El VIH-1 con las sustituciones seleccionadas por raltegravir, T66A/K, Q148H/K y N155H, mostró resistencia cruzada a elvitegravir. No se puede demostrar resistencia in vitro con cobicistat debido a su ausencia de actividad antiviral. La sensibilidad reducida a emtricitabina se asocia con mutaciones M184V/I en la TI del VIH-1. Los aislamientos del VIH-1 con sensibilidad reducida a tenofovir alafenamida expresan una mutación K65R en la TI del VIH-1; además, se ha observado de forma transitoria una mutación K70E en la TI del VIH-1. Los aislamientos del VIH-1 con la mutación K65R muestran una sensibilidad reducida de bajo nivel a abacavir, emtricitabina, tenofovir y lamivudina. Pacientes que no han recibido tratamiento previo: En un análisis combinado, se realizó la genotipificación en los aislamientos del VIH-1 del plasma de pacientes que no habían recibido tratamiento antirretroviral previo tratados con GENVOYA® en los estudios de fase 3GS-US-292-0104 y GS-US-292-0111 con ARN del VIH-1 - 400 copias/mL en el momento del fracaso virológico confirmado, en la semana 144 o en el momento en el que se interrumpió de forma temprana la medicación del estudio. Hasta la semana 144, el desarrollo de una o más mutaciones asociadas con resistencia primaria a elvitegravir, emtricitabina o tenofovir alafenamida fue observado en aislamientos del VIH-1 de 12 de 22 pacientes con datos genotípicos evaluables a partir de aislamientos emparejados al inicio y luego del fracaso del tratamiento con GENVOYA® (12 de 866 pacientes [1.4%]) en comparación con 12 de 20 aislamientos con fracaso al tratamiento en pacientes con datos genotípicos evaluables del grupo de tratamiento con E/C/F/TDF (12 de 867 pacientes [1,4%]). De los aislamientos del VIH-1 de 12 pacientes con desarrollo de resistencia en el grupo tratado con GENVOYA®, las mutaciones que surgieron fueron M184V/I (n = 11) y K65R/N (n = 2) en la TI y T66T/A/I/V (n = 2), E92Q (n = 4), Q148Q/R (n = y N155H (n = 2) en la integrasa. De los aislamientos del VIH-1 de 12 pacientes con desarrollo de resistencia en el grupo tratado con E/C/F/TDF, las mutaciones que surgieron fueron M184V/I (n = 9) y K65R/N (n = 4), yL210W 9n = 1) en la TI y E92Q/V (n = 4), Q148R (n = 2) y N155H/S (n = 3) en la integrasa. La mayoría de los aislamientos del VIH-1 de los pacientes de ambos grupos de tratamiento que desarrollaron mutaciones de resistencia a elvitegravir desarrollaron mutaciones de resistencia tanto a emtricitabina como a elvitegravir. En los análisis fenotípicos de los pacientes de la población de análisis de las resistencias final, 7 de 22 pacientes (32%) tenían aislamientos del VIH-1 con sensibilidad reducida a elvitegravir en el grupo tratado con GENVOYA® en comparación con los aislamientos del VIH-1 de 7 de 20 pacientes (35%) en el grupo tratado con E/C/F/TDF, y los aislamientos del VIH-1 de 8 pacientes (36%) tenían una sensibilidad reducida a emtricitabina en el grupo tratado con GENVOYA® en comparación con los aislamientos del VIH-1 de 7 pacientes (35%) del grupo tratado con E/C/F/TDF. Un paciente del grupo tratado con GENVOYA® (1 de 22 [4,5%]) y 2 pacientes del grupo tratado con E/C/F/TDF (2 de 20 [10%]) mostraron una sensibilidad reducida a tenofovir. En pacientes suprimidos virológicamente: Fueron identificados tres pacientes con resistencia de reciente aparición del VIH-1 a GENVOYA® fue identificado (M184M/I; MI84I+E92G; MI84V+E92Q) hasta la semana 96 en un ensayo clínico de pacientes virológicamente suprimidos que cambiaron desde una pauta que contenía emtricitabina/tenofovir disoproxil fumarato y un tercer fármaco (GS-US-292-0109, n = 959). En pacientes coinfectados por VIH y VHB: En un estudio clínico de pacientes con VIH virológicamente suprimidos coinfectados por hepatitis B crónica que recibieron GENVOYA® durante 48 semanas (GS-US-292-1249, n = 72), 2 pacientes calificaron para el análisis de resistencia. En estos 2 pacientes, no se identificaron sustituciones de aminoácidos asociadas con resistencia a cualquiera de los componentes de GENVOYA® en VIH-1 o VHB. Resistencia cruzada en pacientes infectados por el VIH-1, que no habían recibido tratamiento previo o suprimidos virológicamente: Los virus resistentes a elvitegravir muestran diferentes grados de resistencia cruzada al inhibidor de la transferencia de la cadena de la integrasa raltegravir, dependiendo del tipo y el número de mutaciones. Los virus que expresan las mutaciones T66I/A mantienen la sensibilidad a raltegravir, mientras que la mayoría de los demás patrones mostraron una sensibilidad reducida a raltegravir. Los virus que expresan mutaciones de resistencia a elvitegravir o raltegravir mantienen la sensibilidad a dolutegravir. Los virus resistentes a emtricitabina con la sustitución M184V/I mostraron resistencia cruzada con lamivudina, pero conservaron la sensibilidad a didanosina, estavudina, tenofovir y zidovudina. Las mutaciones K65R y K70E redundan en una sensibilidad reducida a abacavir, didanosina, lamivudina, emtricitabina y tenofovir, pero conservan la sensibilidad a zidovudina.
Indicaciones.
GENVOYA® está indicado para el tratamiento de la infección por el virus de la inmunodeficiencia humana tipo 1 (VIH-1) sin ninguna mutación conocida asociada con resistencia a los inhibidores de la integrasa, emtricitabina o tenofovir, como sigue: En adultos y adolescentes de 12 años de edad o mayores y con un peso corporal de al menos 35 kg. En niños de 6 años de edad o mayores y con un peso corporal de al menos 25 kg para los cuales regímenes alternativos no son adecuados debido a toxicidades. Ver secciones Farmacología clínica, Dosificación y Advertencias - Generales.
Dosificación.
El tratamiento debe ser iniciado por un médico con experiencia en el tratamiento de la infección por el VIH. Dosis recomendada: Adultos y pacientes pediátricos de 6 años de edad y mayores con un peso de al menos 25 kg: Un comprimido que se debe tomar una vez al día con alimentos. Si el paciente omite una dosis de GENVOYA® en el plazo de 18 horas desde el horario normal de administración, debe tomar GENVOYA® lo antes posible con alimentos y continuar la pauta habitual de administración. Si un paciente omite una dosis de GENVOYA® por más de 18 horas, no debe tomar la dosis omitida y simplemente debe continuar la pauta habitual de administración. Si el paciente vomita en el plazo de 1 hora después de tomar GENVOYA®, debe tomar otro comprimido. Pacientes de edad avanzada: No se requiere un ajuste de la dosis de GENVOYA® en pacientes de edad avanzada (ver secciones Propiedades farmacocinéticas y Estudios clínicos). Insuficiencia renal: No se requiere un ajuste de la dosis de GENVOYA® en adultos o adolescentes (de al menos 12 años de edad y al menos 35 kg de peso corporal) con un aclaramiento de creatinina estimado (ClCr) - 30 mL/min. No se dispone de datos para dar recomendaciones de posología en niños menores de 12 años con insuficiencia renal. GENVOYA® no se debe iniciar en pacientes con un ClCr estimado < 30 mL/min, ya que se dispone de datos limitados sobre el uso de GENVOYA® en esta población (ver secciones Propiedades farmacocinéticas y Estudios clínicos). El tratamiento con GENVOYA® se debe suspender en los pacientes cuyo ClCr estimado descienda por debajo de 30 mL/min durante el tratamiento (ver secciones Propiedades farmacocinéticas y Estudios clínicos). Insuficiencia hepática: No se requiere un ajuste de la dosis de GENVOYA® en pacientes con insuficiencia hepática leve (clase A de Child-Pugh) o moderada (clase B de Child-Pugh). No se ha estudiado GENVOYA® en pacientes con insuficiencia hepática grave (clase C de Child-Pugh); por lo tanto, no se recomienda el uso de GENVOYA® en pacientes con insuficiencia hepática grave (ver secciones Propiedades farmacocinéticas y Advertencias - Generales). Población pediátrica: No se ha establecido todavía la seguridad y eficacia de GENVOYA® en niños menores de 6 años de edad o que pesen < 25 kg. No se dispone de datos. Embarazo: El tratamiento con cobicistat y elvitegravir durante el embarazo da como resultado una menor exposición a elvitegravir (ver secciones Advertencias - Generales y Propiedades farmacocinéticas). Por lo tanto, la terapia con GENVOYA® no debe iniciarse durante el embarazo, y las mujeres que quedan embarazadas durante la terapia con GENVOYA® deben cambiarse a un régimen alternativo (ver secciones Advertencias - Generales e Interacciones - Uso en poblaciones específicas) Forma de administración: GENVOYA® se debe tomar por vía oral, una vez al día con alimentos (ver sección Propiedades farmacocinéticas). El comprimido recubierto con película no se debe masticar o triturar. Para pacientes que no pueden tragar el comprimido entero, el comprimido puede ser dividido en dos y se deben tomar las dos mitades una después de la otra, asegurándose que se tome la dosis completa. Formas farmacéuticas y concentraciones: Comprimido recubierto con película, de color verde, en forma de cápsula, de dimensiones 19 mm x 8,5 mm, marcado en una de las caras del comprimido con "GSI" y en la otra cara del comprimido con "510". Cada comprimido contiene 150mg de elvitegravir, 150mg de cobicistat, 200mg de emtricitabina y tenofovir alafenamida fumarato equivalente a 10mg de tenofovir alafenamida.
Contraindicaciones.
Hipersensibilidad a los principios activos o a alguno de los excipientes. La administración concomitante está contraindicada con los medicamentos que son altamente dependientes de CYP3A para la depuración y para los cuales concentraciones plasmáticas elevadas están asociadas con reacciones adversas graves o potencialmente mortales. Por lo tanto, GENVOYA® no debería ser administrado en forma concomitante con medicamentos que incluyen, pero no están limitados a, los siguientes (ver secciones Advertencias - Generales e Interacciones): antagonistas de los receptores adrenérgicos alfa 1: alfuzosina; antiarrítmicos: amiodarona, quinidina; derivados ergóticos: dihidroergotamina, ergometrina, ergotamina; fármacos estimulantes de la motilidad gastrointestinal: cisaprida; inhibidores de la HMG Co-A reductasa: lovastatina, simvastatina; neurolépticos/antipsicóticos: pimozida, lurasidona; inhibidores de la PDE-5: sildenafilo para el tratamiento de la hipertensión arterial pulmonar; sedantes/hipnóticos: midazolam administrado por vía oral, triazolam. Está contraindicada la administración concomitante con medicamentos que son fuertes inductores de CYP3A debido al potencial para la pérdida de la respuesta virológica y posible resistencia a GENVOYA®. Por lo tanto, GENVOYA® no debería ser administrado en forma concomitante con medicamentos que incluyen, pero no están limitados a, los siguientes (ver secciones Advertencias - Generales e Interacciones): antiepilépticos: carbamazepina, fenobarbital, fenitoína; antimicobacterianos: rifampicina; medicamentos a base de plantas: hierba de San Juan (Hypericum perforatum). La administración concomitante con dabigatrán etexilato, un sustrato de la glicoproteína P (P-gp) está contraindicada (ver sección Interacciones).
Reacciones adversas.
Resumen del perfil de seguridad: La evaluación de las reacciones adversas se basa en los datos de seguridad de todos los estudios de fase 2 y 3 en los que 2.396 pacientes recibieron GENVOYA® y por experiencia postcomercial. Las reacciones adversas notificadas con mayor frecuencia en los estudios clínicos a lo largo de 144 semanas fueron náuseas (11%), diarrea (7%) y cefalea (6%) (datos combinados de los estudios clínicos de fase 3 GS-US-292-0104 y GS-US-292-0111 en 866 pacientes adultos sin tratamiento previo que recibieron GENVOYA®). Tabla de reacciones adversas: Las reacciones adversas de la Tabla 7 se muestran según el sistema de clasificación de órganos y frecuencia. Las frecuencias se definen como sigue: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10) y poco frecuentes (≥ 1/1.000 a < 1/100).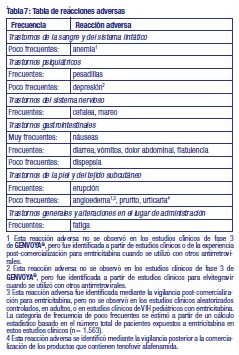
Descripción de las reacciones adversas seleccionadas: Parámetros metabólicos: El peso y los niveles de glucosa y lípidos en la sangre pueden aumentar durante el tratamiento antirretroviral (ver sección Advertencias - Generales). Síndrome de reconstitución inmune: Al inicio de la TARC, en los pacientes infectados por VIH con deficiencia inmune grave, puede aparecer una reacción inflamatoria frente a infecciones oportunistas latentes o asintomáticas. Se han notificado también trastornos autoinmunes (como la enfermedad de Gravesy hepatitis autoinmune); no obstante, el tiempo hasta el inicio notificado es más variable y estos efectos pueden producirse muchos meses después del inicio del tratamiento (ver sección Advertencias - Generales). Osteonecrosis: Se han notificado casos de osteonecrosis, especialmente en pacientes con factores de riesgo generalmente reconocidos, enfermedad avanzada por VIH o exposición prolongada a la TARC. Se desconoce la frecuencia de esta reacción adversa (ver sección Advertencias - Generales). Cambios en la creatinina sérica: Cobicistat aumenta la creatinina sérica debido a la inhibición de la secreción tubular de la creatinina sin afectar a la función glomerular renal. En los estudios clínicos con GENVOYA®, se produjeron aumentos de la creatinina sérica en la semana 2 del tratamiento y se mantuvieron estables a lo largo de 144 semanas. En los pacientes que nunca habían recibido tratamiento, se observó un cambio medio con respecto al valor basal de 0,04 ± 0,12mg/dL (3,5 ± 10,6 mg/L) después de 144 semanas de tratamiento. Los aumentos medios con respecto al valor basal en el grupo tratado con GENVOYA® fueron menores que los observados en el grupo tratado con elvitegravir 150mg/cobicistat 150mg/emtricitabina 200mg/tenofovir disoproxil (en forma de fumarato) 245mg (E/C/F/TDF) en la semana 144 (diferencia -0,04, p < 0,001). Cambios en las pruebas de laboratorio de lípidos: En estudios en pacientes sin terapia antirretroviral previa se observaron aumentos con respecto al valor basal en ambos grupos de tratamiento para los parámetros lipídicos en condiciones de ayuno de colesterol total, colesterol directo ligado a lipoproteínas de baja densidad (LDL) y a lipoproteínas de alta densidad (HDL) y triglicéridos en la semana 1446. La mediana del aumento con respecto al valor basal de dichos parámetros fue mayor en el grupo tratado con GENVOYA® que en el tratado con E/C/F/TDF en la semana 144 (p < 0,001 para la diferencia entre los grupos de tratamiento para el colesterol total en condiciones de ayuno, el colesterol directo ligado a LDL y HDL y los triglicéridos). La mediana (Q1, Q3) del cambio con respecto al valor basal en el cociente colesterol total/colesterol HDL en la semana 144 fue de 0,2 (-0,3; 0,7) en el grupo tratado con GENVOYA® y de 0,1 (-0,4; 0,6) en el grupo tratado con E/C/F/TDF (p < 0,006 para la diferencia entre los grupos de tratamiento). Población pediátrica: La seguridad de GENVOYA® fue evaluada a lo largo de 48 semanas en pacientes adolescentes infectados por VIH-1 de 12 a < 18 años de edad, con un peso corporal ≥ 35 kg, que nunca habían recibido tratamiento (GS-US-292-0106, n = 50), o que estaban virológicamente suprimidos (GS-US-292-1515, n = 50), y en niños virológicamente suprimidos de 8 a < 12 años de edad, con un peso corporal > 25 kg (GS-US-292-0106, n = 23). El perfil de seguridad en pacientes pediátricos que recibieron tratamiento con GENVOYA® era similar al de los adultos. Otras poblaciones especiales: Pacientes con insuficiencia renal: La seguridad de GENVOYA® en 248 pacientes infectados por el VIH-1 que nunca habían recibido tratamiento (n = 6), o bien eran pacientes suprimidos virológicamente (n = 242), con insuficiencia renal leve o moderada (tasa de filtración glomerular estimada mediante el método de Cockcroft-Gault [eTFGCG]: 30-69 mL/min) fue evaluada a lo largo de 144 semanas en un estudio clínico abierto (GS-US-292-0112). El perfil de seguridad de GENVOYA® en pacientes con insuficiencia renal leve o moderada fue similar al de los pacientes con función renal normal (ver sección 8). Pacientes coinfectados por el VIH y el VHB: La seguridad de GENVOYA® fue evaluada en 72 pacientes coinfectados por VIH/VHB que recibían un tratamiento para el VIH en un estudio clínico abierto (GS-US-292-1249), hasta la Semana 48, en la cual los pacientes cambiaron de otro régimen antirretroviral (que incluía tenofovir disoproxil fumarato en 69 de 72 pacientes) a GENVOYA®. En base a estos datos limitados, el perfil de seguridad de GENVOYA® en pacientes con coinfección por VIH/VHB era similar al de los pacientes con monoinfección por VIH-1. Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia al siguiente link: http://sistemas.anmat.gov.ar/aplicaciones_ net/applications/fvg_eventos_adversos_nuevo/index.html o al Departamento de Farmacovigilancia de GADOR SA, enviando un correo electrónico a farmacovigilancia@gador.com.ar, o telefónicamente al 0800 220 2273.
Advertencias.
Generales: A pesar de que se ha probado que la supresión viral con tratamiento antirretroviral eficaz reduce sustancialmente el riesgo de transmisión sexual, no se puede excluir un riesgo residual. Se deben tomar precauciones, conforme a las directrices nacionales, para prevenir la transmisión. Pacientes coinfectados por el VIH y el virus de la hepatitis B o C: Los pacientes con hepatitis B o C crónica, tratados con terapia antirretroviral tienen un riesgo mayor de padecer reacciones adversas hepáticas graves y potencialmente mortales. No se ha establecido la seguridad y eficacia de GENVOYA® en pacientes coinfectados por el VIH-1 y el virus de la hepatitis C (VHC). Tenofovir alafenamida es activo contra el virus de la hepatitis B (VHB). La interrupción del tratamiento con GENVOYA® en pacientes coinfectados por VIH y VHB puede asociarse con exacerbaciones agudas graves de la hepatitis. En pacientes coinfectados por VIH y VHB que interrumpen el tratamiento con GENVOYA® hay que efectuar un seguimiento estrecho, clínico y de laboratorio, durante al menos varios meses después de suspender el tratamiento. Enfermedad hepática: No se ha establecido la seguridad y eficacia de GENVOYA® en pacientes con trastornos hepáticos significativos subyacentes. Los pacientes con insuficiencia hepática preexistente, incluyendo hepatitis crónica activa, tienen una frecuencia aumentada de alteración de la función hepática durante la terapia antirretroviral combinada (TARC) y deben ser monitorizados de acuerdo con las prácticas habituales. Si hay evidencia de empeoramiento de la enfermedad hepática en dichos pacientes, se tendrá que considerar la interrupción o discontinuación del tratamiento. Peso y parámetros metabólicos: Durante el tratamiento antirretroviral se puede producir un aumento en el peso y en los niveles de glucosa y lípidos en la sangre. Tales cambios podrían estar relacionados en parte con el control de la enfermedad y en pa