Gazyva®
ROCHE
Obinutuzumab.
Agente antineoplásico, anticuerpo monoclonal.
Composición.
Cada vial de 50 ml contiene una dosis única de 1.000 mg de obinutuzumab en 40 ml de concentrado líquido (25 mg/ml), en un excipiente compuesto por: L-histidina 57,6 mg, L-histidina clorhidrato monohidrato 89,6 mg, dihidrato de trehalosa 3.632 mg, poloxámero 188: 8 mg y agua para inyectables c.s.p 40 ml.
Farmacología.
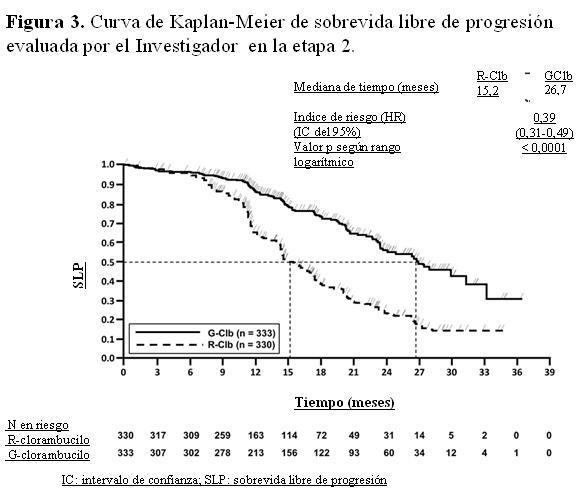
Código ATC: L01XC15. Grupo farmacoterapéutico: Agentes antineoplásicos, anticuerpos monoclonales. Propiedades farmacodinámicas: Mecanismo de acción: Obinutuzumab es un anticuerpo monoclonal recombinante anti-CD20 humanizado Tipo II del isotipo IgG1 modificado mediante glicoingeniería. Está específicamente dirigido al asa extracelular del antígeno transmembrana CD20 en la superficie de linfocitos pre-B y B maduros malignos y no malignos, pero no en células madres hematopoyéticas, células pro-B, células plasmáticas normales u otros tejidos normales. La modificación mediante glicoingeniería de la parte Fc de obinutuzumab aumenta la afinidad por los receptores FccRIII en células efectoras inmunes, tales como células NK (natural killer), macrófagos y monocitos, en comparación con los anticuerpos que no han sido modificados mediante glicoingeniería. En estudios no clínicos, Gazyva induce la muerte celular directa y actúa como mediador de la citotoxicidad celular dependiente de anticuerpos (CCDA) y de la fagocitosis celular dependiente de anticuerpos (FCDA) a través de la incorporación de células efectoras inmunes FccRIII positivas. Además, in vivo obinutuzumab media un bajo grado de citotoxicidad dependiente de complemento (CDC). En comparación con el anticuerpo Tipo I, obinutuzumab, un anticuerpo Tipo II, se caracteriza por una mayor inducción de muerte celular directa con una reducción concomitante de la CDC a una dosis equivalente. Obinutuzumab, anticuerpo modificado por glicoingeniería, se caracteriza por una mayor citotoxicidad celular dependiente de anticuerpos (CCDA) y fagocitosis (FCDA) en comparación con los anticuerpos no modificados por glicoingeniería a una dosis equivalente. En modelos con animales, obinutuzumab media una potente depleción de células B y eficacia antitumoral. En el estudio clínico pivotal BO21004/CLL11, el 91% (40 de 44) de los pacientes evaluables tratados con Gazyva experimentó depleción de células B (definido como recuento de células B CD19+ < 0,07 x 109/l) al finalizar el período de tratamiento y se mantuvo la depleción durante los primeros 6 meses de seguimiento. Se observó una recuperación de las células B dentro de los 12 a 18 meses de seguimiento en el 35% (14 de 40) de los pacientes sin progresión de la enfermedad y en el 13% (5 de 40) con progresión de la enfermedad. Eficacia clínica y seguridad: Leucemia Linfática Crónica: Se llevó a cabo un estudio clínico de Fase III, internacional, multicéntrico, abierto, aleatorizado, en dos etapas y tres grupos de tratamiento (BO21004/CLL11), para investigar la eficacia y la seguridad de Gazyva más clorambucilo (GClb) en comparación con rituximab más clorambucilo (RClb) o clorambucilo (Clb) en monoterapia, en pacientes con leucemia linfática crónica no tratados previamente y con comorbilidades. Previo a la inclusión, los pacientes debían tener LLC CD20+ demostrada, y una o ambas de las siguientes patologías coexistentes: puntuación de comorbilidad de acuerdo con la Escala CIRS mayor a 6 o función renal reducida con un valor de clearance de creatinina (ClCr) < 70 ml/min. Se excluyeron los pacientes con disfunción hepática, pruebas de función hepática Grado 3 según Criterios terminológicos comunes para reacciones adversas del National Cancer Institute (NCI-CTCAE) AST, ALT > 5 veces el LSN durante > 2 semanas; bilirrubina > 3 veces el LSN y disfunción renal (Clcr < 30 ml/min). También se excluyeron aquéllos con una puntuación de 4 en la escala CIRS por insuficiencia en uno o varios órganos individuales o sistemas, a excepción de los sistemas que incluyen ojos, oídos, nariz, garganta y laringe. Un total de 781 pacientes fueron asignados en forma aleatoria en el tratamiento con Gazyva más clorambucilo, rituximab más clorambucilo o clorambucilo en monoterapia, en una proporción de 2:2:1, respectivamente. En la etapa 1a se comparó Gazyva más clorambucilo con clorambucilo en monoterapia en 356 pacientes, y en la etapa 2 Gazyva más clorambucilo con rituximab más clorambucilo en 663 pacientes. Los resultados de eficacia se resumen en la Tabla 1 y en las Figuras 1 a 3. En la mayoría de los pacientes, Gazyva se administró por vía intravenosa con una dosis inicial de 1.000 mg en el día 1, día 8 y día 15 del primer ciclo de tratamiento. A fin de reducir la cantidad de reacciones relacionadas con la infusión en los pacientes, se realizó una modificación y 140 recibieron la primera dosis de Gazyva en 2 días (el día 1 [100 mg] y el día 2 [900 mg]) (véanse Dosificación y Precauciones). En los ciclos de tratamiento posteriores (ciclos 2 a 6), los pacientes recibieron 1.000 mg de Gazyva solamente el día 1. La dosis oral de clorambucilo fue de 0,5 mg/kg de peso corporal el día 1 y el día 15 en todos los ciclos de tratamiento (1 a 6). Los datos demográficos y las características basales estuvieron bien equilibrados entre los distintos grupos de tratamiento. La mayoría de los pacientes eran caucásicos (95%) y de sexo masculino (61%). La mediana de edad fue de 73 años, y un 44% tenía 75 años de edad o más. Inicialmente, el 22% de los pacientes estaba en estadio A de Binet; el 42%, en estadio B de Binet y el 36%, en estadio C de Binet. La mediana de la puntuación de comorbilidad fue de 8 y el 76% de los pacientes incorporados tenía una puntuación de comorbilidad superior a 6. La mediana estimada del Clcr fue de 62 ml/min y el 66% tenía Clcr < 70 ml/min. El 42% de los pacientes incluidos tenían tanto un Clcr < 70 ml/min como una puntuación de comorbilidad mayor de 6. El 34% se incluyó sólo por su puntuación de comorbilidad, y el 23% sólo por su función renal reducida. Las patologías coexistentes notificadas con mayor frecuencia (utilizando un punto de corte de 30% o superior), según la clasificación de órganos del sistema MedDRA, son: trastornos vasculares (73%), trastornos cardíacos (46%), trastornos gastrointestinales (38%), trastornos del metabolismo y de la nutrición (40%), trastornos renales y urinarios (38%), trastornos musculosqueléticos y del tejido conjuntivo (33%).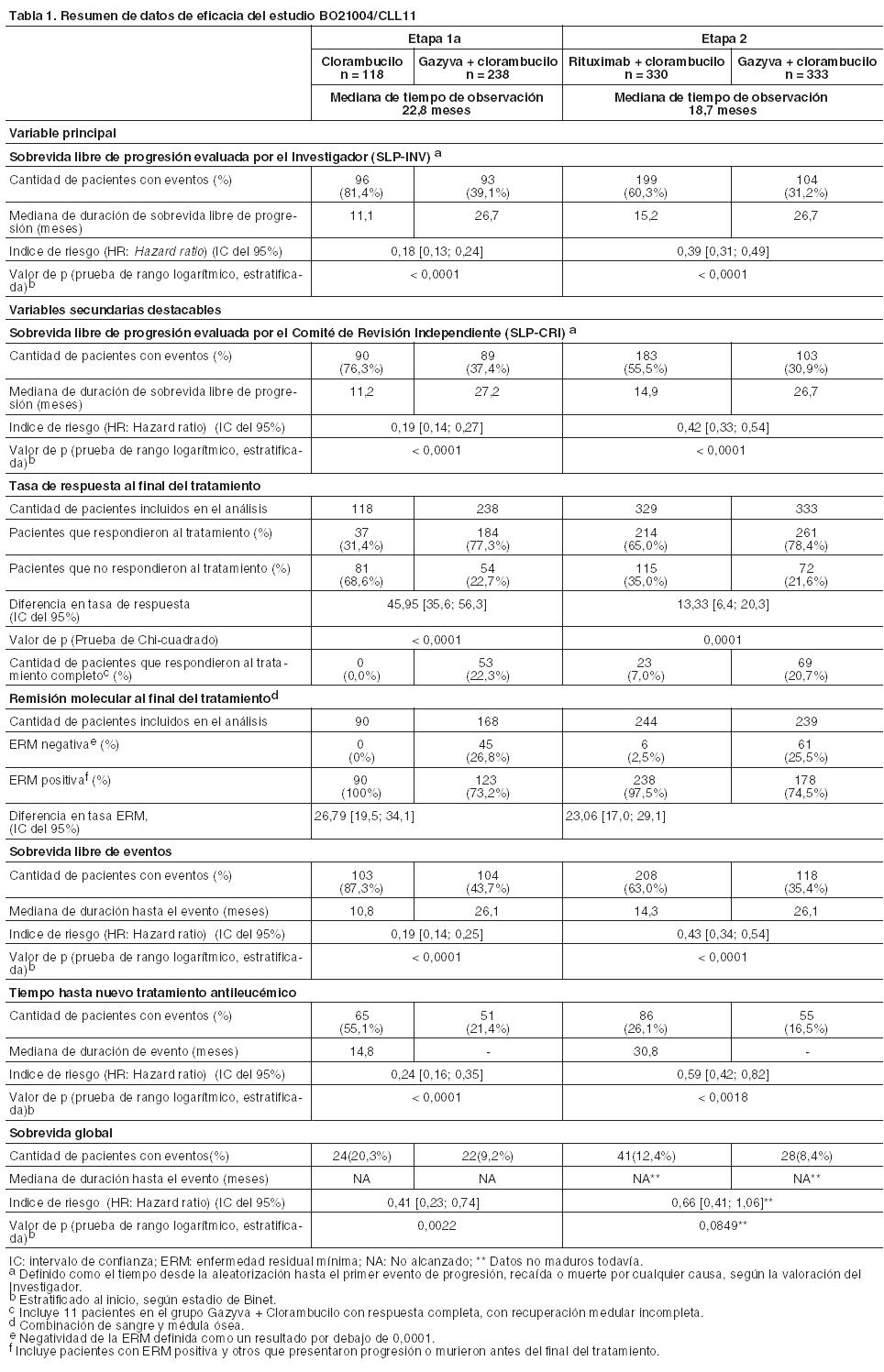
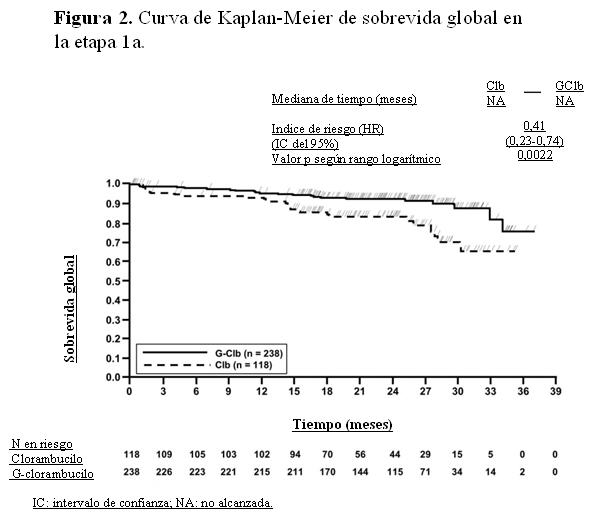
La sobrevida global de la etapa 1a se presenta en la Figura 2. La sobrevida global de la etapa 2 continuará bajo seguimiento. Los resultados de sobrevida libre de progresión del análisis de subgrupos (es decir, sexo, edad, estadios de Binet, ClCr, puntuación de comorbilidad de acuerdo con la Escala CIRS, beta-2-microglobulina, estado de IGVH, anomalías cromosómicas, recuento de linfocitos al inicio) estuvieron de acuerdo con los resultados observados en la población por intención de tratar. El riesgo de progresión de la enfermedad o de muerte fue menor en el grupo tratado con Gazyva más clorambucilo (GClb) que en el que recibió rituximab más clorambucilo (RClb) y en el grupo tratado con clorambucilo en monoterapia (Clb), en todos los subgrupos, excepto en el subgrupo de pacientes con deleción 17p. En el pequeño subgrupo de pacientes con deleción 17p sólo se observó una tendencia positiva comparado con clorambucilo (HR=0,42, p=0,0892); no se registró beneficio comparado con RClb. Por subgrupos, la reducción del riesgo de progresión de la enfermedad o muerte osciló desde 92% a 58% para GClb frente a Clb en monoterapia y 72% a 29% para GClb frente a RClb.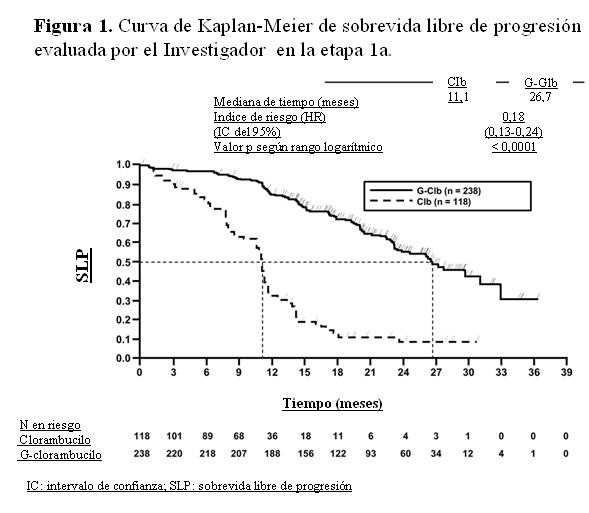
Calidad de vida: En los cuestionarios QLQC30 y QLQ-CLL-16 que fueron completados durante el período de tratamiento, no se observaron diferencias sustanciales en ninguna de las subescalas. Los datos durante el seguimiento son limitados, especialmente los del grupo tratado con clorambucilo en monoterapia. Sin embargo, hasta la fecha no se han detectado variaciones esenciales en la calidad de vida durante el seguimiento. Las evaluaciones de calidad de vida relacionadas con la salud, específicamente en lo relativo al cansancio durante el período de tratamiento, mostraron diferencias que no fueron estadísticamente significativas, lo que sugiere que la adición de Gazyva al régimen con clorambucilo no aumenta la sensación de cansancio en los pacientes. Linfoma no Hodgkin - Linfoma Folicular: En un estudio clínico de Fase III, abierto, multicéntrico, aleatorizado (GAO4753g/GADOLIN), se evaluaron 396 pacientes con Linfoma no Hodgkin indolente (LNHi) que no respondieron durante el tratamiento o que progresaron en los 6 meses siguientes a la última dosis de rituximab o de un régimen con rituximab (incluyendo rituximab en monoterapia como parte del tratamiento de inducción o mantenimiento). Los pacientes fueron aleatorizados en una proporción 1:1 para recibir bendamustina (B) en monoterapia (n=202) o Gazyva en combinación con bendamustina (G+B) (n=194) durante 6 ciclos, cada uno de 28 días de duración. Los pacientes del grupo G+B que no tuvieron progresión de la enfermedad (es decir, con respuesta completa (RC), respuesta parcial (RP) o enfermedad estable (EE)) al final de la fase de inducción, continuaron recibiendo Gazyva en mantenimiento una vez cada dos meses durante dos años o hasta progresión de la enfermedad (lo que ocurriera primero). Los pacientes fueron estratificados según la región, subtipo LNHi (folicular frente a no folicular), tipo rituximab refractario (ya sea refractario a la monoterapia previa con rituximab o a rituximab en combinación con monoterapia) y número de tratamientos previos (≤2 frente a > 2). Los datos demográficos y las características basales estuvieron bien equilibrados (mediana de edad de 63 años, la mayoría de los pacientes eran caucásicos [88%] y de sexo masculino [58%]). La mayor parte tenía linfoma folicular (81%). La mediana de tiempo desde el diagnóstico inicial fue de 3 años y la mediana del número de tratamientos previos fue de 2 (rango 1 a 10); el 44% de los pacientes había recibido un tratamiento previo y el 34% dos tratamientos previos. Gazyva se administró por infusión intravenosa como una dosis única de 1.000 mg en el día 1, día 8 y día 15 del ciclo 1, el día 1 de los ciclos 2-6, y en pacientes que no tuvieron progresión de la enfermedad, una vez cada dos meses durante dos años o hasta progresión de la enfermedad (lo que ocurriera primero). Bendamustina se administró en forma intravenosa, los días 1 y 2 para todos los ciclos de tratamiento (ciclos 1-6) a razón de 90 mg/m2/día en combinación con Gazyva o 120 mg/m2/día en monoterapia. En pacientes tratados con G+B, el 79,4% de los pacientes recibió los seis ciclos de tratamiento en comparación con el 66,7% de los del grupo B. El análisis principal basado en la evaluación del Comité de Revisión Independiente (CRI) mostró una reducción estadísticamente significativa del 45% en el riesgo de progresión de la enfermedad (PE) o muerte, en pacientes con LNHi que recibieron G+B seguido de Gazyva en mantenimiento, en comparación con aquellos tratados con bendamustina en monoterapia. La reducción en el riesgo de progresión de la enfermedad o muerte observada en la población LNHi está impulsada por el subgrupo de pacientes con LF. La mayoría de los pacientes del estudio GAO4753g/GADOLIN tenía linfoma folicular (LF) (81,1%). Los resultados de eficacia de la población con linfoma folicular se muestran en la Tabla 2. El 11,6% de los pacientes tenía linfoma de la zona marginal (LZM) y el 7,1% linfoma linfocítico pequeño (LLP).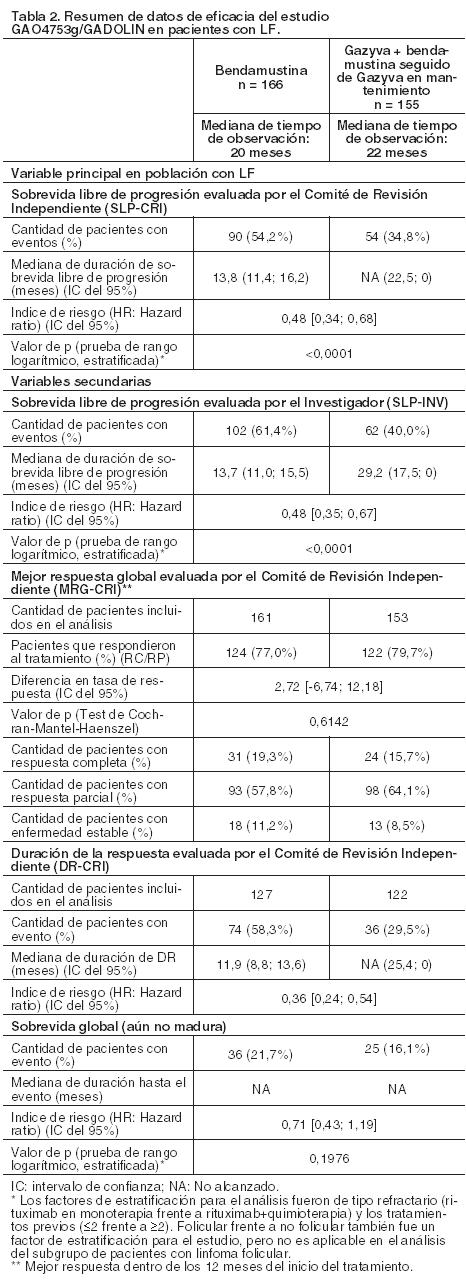
En la población no-LF el índice de riesgo (HR) de la SLP evaluada por el CRI fue 0,94 [IC del 95%: 0,49; 1,90]. No se pueden establecer conclusiones definitivas sobre la eficacia en las subpoblaciones con LZM y LLP.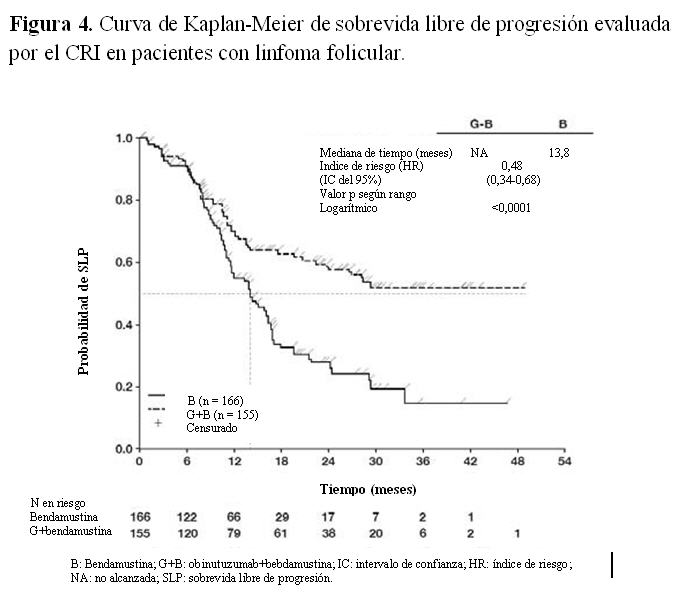
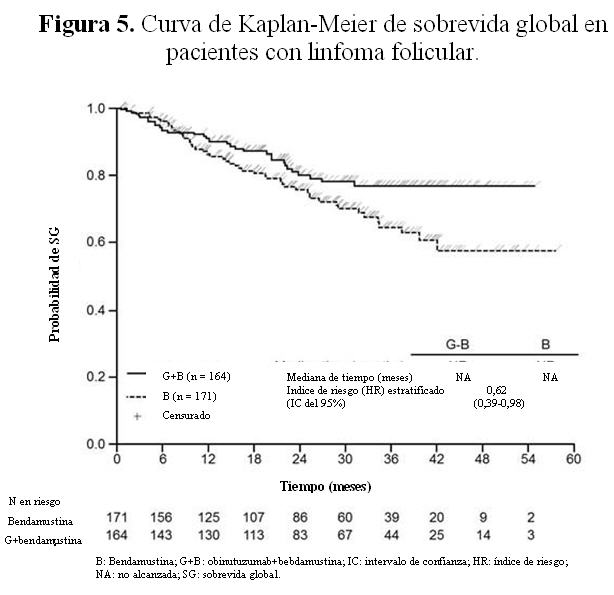
Se realizó un análisis post hoc 8 meses después del corte de datos del análisis primario. Con una mediana de observación de 24,1 meses en pacientes con linfoma folicular, 48 (28,1%) en el grupo B y 30 (18,3%) en el grupo G+B, habían fallecido. En este análisis post hoc, la mejora observada en la SG con G+B fue respaldada por un índice de riesgo (HR) estratificado para la SG de 0,62 (IC del 95%: 0,39; 0,98). La mediana de sobrevida global (SG) aún no ha sido alcanzada en ningún grupo. Los resultados de SLP en el análisis post hoc son coherentes con el análisis primario y su significación no varía, y el perfil de seguridad es análogo con el análisis primario. Resultados de los análisis de los subgrupos: En general los resultados de los análisis de los subgrupos fueron coherentes con los observados en la población con LF, avalados por la solidez de los resultados globales.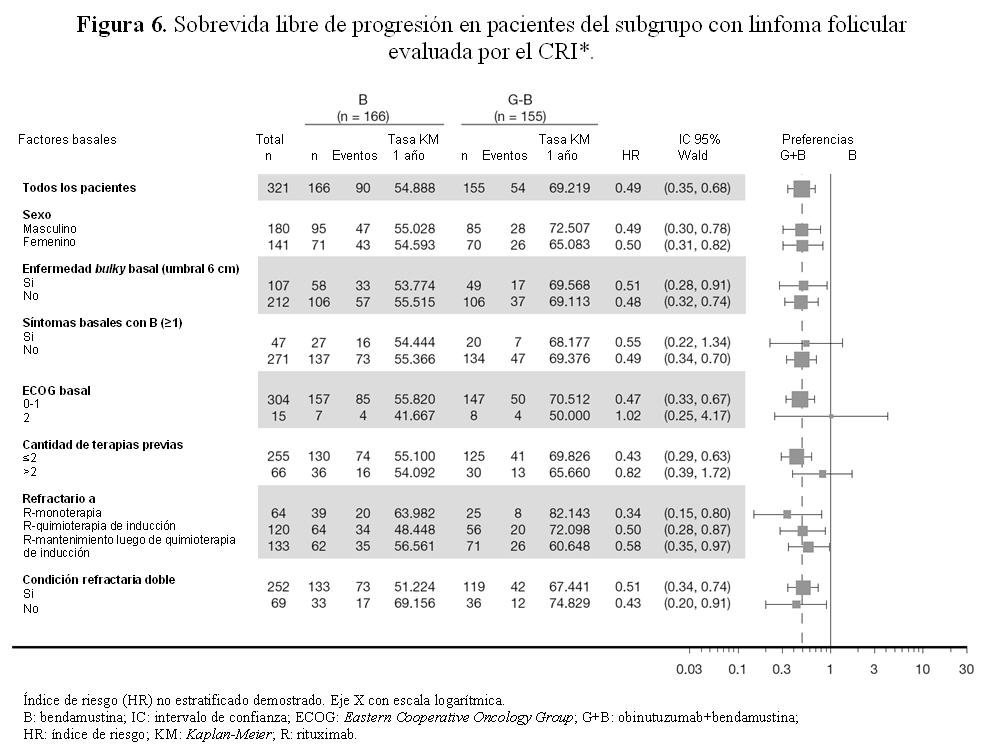
Resultados notificados por el paciente: De acuerdo con el cuestionario FACT-Lym y la escala del índice EQ-5D recogidos durante el tratamiento y durante los períodos de seguimiento, la calidad de vida relacionada con la salud en el estudio pivotal se mantuvo en general sin ninguna diferencia significativa entre los grupos. No obstante, la adición de Gazyva con bendamustina en pacientes con LF retrasó el tiempo hasta el empeoramiento de la calidad de vida vinculada con la salud, medido por la puntuación TOI FACT-Lym en 2,2 meses (mediana de 5,6 frente a 7,8 meses en B y G+B respectivamente, HR = 0,83, IC del 95%: 0,60; 1,13). Inmunogenicidad: Los resultados de los ensayos de inmunogenicidad dependen considerablemente de varios factores, como la sensibilidad y la especificidad del ensayo, la metodología y la solidez del ensayo frente a las cantidades de Gazyva/anticuerpo presentes en la circulación, la manipulación de las muestras, el momento de la recolección de las muestras, la medicación concomitante y enfermedades subyacentes. Por estas razones, la comparación de la incidencia de anticuerpos a Gazyva con la de anticuerpos a otros medicamentos puede resultar equívoca. Los pacientes que fueron parte del estudio pivotal BO21004/CLL11 fueron evaluados en múltiples puntos de tiempo para determinar los niveles de anticuerpos antiterapéuticos (ATA) a Gazyva. En aquéllos tratados con Gazyva, 8 de 140 pacientes en la fase aleatorizada y 2 de 6 en la fase preinclusión presentaron resultados positivos para ATA a los 12 meses de seguimiento. De éstos, ninguno experimentó reacciones anafilácticas o de hipersensibilidad que se consideraran relacionadas con ATA, ni se vio afectada su respuesta clínica. En el estudio pivotal para LNHi, GAO4753g, dos pacientes del grupo G+B tuvieron una evaluación HAHA (Anticuerpo Humano Anti-Humano) positiva al inicio y experimentaron RRI. Ningún paciente desarrolló HAHA a Gazyva durante o después del tratamiento con Gazyva. Población pediátrica: La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Gazyva en los diferentes grupos de la población pediátrica con leucemia linfática crónica y linfoma folicular (véase Dosificación, Población pediátrica). Propiedades farmacocinéticas: Se desarrolló un modelo farmacocinético poblacional para analizar los datos farmacocinéticos en 469 pacientes con LNHi, 342 con LLC y 130 con DBLC tratados con obinutuzumab en monoterapia o en combinación con quimioterapia en estudios de Fases I, II y III. Absorción: Obinutuzumab es administrado por vía intravenosa. No se han realizado estudios con otras vías de administración. A partir del modelo farmacocinético poblacional, después de la infusión del día 1 del ciclo 6 en pacientes con LLC, el valor medio estimado de Cmáx fue de 465,7 mg/ml y el del ABC(t) de 8.961 mg•día/ml; en los pacientes con LNHi la mediana de Cmáx estimada fue de 539,3 mg/ml y el valor del ABC(t) de 10.956 mg•día/ml. Distribución: Después de la administración intravenosa, el volumen de distribución en el compartimiento central, 2,98 litros en pacientes con LLC y 2,97 litros en pacientes con LNHi, se aproxima al volumen sérico, lo que indica que la distribución está restringida principalmente al plasma y al líquido intersticial. Biotransformación: No se ha estudiado directamente el metabolismo de obinutuzumab. Los anticuerpos se eliminan principalmente por catabolismo. Eliminación: El clearance de obinutuzumab es de aproximadamente 0,11 litros/día en pacientes con LLC y de 0,08 litros/día en aquéllos con LNHi con un promedio de vida media de eliminación de 26,4 días en pacientes con LLC y de 36,8 días en quienes tienen LNHi. La eliminación de obinutuzumab comprende dos vías paralelas que describen el clearance, una vía lineal y otra no lineal que cambia en función del tiempo. Durante el tratamiento inicial, predomina la vía de clearance de tiempo variable no lineal y es consecuentemente la vía de clearance principal. A medida que el tratamiento continúa, el impacto de esta vía disminuye y predomina la vía de clearance lineal. Esto es indicativo de la distribución del fármaco de acuerdo con el objetivo (DMAO), donde la abundancia inicial de células CD20 causa la eliminación rápida de obinutuzumab de la circulación. No obstante, una vez que la mayoría de las células CD20 se une con obinutuzumab, el impacto de la DMAO en la farmacocinética es minimizado. Relaciones farmacocinéticas / farmacodinámicas: En el análisis farmacocinético poblacional, se comprobó que el sexo es una covariable que explica parte de la variabilidad entre pacientes, con un clearance en estado estacionario 18% mayor (CLee) y un volumen de distribución (V) 19% mayor en pacientes de sexo masculino. No obstante, los resultados del análisis poblacional han mostrado que las diferencias en exposición no son significativas, en pacientes con LLC con una mediana estimada de ABC y Cmáx de 11.282 mg•día/ml y 578,9 mg/ml en pacientes de sexo femenino y de 8.451 mg•día/ml y 432,5 mg/ml en los de sexo masculino, respectivamente, en el ciclo 6; y en aquéllos con LNHi con una mediana estimada de ABC y Cmáx de 13.172 mg•día/ml y 635,7 mg/ml en pacientes de sexo femenino y de 9.769 mg•día/ml y 481,3 mg/ml en los de sexo masculino, respectivamente, lo que indica que no es necesario ajustar la dosis según el sexo del paciente. Pacientes pediátricos: No se han realizado estudios para investigar la farmacocinética de obinutuzumab en esta población. Pacientes de edad avanzada: El análisis farmacocinético poblacional de obinutuzumab mostró que la edad no afecta su farmacocinética. No se observaron diferencias significativas en la farmacocinética de obinutuzumab entre pacientes menores de 65 años de edad (n=375), entre 65 y 75 años (n=265) y mayores de 75 años (n=171). Pacientes con insuficiencia renal: El análisis farmacocinético poblacional de obinutuzumab demostró que el clearance de creatinina no afecta a su farmacocinética. La farmacocinética de obinutuzumab en pacientes con clearance de creatinina leve (Clcr 50 a 89 ml/min, n=464) o insuficiencia renal moderada (Clcr 30 a 49 ml/min, n=106) fue similar a la de pacientes con función renal normal (Clcr ≥90 ml/min, n=383). Los datos farmacocinéticos en aquéllos con insuficiencia renal grave (Clcr 15 a 29 ml/min) son limitados (n=8); por lo tanto, no es posible recomendar dosis específicas. Pacientes con insuficiencia hepática: No se ha realizado ningún estudio farmacocinético formal en pacientes con insuficiencia hepática. Datos preclínicos sobre seguridad: No se han llevado a cabo estudios para establecer el potencial carcinogénico de obinutuzumab. No se efectuaron estudios específicos en animales para evaluar el efecto de obinutuzumab en la fertilidad. En ensayos de toxicidad con dosis repetidas no se observaron efectos adversos en los órganos reproductivos de monos cynomolgus machos y hembras. Un estudio de toxicidad mejorado sobre el desarrollo pre y posnatal en monos cynomolgus preñados no mostró evidencia de efectos teratogénicos. Sin embargo, una dosis semanal de obinutuzumab desde el día 20 posterior al coito hasta el parto dio como resultado una depleción completa de células B en las crías con dosis intravenosas semanales de obinutuzumab de 25 y 50 mg/kg (2 a 5 veces la exposición clínica basada en la Cmáx y el ABC). La exposición de las crías en el día 28 posparto sugiere que obinutuzumab puede atravesar la barrera placentaria. Las concentraciones séricas en lactantes en el día 28 posterior al parto se encontraban en el intervalo de concentraciones del suero materno, mientras que las concentraciones en la leche materna el mismo día eran muy bajas (menos de 0,5% de los niveles séricos maternos correspondientes), lo cual sugiere que la exposición de las crías se debe haber producido en el útero. Los recuentos de células B regresaron a niveles normales y la función inmunológica fue recuperada dentro de los 6 meses posteriores al parto. En un estudio de 26 semanas de duración realizado en monos cynomolgus, se registraron reacciones de hipersensibilidad y se atribuyeron al reconocimiento como extraño del anticuerpo humanizado (basado en una exposición clínica de 0,7 a 6 veces sobre los valores de Cmáx y ABC en estado estacionario después de la administración semanal de 5, 25 y 50 mg/kg). Los hallazgos incluían reacciones anafilácticas o anafilactoides agudas y una mayor prevalencia de inflamación sistémica e infiltrados compatibles con reacciones de hipersensibilidad mediadas por el complejo inmune, como arteritis/periarteritis, glomerulonefritis e inflamación serosa/adventicial. Estas reacciones llevaron a la interrupción no programada del tratamiento con obinutuzumab de 6/36 animales durante las fases de dosificación y recuperación; estos cambios fueron parcialmente reversibles. No se verificó toxicidad renal con relación causal atribuible a obinutuzumab en seres humanos.
Indicaciones.
Leucemia Linfática Crónica (LLC): Gazyva está indicado, en combinación con clorambucilo, para el tratamiento de pacientes adultos con leucemia linfática crónica (LLC), no tratados previamente y con comorbilidades para los que el tratamiento con fludarabina a dosis plena no es adecuado (véase Farmacología; Propiedades farmacodinámicas). Linfoma Folicular (LF): Gazyva está indicado en combinación con bendamustina, seguido de un tratamiento de mantenimiento con Gazyva (que no supere los dos años), para el tratamiento de pacientes con linfoma folicular (LF), que no hayan respondido a un tratamiento con rituximab, o a un esquema terapéutico que contenga rituximab, o que hayan presentado progresión de la enfermedad (PE) durante o después de dicho tratamiento.
Dosificación.
El reemplazo por cualquier otro agente biológico requiere el consentimiento del médico prescriptor. Gazyva debe ser administrado bajo la estrecha supervisión de un médico experimentado, y en un entorno que disponga en forma inmediata de un equipo completo de reanimación. Posología: Profilaxis y premedicación para Síndrome de Lisis Tumoral (SLT): Los pacientes con una alta carga tumoral y/o un recuento alto de linfocitos en circulación ( > 25 x 109/l) y/o insuficiencia renal (Clcr < 70 ml/min), se consideran en riesgo de SLT y deben recibir profilaxis. Dicha profilaxis debe constar de una adecuada hidratación y administración de uricostáticos (por ejemplo, alopurinol), o un tratamiento alternativo adecuado como urato oxidasa (por ejemplo, rasburicasa), empezando de 12 a 24 horas antes de iniciar la infusión de Gazyva de acuerdo con la práctica habitual (ver Precauciones). Los pacientes deben continuar recibiendo profilaxis repetidas antes de cada siguiente infusión, si se considera apropiado. Profilaxis y premedicación para Reacciones Relacionadas con la Infusión (RRI): La premedicación para reducir el riesgo de las RRI se describe en las Tablas 3 y 4 (ver también Precauciones). La premedicación con corticosteroides está recomendada en pacientes con LF y es obligatoria para pacientes con LLC en el primer ciclo (ver Tabla 3). La premedicación para infusiones posteriores y otra premedicación se deben administrar como se describe a continuación. Durante las infusiones intravenosas de Gazyva, el paciente puede presentar hipotensión como síntoma de RRI. Por lo tanto, se debe considerar la suspensión de los tratamientos antihipertensivos desde 12 horas previas a la infusión de Gazyva, durante el procedimiento y la primera hora posterior a la administración (ver Precauciones).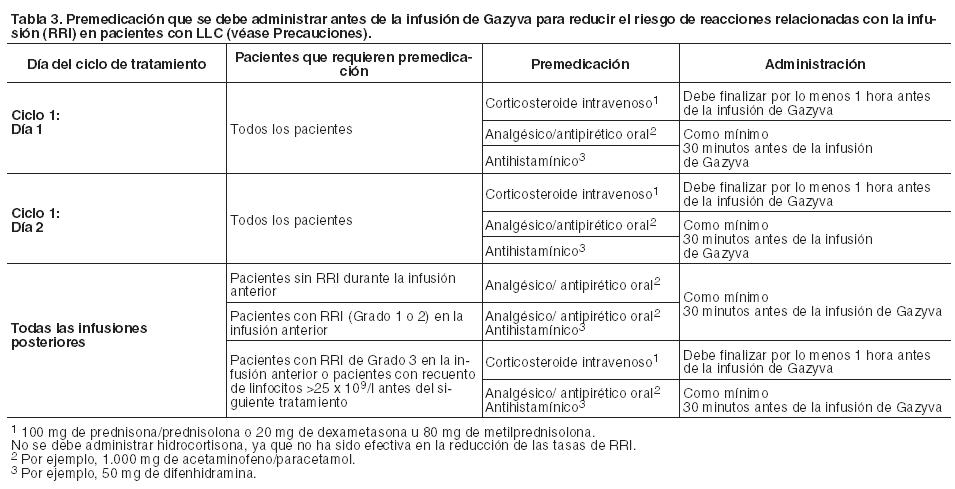
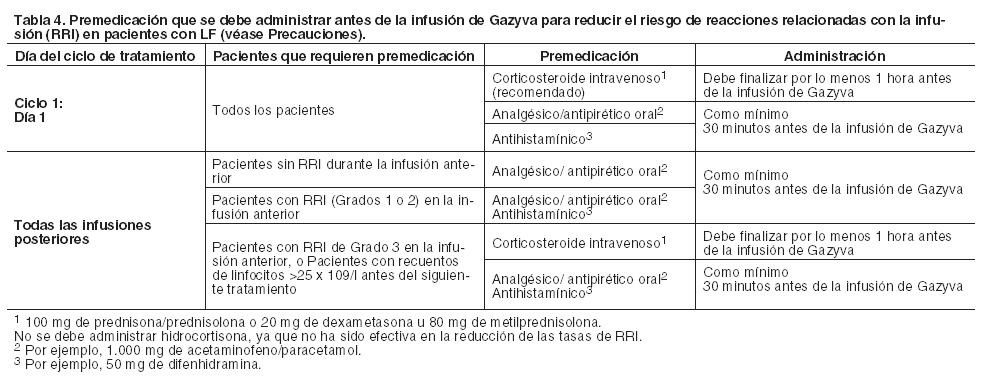
Dosis: Leucemia Linfática Crónica (LLC, en combinación con clorambucilo) Las dosis recomendadas de Gazyva en combinación con clorambucilo para pacientes con LLC se indican en la Tabla 5; para mayor información sobre la dosis de clorambucilo ver Farmacología, Propiedades farmacodinámicas. Ciclo 1: La dosis recomendada de Gazyva asociada con clorambucilo es de 1.000 mg, administrados el día 1 y el día 2 (o continuación del día 1), y en el día 8 y día 15 del primer ciclo de tratamiento de 28 días. Se deben preparar dos bolsas para la primera dosis (100 mg para la primera infusión y 900 mg para la segunda infusión). Si la dosis de 100 mg se completa sin modificarse la velocidad de infusión o sin interrupciones, la dosis de 900 mg se podrá administrar el mismo día (sin retraso en la dosis), siempre que estén dadas las condiciones, el tiempo adecuado y la supervisión médica estén disponibles durante toda la infusión. Si durante los primeros 100 mg se produjera algún cambio en la velocidad de infusión o interrupción, la infusión de 900 mg se debe administrar al día siguiente. Ciclos 2 al 6: La dosis recomendada de Gazyva en combinación con clorambucilo es de 1.000 mg administrada en el día 1 de cada ciclo.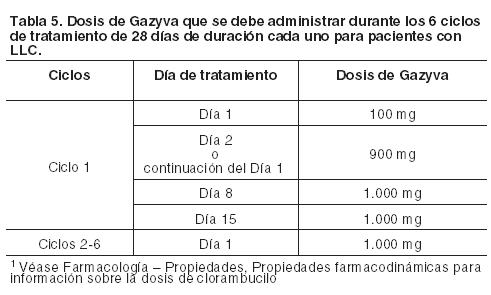
Duración del tratamiento: Seis ciclos de tratamiento, de 28 días de duración cada uno. Retrasos y omisiones de dosis Si se omite una dosis prevista de Gazyva, la misma debe ser administrada lo antes posible; no se debe esperar hasta el momento de la próxima dosis programada. El intervalo de tratamiento predeterminado para Gazyva debe ser mantenido entre las dosis. Linfoma Folicular (LF): Las dosis recomendadas de Gazyva en combinación con bendamustina para pacientes con LF se indican en la Tabla 6; para mayor información sobre la dosis de bendamustina ver Farmacología, Propiedades farmacodinámicas: Fase de inducción (en combinación con bendamustina): Ciclo 1: La dosis recomendada de Gazyva en combinación con bendamustina es de 1.000 mg administrados el día 1, día 8 y día 15 del primer ciclo de tratamiento de 28 días. Ciclos 2 al 6: La dosis recomendada de Gazyva en combinación con bendamustina es de 1.000 mg administrados en el día 1 de cada ciclo de tratamiento de 28 días. Fase de mantenimiento: Los pacientes que respondan al tratamiento de inducción (es decir, los 6 ciclos iniciales) con Gazyva en combinación con bendamustina o tengan enfermedad estable, deben continuar recibiendo Gazyva 1.000 mg en monoterapia, como tratamiento de mantenimiento una vez cada 2 meses, durante 2 años o hasta progresión de la enfermedad (lo que ocurra primero).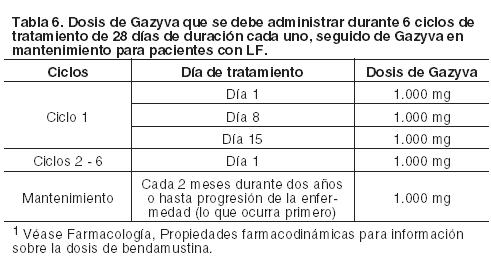
Duración del tratamiento: Seis ciclos de tratamiento, de 28 días de duración cada uno, seguido de tratamiento de mantenimiento una vez cada dos meses durante dos años o hasta progresión de la enfermedad (lo que ocurra primero). Retrasos u omisiones de dosis: Si se omite una dosis prevista de Gazyva, la misma debe ser administrada lo antes posible; no se debe esperar hasta el momento de la próxima dosis programada. Durante la fase de inducción, el intervalo de tratamiento predeterminado para Gazyva debe ser mantenido entre las dosis. Durante la fase de mantenimiento, conservar el calendario de dosificación original para dosis posteriores. Modificación de la dosis durante el tratamiento (para todas las indicaciones): No se recomiendan reducciones de la dosis de Gazyva. Para el manejo de las reacciones adversas sintomáticas (incluidas las RRI), véase Manejo de las RRI o Precauciones). Poblaciones especiales: Pacientes pediátricos: No se ha establecido la seguridad y eficacia de Gazyva en niños y adolescentes menores de 18 años de edad. No se dispone de datos al respecto. Pacientes de edad avanzada: No se requiere ajustar la dosis en pacientes ancianos (ver Farmacología; Propiedades farmacocinéticas). Pacientes con insuficiencia renal: No es necesario ajustar la dosis en pacientes con insuficiencia renal de leve a moderada (ver Farmacología; Propiedades farmacocinéticas). No se ha establecido la seguridad y eficacia de Gazyva en pacientes con insuficiencia renal grave (clearance de creatinina < 30 ml/min). Pacientes con insuficiencia hepática: No se ha establecido la seguridad y eficacia de Gazyva en pacientes con insuficiencia hepática. No se puede realizar una recomendación posológica específica. Formas de administración: Gazyva se administra por vía intravenosa. Se debe administrar después de la dilución como infusión intravenosa utilizando una vía específica. Las infusiones de Gazyva no se deben administrar en infusión rápida o en bolo intravenoso. Las instrucciones para la dilución de Gazyva previa a la administración se encuentran especificadas en Precauciones especiales de eliminación y otras manipulaciones; instrucciones para la dilución. En las Tablas 7 y 8 se detallan las instrucciones sobre la velocidad de infusión.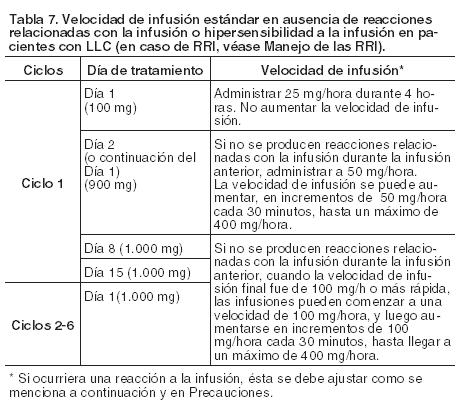
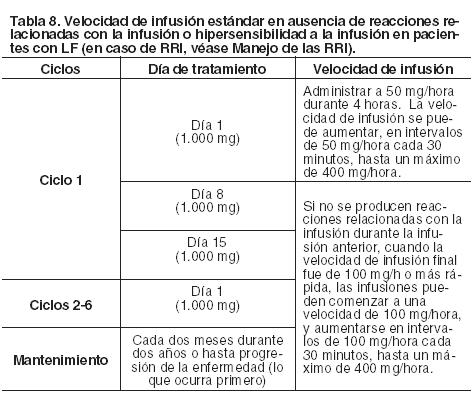
Manejo de las RRI (todas las indicaciones): El manejo de las reacciones relacionadas con la infusión (RRI) puede requerir la interrupción temporaria del tratamiento, la reducción en la velocidad de infusión o la discontinuación de Gazyva, como se especifica a continuación (ver también Precauciones). Grado 4 (potencialmente mortal): Detener la infusión e interrumpir permanentemente el tratamiento. Grado 3 (grave): Interrumpir la infusión temporalmente y tratar los síntomas. Una vez resueltos los mismos, reiniciar la infusión a una velocidad que no exceda la mitad de la velocidad previa (es decir, la utilizada en el momento en que ocurrió la reacción relacionada con la infusión) y, si el paciente no experimenta ningún tipo de síntoma de RRI, se puede retomar el incremento de la velocidad de infusión en la medida y en los intervalos apropiados para la dosis del tratamiento (véanse las Tablas 7 y 8). Para pacientes con LLC que reciben la dosis del día 1 (ciclo 1) dividida en dos días, la velocidad de infusión del día 1 se puede aumentar nuevamente hasta 25 mg/hora después de 1 hora, como máximo. Detener la infusión e interrumpir permanentemente el tratamiento si los pacientes sufren una segunda RRI de Grado 3. Grados 1-2 (leve a moderada): Se puede reducir la velocidad de infusión y tratar los síntomas. Una vez resueltos los mismos, continuar la infusión y, si el paciente no experimenta ningún síntoma de RRI, se puede retomar el incremento de la velocidad de infusión en la medida y en los intervalos apropiados para la dosis del tratamiento (ver las Tablas 7 y 8). Para pacientes con LLC que reciben la dosis del día 1 (ciclo 1) dividida en dos días, la velocidad de infusión del día 1 se puede aumentar nuevamente hasta 25 mg/hora después de 1 hora, como máximo.
Contraindicaciones.
Gazyva está contraindicado en pacientes con hipersensibilidad conocida (mediada por IgE) a obinutuzumab o a cualquiera de los excipientes.
Reacciones adversas.
Resumen del perfil de seguridad: Las reacciones adversas al medicamento (RAM) descritas a continuación, se identificaron durante el tratamiento y el seguimiento, en dos estudios clínicos pivotales, BO21004/CLL11 (n=781) y GAO4753g (n=396), en pacientes con LLC no tratados previamente y en pacientes con LNHi (el 81,1% de los pacientes tenía LF) que no habían respondido o habían progresado durante o hasta 6 meses después del tratamiento con rituximab o con un régimen con rituximab. En estos ensayos se estudió Gazyva en combinación con diferentes agentes quimioterapéuticos (clorambucilo para LLC, bendamustina para LNHi) y en monoterapia de mantenimiento (sólo en LNHi). El protocolo del estudio GAO4753g define pacientes con LNHi incluyendo LF como población del estudio. Por ello, con el fin de proporcionar la información de seguridad más completa, se ha realizado el análisis de las reacciones adversas que se presentan a continuación en toda la población de estudio (es decir, LNHi). En la Tabla 7 se resumen las RAM que se manifestaron con mayor incidencia (diferencia de ≥2%) en pacientes con LLC tratados con Gazyva más clorambucilo, en comparación con aquéllos que recibieron clorambucilo en monoterapia o con rituximab más clorambucilo (ensayo BO21004/CLL11) y en pacientes con LNHi tratados con Gazyva más bendamustina seguido de Gazyva en mantenimiento en algunos de ellos, en comparación con bendamustina sola (estudio GAO4753g). Las frecuencias se clasifican en las siguientes categorías: muy frecuentes (≥1/10), frecuentes (≥1/100 a < 1/10), poco frecuentes (≥1/1.000 a < 1/100), raras (≥1/10.000 a < 1/1.000) y muy raras ( < 1/10.000). Las reacciones adversas se incluyen en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Tabla de reacciones adversas: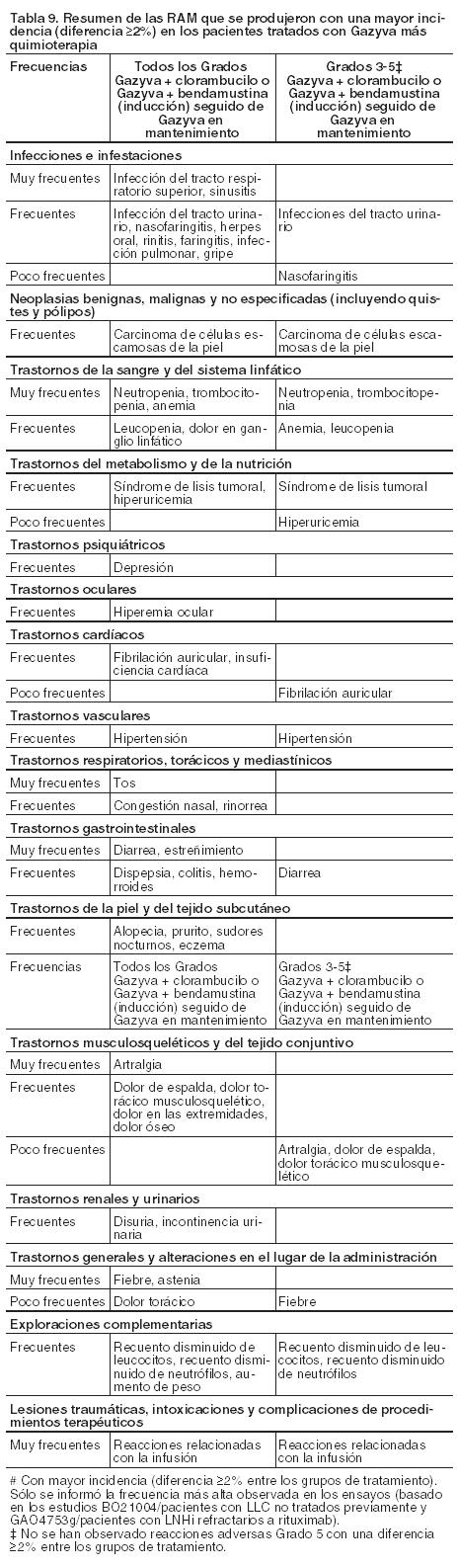
En el estudio GAO4753g, los pacientes del grupo bendamustina (B) sólo recibieron 6 meses de tratamiento de inducción, mientras que los del grupo Gazyva más bendamustina (G+B), después del tratamiento de inducción, continuaron con un tratamiento de mantenimiento con Gazyva. Durante el período de mantenimiento en el estudio GAO4753g, las reacciones adversas más frecuentes fueron tos (15%), infección del tracto respiratorio superior (12%), neutropenia (11%), sinusitis (10%), diarrea (8%), reacciones relacionadas con la infusión (8%), náuseas (8%), fatiga (8%), bronquitis (7%), artralgia (7%), fiebre (6%), nasofaringitis (6%) e infecciones del tracto urinario (6%). Las reacciones adversas más frecuentes de Grados 3-5 fueron neutropenia (10%) y anemia, neutropenia febril, trombocitopenia, sepsis, infección del tracto respiratorio superior e infecciones del tracto urinario (todas al 1%). El perfil de reacciones adversas del subgrupo de pacientes con LF fue coherente con la población total LNHi. Descripción de reacciones adversas seleccionadas: Reacciones relacionadas con la infusión (RRI): Los síntomas asociados con RRI informados más frecuentemente (≥5%) fueron náuseas, fatiga, escalofríos, hipotensión, fiebre, vómitos, disnea, eritema, hipertensión, cefalea, taquicardia, mareos y diarrea. También se notificaron síntomas respiratorios y cardíacos, como broncospasmo, irritación de laringe y garganta, sibilancia, edema laríngeo y fibrilación auricular (ver Precauciones). Leucemia Linfática Crónica: La incidencia de RRI fue mayor en el grupo de Gazyva más clorambucilo en comparación con el de rituximab más clorambucilo. La incidencia de las RRI fue del 65% durante la infusión de los primeros 1.000 mg de Gazyva (el 20% de los pacientes experimentó RRI de Grados 3-5, sin eventos fatales informados). En general, el 7% tuvo una RRI que condujo a la discontinuación de Gazyva. La incidencia de RRI en las infusiones siguientes fue del 3% con la segunda dosis de 1.000 mg y del 1% con las dosis siguientes. No se notificaron RRI Grados 3-5 después de las primeras infusiones de 1.000 mg del ciclo 1. Se observó una reducción en la incidencia de las RRI de todos los Grados, en los pacientes para los que se tomaron las medidas necesarias para la prevención de RRI según se describe en Dosificación (dosis adecuadas de corticosteroides, analgésico/antihistamínico oral, omisión de la medicación antihipertensiva en la mañana de la primera infusión y administración de la dosis del día 1 del ciclo 1 durante 2 días). Las tasas de RRI de Grados 3-4 (que se verificaron en un número relativamente pequeño de pacientes) fueron similares antes y después de la implementación de las medidas de prevención. Linfoma no Hodgkin indolente, incluido Linfoma Folicular: En el ciclo 1, la incidencia global de RRI fue mayor en los pacientes tratados con Gazyva más bendamustina (G+B) (55%) en comparación con aquellos que recibieron bendamustina (B) en monoterapia (42%) (se informaron RRI de Grados 3-5 en el 9% y el 2% de los pacientes, respectivamente y no se notificaron eventos fatales). En pacientes tratados con G+B, la incidencia de RRI fue mayor el día 1 (38%) y disminuyó gradualmente los días 2, 8 y 15 (25%, 7% y 4%, respectivamente). Durante el ciclo 2, la incidencia de RRI fue menor en los pacientes tratados con G+B (24%) en comparación con aquellos tratados con B en monoterapia (32%). La incidencia de RRI en infusiones posteriores fue comparable en ambos grupos y disminuyó con cada ciclo. También se observaron RRI en el 8% de los pacientes durante el período de mantenimiento con Gazyva. En general, el 3% de los mismos experimentó RRI, dando lugar a la interrupción de Gazyva. Neutropenia e infecciones: Leucemia Linfática Crónica: La incidencia de neutropenia fue mayor en el grupo tratado con Gazyva más clorambucilo (41%) que en el que recibió rituximab más clorambucilo, con la neutropenia resuelta de manera espontánea o a través del uso de factores estimulantes de colonias de granulocitos. La incidencia de infección fue del 38% en el grupo de Gazyva más clorambucilo y del 37% en el de rituximab más clorambucilo (se informaron eventos de Grados 3-5 en el 12% y el 14% de los pacientes, respectivamente, y eventos fatales en < 1% en ambos grupos de tratamiento). También se comunicaron casos de neutropenia prolongada (2% en el grupo tratado con Gazyva más clorambucilo y 4% en el de rituximab más clorambucilo) y neutropenia de inicio tardío (16% en el grupo tratado con Gazyva más clorambucilo y 12% en el de rituximab más clorambucilo) (ver Precauciones). Linfoma no Hodgkin indolente, incluido Linfoma Folicular: La incidencia de neutropenia fue mayor en el grupo tratado con Gazyva más bendamustina (G+B) en comparación con el grupo tratado con bendamustina (B) en monoterapia (38% y 32%, respectivamente). La incidencia de infección fue del 66% en el grupo G+B y 57% en el grupo B (se informaron eventos de Grados 3-5 en el 18% y 17% de los pacientes, respectivamente, y eventos fatales en 5 (3%) en el grupo G+B y 7 (4%) en el grupo B). También se han notificado casos de neutropenia prolongad