Fuzeon®
ROCHE
Enfuvirtida.
Antiviral de uso sistémico, inhibidor de la fusión.
Composición.
Cada ml de solución reconstituida contiene como principio activo 90 mg de enfuvirtida y como excipientes: carbonato de sodio anhidro 2,39 mg, manitol 22,5 mg, hidróxido de sodio c.s., ácido clorhídrico c.s. en 1 ml de agua para inyectables. Viales de polvo liofilizado de color blanco a blanquecino. Disolvente: Cada vial de 2 ml contiene agua para preparaciones inyectables.
Farmacología.
Código ATC: J05AX07. Grupo farmacoterapéutico: Antiviral de uso sistémico, otros antivirales: inhibidor de la fusión. Propiedades farmacodinámicas: Mecanismo de acción: La enfuvirtida es un miembro de la clase terapéutica denominada inhibidores de la fusión. Se trata de un inhibidor de la reordenación estructural de la gp41 de VIH-1 y actúa uniéndose extracelularmente a esta proteína del virus de manera específica, bloqueando la fusión entre la membrana del virus y la membrana de la célula diana, previniendo la entrada del ARN viral en dicha célula. Actividad antiviral in vitro: La susceptibilidad basal a la enfuvirtida, medida en 612 VIH recombinantes que contenían genes env, procedentes de muestras de ARN del VIH de pacientes de ensayos en Fase III, resultó en una media geométrica EC50 de 0,259 mg/ml (media geométrica + 2DE = 1,96 mg/ml) en un ensayo de entrada de recombinación de genotipos VIH. Además, la enfuvirtida inhibió la fusión entre células mediada por la cubierta del VIH-1. Los estudios de combinación de enfuvirtida con miembros representativos de las distintas clases antirretrovirales mostraron una actividad antiviral de aditiva a sinérgica y la ausencia de antagonismo. No se ha establecido ninguna relación entre la sensibilidad in vitro del VIH-1 a la enfuvirtida y la inhibición de la replicación de VIH-1 en la especie humana. Resistencia a los antirretrovirales: La supresión viral incompleta puede llevar al desarrollo de resistencia farmacológica a uno o más componentes del tratamiento. Resistencia in vitro a la enfuvirtida: Se han seleccionado cepas de VIH-1 in vitro con menor sensibilidad a la enfuvirtida que contienen sustituciones en los aminoácidos (aa) 36-38 del ectodominio gp41. Estos cambios se correlacionan con varios niveles de disminución en la sensibilidad a la enfuvirtida de cepas de VIH con mutación específica de sitio. Resistencia in vivo a la enfuvirtida: Se observó una reducción de la susceptibilidad a la enfuvirtida de más de 4 veces al comparar los recombinantes VIH que contenían genes env, obtenidos a partir de muestras de ARN del VIH tomadas antes de la semana 24, y procedentes de 187 pacientes participantes en los ensayos clínicos en Fase III, frente a las muestras pretratamiento correspondientes. De éstos, 185 (98,9%) genes env portaban sustituciones específicas en la región del aa 36-45 de la gp41. Las sustituciones observadas, en orden decreciente de frecuencia, se localizaron en los aminoácidos de las posiciones 38, 43, 36, 40, 42 y 45. Cada sustitución única específica en estos residuos de la gp41 originó una disminución gradual en la susceptibilidad viral recombinante a la enfuvirtida en comparación con las condiciones iniciales. Los cambios de la media geométrica oscilaban entre 15,2 veces para V38M y 41,6 veces para V38A. Los ejemplos de sustituciones múltiples fueron insuficientes para determinar un patrón de sustituciones consistente o su efecto en la susceptibilidad viral a la enfuvirtida. No se ha establecido ninguna relación entre estas sustituciones y la eficacia in vivo del tratamiento con enfuvirtida. La disminución de la sensibilidad viral se correlacionaba con el grado de resistencia pretratamiento a la terapia optimizada (véase Tabla 2). Resistencia cruzada: Como consecuencia de la nueva diana viral, la enfuvirtida se muestra igual de eficaz in vitro frente a las cepas salvajes de laboratorio y clínicas que frente a aquellas con resistencia a 1, 2 ó 3 clases de antirretrovirales diferentes (inhibidores de la transcriptasa inversa análogos de nucleósidos, inhibidores de la transcriptasa inversa no análogos de nucleósidos e inhibidores de la proteasa). A la inversa, las mutaciones de los aminoácidos 36-45 de la gp41, que confieren resistencia a la enfuvirtida, no deberían dar resistencia cruzada a otras clases de antirretrovirales. Eficacia clínica: Ensayos en pacientes pretratados con antirretrovirales: La actividad clínica de Fuzeon (en combinación con otros antirretrovirales) sobre los valores plasmáticos del ARN del VIH y recuento de linfocitos CD4 se ha investigado en dos ensayos aleatorizados, multicéntricos y controlados (TORO 1 y TORO 2) de 48 semanas de duración. La población por intención de tratar comprendía 995 pacientes. Los datos demográficos de los pacientes de cada grupo, Fuzeon más Tratamiento Optimizado y Tratamiento Optimizado, fueron una mediana basal de ARN del VIH-1 de 5,2 log10 copias/ml y 5,1 log10 copias/ml y una mediana basal del recuento de células CD4 de 88 células/mm3 y 97 células/mm3, respectivamente. Los pacientes tuvieron una exposición previa a una mediana de 12 antirretrovirales durante una mediana de 7 años. Todos recibieron un Tratamiento Optimizado que contenía de 3 a 5 agentes antirretrovirales seleccionados de acuerdo con la historia terapéutica previa de cada paciente, así como con los resultados basales de resistencias virales genotípicas y fenotípicas. La proporción de pacientes que alcanzaron una carga viral de < 400 copias/ml en la semana 48 fue del 30,4% entre los tratados con el régimen de Fuzeon más Tratamiento Optimizado comparado con el 12% entre los que recibieron Tratamiento Optimizado solo. El principal aumento de recuento de células CD4 fue mayor en los pacientes en el régimen de Fuzeon más Tratamiento Optimizado que en los que se administró Tratamiento Optimizado solo (véase Tabla 1).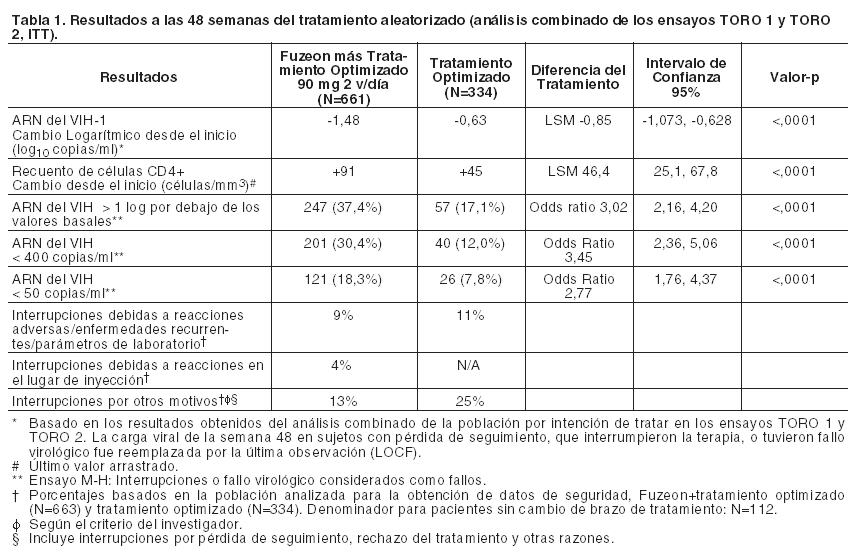
La terapia de Fuzeon más Tratamiento Optimizado se asoció con una mayor proporción de pacientes que alcanzaron < 400 copias/ml (o < 50 copias/ml) en todos los subgrupos establecidos según los CD4 basales, el ARN del VIH-1 basal, según el número previo de antirretrovirales (ARVs) o de ARVs activos en el régimen Tratamiento Optimizado. Sin embargo, los pacientes con valores iniciales de CD4 > 100 células/mm3, valores iniciales de ARN del VIH-1 < 5,0 log10 copias/ml, ≤ 10 ARVs previos, y/u otros ARVs activos en su régimen Tratamiento Optimizado tuvieron más probabilidades de alcanzar un ARN del VIH-1 de < 400 copias/ml (o < 50 copias/ml) en cualquiera de los tratamientos (véase Tabla 2). 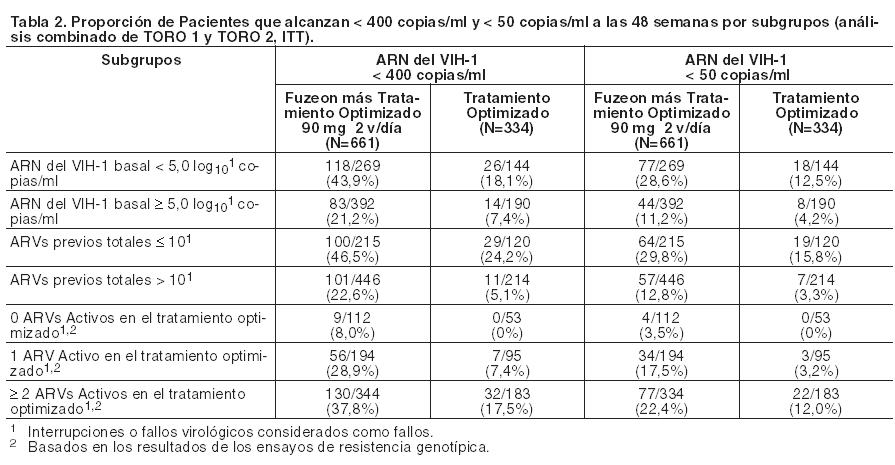
Propiedades farmacocinéticas: Las propiedades farmacocinéticas de la enfuvirtida se han examinado en adultos y niños infectados por el VIH-1. Absorción: La biodisponibilidad absoluta después de la administración subcutánea de 90 mg de enfuvirtida en el abdomen resultó de 84,3 ± 15,5%. La Cmáx media (± DE) fue de 4,59 ± 1,5 mg/ml, y el área bajo la curva (ABC) de 55,8 ± 12,1 mg•h/ml. La absorción subcutánea de la enfuvirtida es proporcional a la dosis administrada en el intervalo de 45 a 180 mg. La absorción subcutánea de la dosis de 90 mg es comparable cuando se inyecta en el abdomen, el muslo o el brazo. El valor medio de concentración plasmática mínima en estado estacionario varió entre 2,6 y 3,4 mg/ml en cuatro ensayos diferentes (N= 9 a 12). Distribución: El volumen de distribución en el estado estacionario después de la administración intravenosa de una dosis de 90 mg de enfuvirtida fue de 5,5 ± 1,1 litros. La enfuvirtida se une en un 92% a las proteínas del plasma infectado por el VIH, en un intervalo de concentraciones plasmáticas de 2 a 10 mg/ml. La enfuvirtida se une sobre todo a la albúmina y, en menor medida, a la glicoproteína ácida a-1. En los estudios in vitro, la enfuvirtida no fue desplazada de su lugar de unión por otros fármacos, ni tampoco desplazaba a otros fármacos de sus lugares de unión. Se han notificado niveles insignificantes de enfuvirtida en el líquido cefalorraquídeo en pacientes con VIH. Biotransformación: La enfuvirtida, como péptido que es, se cataboliza en sus aminoácidos constituyentes; luego, los aminoácidos se reciclan dentro del organismo. Los estudios in vitro con microsomas humanos y los realizados in vivo señalan que la enfuvirtida no inhibe las enzimas del citocromo P450. En estudios in vitro con microsomas y hepatocitos humanos, la hidrólisis del grupo amida de la fenilalanina carboxiterminal determina un metabolito desamidado, cuya formación no depende del NADPH. Este metabolito se detecta en el plasma humano después de administrar la enfuvirtida y con un valor del ABC que varía entre el 2,4 y el 15% del ABC de la enfuvirtida. Eliminación: El clearance de la enfuvirtida después de la administración intravenosa de 90 mg fue de 1,4 ± 0,28 l/h y la vida media de eliminación de 3,2 ± 0,42 horas. Después de administrar una dosis subcutánea de 90 mg de enfuvirtida, la vida media de eliminación (± DE) es de 3,8 ± 0,6 horas. No se han realizado estudios de balance de masa en seres humanos para determinar la(s) ruta(s) de eliminación de la enfuvirtida. Farmacocinética en poblaciones especiales: Pacientes pediátricos: Se ha investigado la farmacocinética de la enfuvirtida en 37 niños. La dosis de 2 mg/kg aplicada dos veces por día (con un máximo de 90 mg, dos veces por día) proporcionó concentraciones plasmáticas de enfuvirtida similares a las de los pacientes adultos tratados con 90 mg, dos veces por día. Los valores obtenidos entre 25 niños de 5 a 16 años que recibieron una dosis de 2 mg/kg, dos veces por día, inyectada en el brazo, cara anterior del muslo o abdomen fueron estos: mediana del ABC en el estado estacionario de 54,3 ± 23,5 mg•h/ml, Cmáx de 6,14 ± 2,48 mg/ml, y Cmín de 2,93 ± 1,55 mg/ml. Pacientes de edad avanzada: La farmacocinética de la enfuvirtida tampoco se ha investigado de manera formal entre personas de 65 ó más años. Pacientes con insuficiencia renal: El análisis de los datos de la concentración plasmática de los pacientes que intervinieron en los ensayos clínicos revela que el clearance de la enfuvirtida no se ve afectado con ningún efecto clínico relevante en pacientes con insuficiencia renal de leve a moderada. En un ensayo en pacientes con insuficiencia renal, el ABC de enfuvirtida aumentó por término medio un 43 - 62 % en aquéllos con insuficiencia renal grave y en los que se hallaban en el estado final de la enfermedad renal, en comparación con los que tenían función renal normal. La hemodiálisis no modifica considerablemente el clearance de enfuvirtida. Se eliminó menos de un 13 % de la dosis durante la hemodiálisis. No se requiere ajuste de dosis en los pacientes con la función renal afectada. Pacientes con insuficiencia hepática: No se ha investigado la farmacocinética de la enfuvirtida en pacientes con alteración de la función hepática. Pacientes según su sexo y peso corporal: El análisis de los datos de la concentración plasmática de los pacientes de los ensayos clínicos puso de relieve que el clearance de la enfuvirtida es un 20% inferior en el sexo femenino que en el masculino, con independencia del peso, y aumenta según lo hace el peso corporal, con independencia del sexo (20% superior para personas de 100 kg y 20% inferior para pacientes de 40 kg, en relación con un paciente prototipo de 70 kg). Sin embargo, estas variaciones carecen de significado clínico y no se precisa ningún ajuste posológico. Pacientes según su etnia: El análisis de los datos de la concentración plasmática en los pacientes de los ensayos clínicos, indica que el clearance de la enfuvirtida no difiere entre afroamericanos comparado con caucásicos. Los demás estudios farmacocinéticos, tampoco señalan diferencias entre los asiáticos y los caucásicos, una vez ajustada la exposición según el peso corporal. Datos preclínicos sobre seguridad: Los datos en los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad y desarrollo embrionario tardío. No se han realizado estudios de carcinogenicidad a largo plazo en animales. En los estudios con cobayas se observó que la enfuvirtida induce una hipersensibilidad tardía por contacto. En un modelo con ratas para estudiar la resistencia a la infección por influenza, se observó una alteración en la producción de IFN-c (Interferón-gamma). La resistencia a la infección por influenza y estreptococo en ratas tan sólo estuvo débilmente comprometida. Se desconoce la relevancia clínica de estos hallazgos.
Indicaciones.
Fuzeon está indicado en combinación con otros medicamentos antirretrovirales en el tratamiento de pacientes infectados por el VIH-1 que han recibido tratamiento, y a los que les han fallado los tratamientos con al menos un medicamento de cada una de las siguientes clases antirretrovirales: inhibidores de la proteasa, inhibidores de la transcriptasa inversa no análogos de nucleósidos e inhibidores de la transcriptasa inversa análogos de nucleósidos, o que han tenido intolerancia a tratamientos antirretrovirales previos (véase Farmacología, Propiedades farmacodinámicas). Para decidir el nuevo tratamiento en los pacientes que hayan fallado a un tratamiento antirretroviral, hay que prestar atención especial a la historia terapéutica de cada paciente y a los patrones de mutaciones asociados con los distintos medicamentos. Se deberían realizar tests de resistencias siempre que fuera posible (véanse Farmacología, Propiedades farmacodinámicas y Precauciones).
Dosificación.
Fuzeon debe ser indicado por médicos con experiencia en el tratamiento de la infección causada por el VIH. Posología: Pacientes adultos y adolescentes ≥ 16 años: La dosis recomendada de Fuzeon es de 90 mg, dos veces por día, en inyección subcutánea en el brazo, la cara anterior del muslo o el abdomen. En caso de que se olvide una dosis de Fuzeon, se debe instruir a los pacientes a administrarse la dosis lo antes posible, si quedan menos de 6 horas antes de la siguiente dosis habitual, se debe omitir la dosis olvidada. Niños ≥ 6 años y adolescentes: La experiencia en niños es limitada (véase Farmacología, Propiedades farmacocinéticas). La pauta posológica que se utilizó en los ensayos clínicos es la que se indica en la Tabla 3.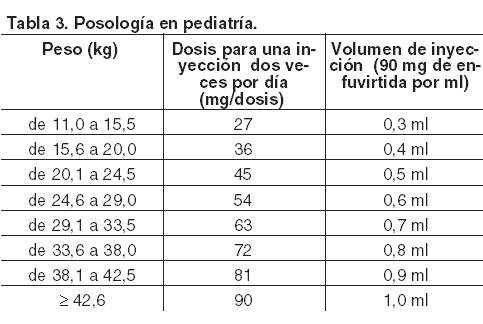
Fuzeon no está recomendado para su uso en niños menores de 6 años, debido a que los datos sobre seguridad y eficacia disponibles no son suficientes (véase Farmacología, Propiedades farmacocinéticas). Pacientes de edad avanzada: No hay experiencia en pacientes mayores de 65 años. Pacientes con insuficiencia renal: No es necesario ajustar la dosis de los pacientes con insuficiencia renal, incluyendo aquellos tratados con diálisis (véanse Precauciones y Farmacología, Propiedades farmacocinéticas). Pacientes con insuficiencia hepática: No se dispone de datos para establecer recomendaciones posológicas para los pacientes con insuficiencia hepática (véanse Precauciones y Farmacología, Propiedades farmacocinéticas). Formas de administración: Fuzeon sólo debe administrarse en inyección subcutánea. Para consultar las instrucciones de reconstitución antes de la administración, véase Precauciones especiales de eliminación y otras manipulaciones.
Contraindicaciones.
Fuzeon está contraindicado en pacientes con hipersensibilidad conocida a la enfuvirtida o a cualquiera de sus excipientes.
Reacciones adversas.
Resumen del perfil de seguridad: La información de seguridad se refiere principalmente a los datos de 48 semanas de los ensayos TORO 1 y TORO 2 combinados (véase Farmacología, Propiedades Farmacodinámicas). Los resultados de seguridad se expresan como el número de pacientes con una reacción adversa por cada 100 pacientes-año de exposición (a excepción de las reacciones en el lugar de inyección). Los acontecimientos notificados más frecuentemente fueron reacciones en el lugar de inyección, diarrea y náuseas. La adición de Fuzeon a la terapia antirretroviral previa generalmente no aumenta la frecuencia o la gravedad de la mayoría de las reacciones adversas. Tabla de reacciones adversas: La Tabla 5 muestra los acontecimientos observados en una proporción mayor entre pacientes tratados con Fuzeon más Tratamiento Optimizado que entre los tratados sólo con régimen de Tratamiento Optimizado; este incremento, ajustado por la exposición, fue de al menos 2 pacientes con acontecimiento por cada 100 pacientes-año. Se observó un incremento estadísticamente significativo para neumonía y linfodenopatía. La mayoría de las reacciones adversas fueron de intensidad leve o moderada. Las reacciones adversas se ordenan según el sistema de clasificación de órganos de MedDRA y por categoría de frecuencia. Las categorías de frecuencia se definen como: muy frecuentes (≥1/10); frecuentes (≥1/100 a < 1/10); poco frecuentes (≥1/1.000 a < 1/100); raras (≥1/10.000 a < 1/1.000); muy raras ( < 1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).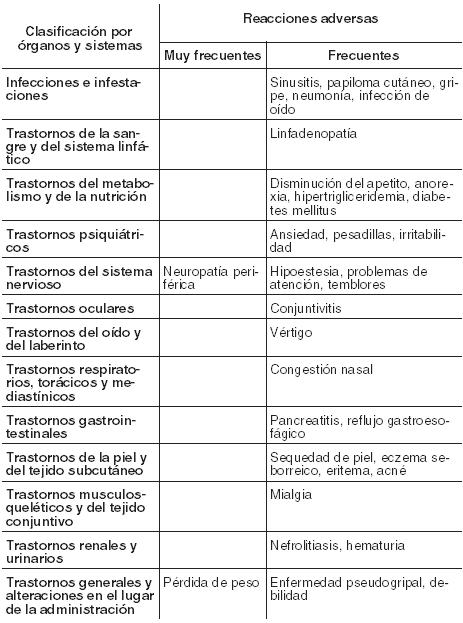
Descripción de reacciones adversas seleccionadas: Reacciones en el lugar de inyección: Las reacciones en el lugar de inyección (RLIs) fueron los eventos adversos más frecuentes y ocurrieron en el 98% de los pacientes (Tabla 6). La inmensa mayoría de las RLIs se manifestó en la primera semana de tratamiento con Fuzeon, y se asoció con dolor o molestias de intensidad leve o moderada en el lugar de inyección, que no limitaron las actividades habituales de los pacientes. La intensidad del dolor o de las molestias asociadas con las RLIs no aumentaron en el curso del tratamiento. Por lo general, la duración de los signos y los síntomas fue igual o menor de 7 días. Las infecciones en el lugar de inyección (incluidos los abscesos y la celulitis) se produjeron en el 1,5% de los pacientes.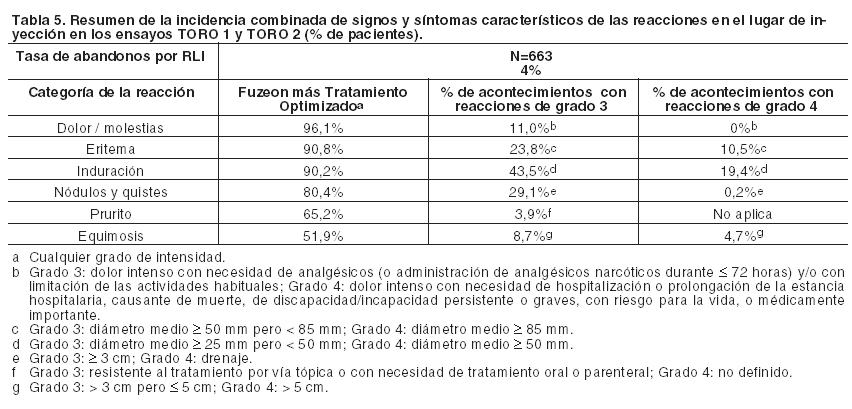
Otras reacciones adversas: Generalmente la adición de Fuzeon al tratamiento antirretroviral optimizado no aumentó la frecuencia ni la gravedad de la mayoría de las reacciones adversas. Los acontecimientos comunicados con más frecuencia y que ocurrieron durante los ensayos TORO 1 y TORO 2 fueron diarrea (38 pacientes con acontecimiento entre los tratados con Fuzeon más Tratamiento Optimizado por cada 100 pacientes-año versus 73 con acontecimiento entre los tratados con Tratamiento Optimizado por cada 100 pacientes-año) y náuseas (27 pacientes con acontecimiento entre los tratados con Fuzeon más Tratamiento Optimizado por cada 100 pacientes-año versus 50 con acontecimiento entre los tratados con Tratamiento Optimizado por cada 100 pacientes-año). Además, se observó un pequeño número de reacciones de hipersensibilidad, atribuidas a la enfuvirtida y, en algunos casos recidivaron tras la reexposición (véase Precauciones y advertencias). Al inicio de la terapia antirretroviral combinada, en los pacientes infectados por VIH con deficiencia inmune grave, puede aparecer una respuesta inflamatoria frente a infecciones oportunistas latentes o asintomáticas (véase Precauciones). También se informaron trastornos autoinmunitarios, como por ejemplo, enfermedad de Graves; sin embargo, el tiempo notificado hasta su aparición es más variable y estos eventos pueden suceder muchos meses después del inicio del tratamiento (véase Precauciones). Se han notificado casos de osteonecrosis, especialmente en pacientes con factores de riesgo generalmente reconocidos, enfermedad avanzada por VIH o exposición prolongada al tratamiento antirretroviral combinado (TARC). Se desconoce la frecuencia de esta reacción adversa (véase Precauciones). Las siguientes reacciones adversas se informaron asimismo en el análisis de 24 semanas de los dos estudios fundamentales con una incidencia > 2% y una frecuencia mayor en los pacientes tratados con Fuzeon más Tratamiento Optimizado que en los que habían recibido el régimen de Tratamiento Optimizado solo. No se ha establecido una relación causal entre estos eventos adversos y Fuzeon. Infecciones e infestaciones: candidiasis oral, herpes simple, foliculitis. Trastornos psiquiátricos: insomnio, depresión. Trastornos neurológicos: cefalea, mareo (excluido vértigo), disgeusia. Trastornos respiratorios, torácicos y mediastínicos: tos. Trastornos gastrointestinales: dolor epigástrico, estreñimiento, dolor faringolaríngeo. Trastornos de la piel y del tejido subcutáneo: prurito, sudores nocturnos, sudoración aumentada. Trastornos musculosqueléticos y del tejido conjuntivo: artralgia, dorsalgia, dolores en las extremidades, calambres musculares. Trastornos generales y alteraciones en el lugar de la administración: astenia. Se registró una tasa más alta de neumonía bacteriana (se incluyó en el análisis la bronconeumonía y acontecimientos relacionados) entre los tratados con Fuzeon más Tratamiento Optimizado en los estudios TORO 1 y TORO 2 que en el grupo de control con el régimen de Tratamiento Optimizado solo (6,6 y 0,6 pacientes con episodios de neumonía por 100 pacientes-año, respectivamente). Los factores de riesgo de neumonía fueron los siguientes: recuento basal de linfocitos CD4 bajo, carga vírica basal alta, uso de drogas por vía intravenosa, tabaquismo y antecedentes de enfermedad pulmonar. Dado que no estaba claro si la tasa mayor de neumonía guardaba una relación con Fuzeon, se realizó un estudio observacional en pacientes infectados por el VIH (grupo de Fuzeon: 2.045 pacientes-año de observación; grupo comparativo: 3.501 pacientes-año de observación) con el fin de evaluar minuciosamente el riesgo de neumonía por Fuzeon controlando otros factores de riesgo conocidos. En este estudio observacional a gran escala no se demostró ninguna diferencia significativa en el riesgo de neumonía entre los pacientes tratados y los no tratados con Fuzeon después de ajustar los grupos de comparación en cuanto a efectos de factores de riesgo desequilibrados. El índice de riesgo ajustado de neumonía era de 0,989 para neumonía confirmada solamente y de 1.228 para neumonía confirmada o probable, siendo el límite inferior del intervalo de confianza del 95% de 0,437 y 0,862, respectivamente. Población pediátrica: Se estudió Fuzeon en 69 niños y adolescentes de 4 a 16 años, con una exposición al medicamento de entre 1 dosis y > 48 semanas de tratamiento. Los eventos adversos registrados durante los ensayos clínicos eran similares a los observados en pacientes adultos. Alteraciones de laboratorio: La mayoría de los pacientes no experimentó cambios en el grado de toxicidad de ninguno de los parámetros de laboratorio a lo largo del estudio, a excepción de los indicados en la Tabla 7. Durante la semana 48, la eosinofilia [recuento mayor al Límite Superior de Normalidad (LSN) > 0,7 x 109/l] se dio en mayor porcentaje entre el grupo de los pacientes tratados con Fuzeon (12,4 pacientes con acontecimiento por cada 100 pacientes-año) que en los que recibieron solamente Tratamiento Optimizado (5,6 pacientes con acontecimiento por cada 100 pacientes-año). Si se aplica un umbral de eosinofilia más alto ( > 1,4 x 109/l), la tasa ajustada por la exposición del paciente es igual en ambos grupos (1,8 pacientes con eosinofilia por 100 pacientes-año).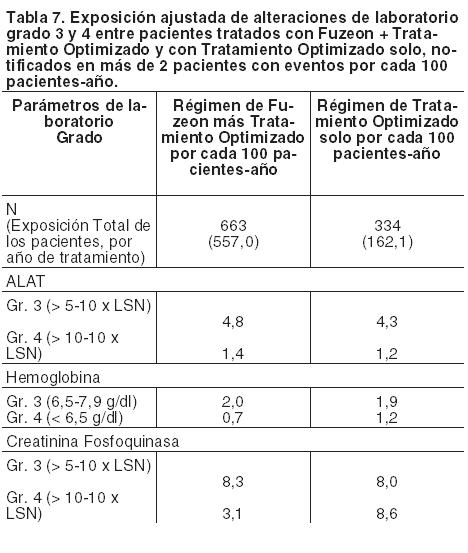
Se notificaron las siguientes alteraciones analíticas en el análisis de 24 semanas de los dos ensayos fundamentales con una incidencia > 2% y una frecuencia mayor en los pacientes tratados con Fuzeon más Tratamiento Optimizado que en los que habían recibido Tratamiento Optimizado solo. No se ha establecido una relación causal entre estos eventos adversos y Fuzeon. Exploraciones complementarias: valores elevados de gamma-glutamiltransferasa, amilasa, lipasa, y AST. Información poscomercialización: Trastornos de la piel y del tejido subcutáneo: amiloidosis cutánea en el sitio de inyección. Comunicación de reportes de reacciones adversas:Es importante comunicar las presuntas reacciones adversas después de la autorización del medicamento. Esto permite la monitorización continua de la relación riesgo/beneficio. Se solicita a los profesionales de la salud informar de cualquier sospecha de eventos adversos asociados con el uso de Fuzeon® al Área de Farmacovigilancia de Roche al siguiente teléfono 0800-77-ROCHE (76243). En forma alternativa, esta información puede ser reportada ante ANMAT. Ante cualquier inconveniente con el producto, el paciente puede llenar la ficha que está en la Página Web de la ANMAT: http://www.anmat.gov.ar/farmacovigilancia/Notificar.asp o llamar a ANMAT responde al 0800-333-1234.
Precauciones.
Fuzeon debe administrarse como parte de un tratamiento combinado. También, se deben consultar los Prospectos Información para Profesionales de los otros medicamentos antirretrovirales que se empleen en la combinación. Al igual que otros antirretrovirales, la enfuvirtida debe asociarse lo más adecuadamente posible con otros antirretrovirales a los cuales el virus del paciente sea sensible (véase Farmacología, Propiedades farmacodinámicas). Se debe informar a los pacientes que Fuzeon no cura la infección por el VIH-1. A pesar de que se ha probado que la supresión viral con tratamiento antirretroviral eficaz reduce sustancialmente el riesgo de transmisión sexual, no se puede excluir un riesgo residual. Se deben tomar las medidas apropiadas de precaución, para prevenir la transmisión. Los estudios en animales han demostrado que enfuvirtida puede afectar a algunas de las funciones inmunes (véase Farmacología, Datos preclínicos sobre seguridad). En ensayos clínicos se ha comprobado un aumento de la incidencia de algunas infecciones bacterianas, de manera más notable de neumonía, en pacientes tratados con Fuzeon; sin embargo, un incremento del riesgo de neumonía bacteriana relacionado con el uso de Fuzeon no ha sido confirmado mediante datos epidemiológicos posteriores. El tratamiento con enfuvirtida se ha asociado ocasionalmente con reacciones de hipersensibilidad y en raras circunstancias éstas se han repetido con la reexposición. Las complicaciones comprenden rash, fiebre, náuseas y vómitos, escalofríos, rigidez, tensión sanguínea baja y transaminasas hepáticas elevadas en suero en varias combinaciones, y posible reacción primaria del complejo inmune, dificultad respiratoria y glomerulonefritis. Los pacientes que presenten signos o síntomas de una reacción de hipersensibilidad sistémica suspenderán la administración de enfuvirtida y acudirán al médico para su evaluación de inmediato. El tratamiento con enfuvirtida no se debe reiniciar tras la aparición de signos y síntomas sistémicos característicos de una reacción de hipersensibilidad que se considere como relacionada con enfuvirtida. No se han identificado los factores de riesgo que pronostican la aparición o la gravedad de la hipersensibilidad a la enfuvirtida. Enfermedad hepática: La seguridad y la eficacia de la enfuvirtida no se han estudiado de manera específica en pacientes con trastornos hepáticos subyacentes significativos. Aquéllos con hepatitis crónica B y C, en tratamiento con antirretrovirales tienen mayor riesgo de experimentar acontecimientos adversos hepáticos graves o potencialmente mortales. Algunos pacientes que participaron en los ensayos en Fase III estaban coinfectados por la hepatitis B/C. En ellos la adición de Fuzeon no aumentó la incidencia de acontecimientos hepáticos. En caso de tratamiento antirretroviral concomitante para la hepatitis B o C, se deberá consultar también el Prospecto Información para Profesionales del producto relevante para estos medicamentos. La administración de Fuzeon a sujetos no infectados por el VIH-1 puede provocar la formación de anticuerpos antienfuvirtida que reaccionen en forma cruzada con el antígeno gp41 del VIH. Esto podría ocasionar un resultado falso positivo del test de ELISA de anticuerpos anti-VIH. No hay experiencia en pacientes con la función hepática disminuida. Los datos son limitados en pacientes con insuficiencia renal de moderada a grave y en dializados. Fuzeon se debe emplear con precaución en estas poblaciones (véanse Dosificación y Farmacología, Propiedades farmacocinéticas). Síndrome de Reconstitución Inmune: Cuando se instaura una terapia antirretroviral combinada en pacientes infectados por VIH con deficiencia inmune grave, puede aparecer una respuesta inflamatoria frente a patógenos oportunistas latentes o asintomáticos, y provocar situaciones clínicas graves, o un empeoramiento de los síntomas. Normalmente estas reacciones se han observado en las primeras semanas o meses, después del inicio de la terapia antirretroviral combinada. Algunos ejemplos relevantes de estas manifestaciones son: retinitis por citomegalovirus, infecciones micobacterianas generalizadas y/o localizadas y neumonía por Pneumocystis carinii. Se debe evaluar cualquier síntoma inflamatorio y establecer un tratamiento cuando sea necesario. También se han comunicado trastornos autoinmunitarios (como por ejemplo, la enfermedad de Graves y síndrome de Guillain-Barré) durante la reconstitución inmune; sin embargo, el tiempo notificado hasta su aparición es más variable y estos acontecimientos pueden suceder muchos meses después del inicio del tratamiento. Osteonecrosis: Se han informado casos de osteonecrosis, especialmente en pacientes con infección avanzada por VIH y/o exposición prolongada al tratamiento antirretroviral combinado (TARC), aunque se considera que la etiología es multifactorial (incluyendo uso de corticosteroides, consumo de alcohol, inmunodepresión grave, índice de masa corporal elevado). Se debe aconsejar a los pacientes que consulten con el médico si experimentan molestias o dolor articular, rigidez articular o dificultad para moverse. Efectos sobre la capacidad para conducir y utilizar máquinas: No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas. No existen pruebas de que la enfuvirtida pueda alterar la aptitud del paciente para conducir o utilizar máquinas; no obstante, hay que tener en cuenta el perfil de reacciones adversas de la enfuvirtida (véase Reacciones adversas). Fertilidad, embarazo y lactancia: Embarazo: No se han realizado estudios adecuados y bien controlados en mujeres embarazadas. Los ensayos en animales no muestran efectos dañinos sobre el desarrollo fetal. La enfuvirtida debe utilizarse durante el embarazo sólo si el beneficio potencial justifica el riesgo potencial para el feto. Lactancia: No se sabe si la enfuvirtida se excreta por la leche humana. Se debe informar a las madres que no deben dar el pecho si están recibiendo enfuvirtida, debido a la posibilidad de transmisión del VIH y de cualquier posible efecto adverso en los lactantes.
Interacciones.
Los estudios de interacciones se han realizado sólo en adultos. No se esperan interacciones farmacocinéticas con repercusión clínica entre la enfuvirtida y los otros medicamentos que se administran en forma simultánea y se metabolizan por las enzimas del citocromo P450. Efecto de la enfuvirtida sobre el metabolismo de medicamentos concomitantes: Según los resultados de un estudio in vitro con microsomas humanos, la enfuvirtida no inhibe las enzimas CYP450 y, en consecuencia, no altera el metabolismo de los medicamentos metabolizados por estas enzimas. Según un estudio in vivo en el metabolismo humano, la enfuvirtida, en las dosis recomendadas de 90 mg dos veces por día, no inhibe el metabolismo de los sustratos de las isoenzimas CYP3A4 (dapsona), la CYP2D6 (debrisoquina), la CYP1A2 (cafeína), la CYP2C19 (mefenitoína) y la CYP2E1 (clorzoxazona). Efecto de medicamentos concomitantes sobre el metabolismo de la enfuvirtida: Según estudios independientes de interacción farmacocinética, la coadministración de ritonavir (inhibidor potente de la CYP3A4) o saquinavir en combinación con una dosis potenciada de ritonavir o rifampicina (inductor potente de la CYP3A4) no produjo cambios clínicos significativos en la farmacocinética de la enfuvirtida.
Incompatibilidades.
Este medicamento no debe mezclarse con otros medicamentos, excepto los mencionados en "Precauciones especiales de eliminación y otras manipulaciones".
Conservación.
Polvo: Conservar a temperatura inferior a 30°C. Para las condiciones de conservación del medicamento reconstituido, véase "Período de validez". Disolvente: No requiere condiciones especiales de conservación. Precauciones especiales de eliminación y otras manipulaciones: La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local. Los pacientes deben recibir instrucciones de los profesionales sanitarios sobre el uso y la administración de Fuzeon antes de utilizarlo por primera vez. Fuzeon sólo debe reconstituirse con 1,1 ml de agua para preparaciones inyectables. Los pacientes deben aprender a añadir el agua para preparación inyectable y golpear el vial suavemente con la yema de los dedos hasta que el polvo empiece a disolverse. Nunca se debe agitar el vial ni invertirlo para mezclarlo, pues esto provocará que se produzca demasiada espuma. Una vez que el polvo empiece a disolverse se puede dejar reposar el vial para permitir la completa disolución. El polvo puede tardar hasta 45 minutos en disolverse. El paciente puede hacer rodar el vial suavemente entre sus manos después de añadir el agua para preparación inyectable hasta que el polvo esté completamente disuelto, lo que puede reducir el tiempo que tarda éste en disolverse. Antes de retirar la solución para su administración, el paciente debe realizar una inspección visual del vial para verificar que se ha disuelto todo el contenido, que la solución es transparente y que no presenta burbujas ni partículas. Si se observan partículas en suspensión, no debe utilizarse el vial, sino que se debe desechar o devolver a la farmacia. Los viales de disolvente contienen 2 ml de agua para preparación inyectable, de los cuales 1,1 ml deben extraerse para la reconstitución del polvo. Se debe informar a los pacientes que desechen el volumen restante de los viales de disolvente. Fuzeon no contiene conservantes. Una vez reconstituido, debe inyectarse de inmediato. Si no se puede inyectar enseguida la solución reconstituida, debe mantenerse en heladera y utilizarse antes de 24 horas. La solución reconstituida y refrigerada debe llevarse a la temperatura ambiente antes de su inyección. 1 ml de solución reconstituida se inyecta por vía subcutánea en el brazo, abdomen o cara anterior del muslo. La inyección debe realizarse en un sitio diferente al de la inyección previa y donde no se haya producido una reacción en el lugar de inyección. Cada vial sirve para un solo uso; se deben desechar las porciones no utilizadas. Este medicamento no debe ser utilizado después de la fecha de vencimiento indicada en el envase. Este medicamento debe ser usado exclusivamente bajo prescripción y vigilancia médica y no puede repetirse sin nueva receta médica. Mantenga los medicamentos fuera del alcance de los niños. Período de validez: Período de validez después de la reconstitución: Después de su reconstitución, el producto debe emplearse inmediatamente. Si no se utiliza en forma inmediata, debe conservarse en heladera (entre 2°C y 8°C) por un período máximo de 24 horas.
Sobredosificación.
No se han notificado casos de sobredosis. La dosis máxima administrada a 12 pacientes fue de 180 mg en inyección subcutánea única durante un ensayo clínico. Estos pacientes no experimentaron ninguna reacción adversa que no se observara ya con las dosis recomendadas. En un estudio correspondiente al Programa de Acceso Precoz, se administró en una ocasión 180 mg de Fuzeon a un paciente como dosis única, sin que manifestara ningún efecto adverso. No se conoce ningún antídoto específico para tratar la sobredosis de enfuvirtida. El tratamiento de la sobredosis consistiría en las medidas de apoyo habituales. Ante la eventualidad de una sobredosificación concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Dr. Ricardo Gutiérrez: 4962-6666/2247; Policlínico Dr. G. A. Posadas: 4654-6648; 4658-7777; Hospital General de Niños Dr. Pedro de Elizalde: 4300-2115; 4363-2100/2200 Interno 6217.
Presentación.
Envase con: Viales de 3 ml con polvo para inyectable: 60. Viales de 2 ml con disolvente (agua para inyección): 60. Jeringas descartables de 3 ml con protector de seguridad: 60. Jeringas descartables de 1 ml con protector de seguridad: 60. Toallitas empapadas en alcohol: 180.
Revisión.
Enero 2016. Aprobación: 04/04/2016. Disp. ANMAT N° 3.329 (RI+EMA+ANMAT C004/2013 y rcp+Shpe+CDS: 5.0C+6.0C).