ENTRESTOTM
SIEGFRIED
Antagonista de la Angiotensina II, otras combinaciones. Código ATC: C09.
Composición.
Cada Comprimido Recubierto de ENTRESTO 50 mg contiene: Sacubitrilo 24 mg; Valsartán 26 mg. Excipientes. Cada Comprimido Recubierto de ENTRESTO 100 mg contiene: Sacubitrilo 49 mg; Valsartán 51 mg. Excipientes. Cada Comprimido Recubierto de ENTRESTO 200 mg contiene: Sacubitrilo 97 mg. Valsartán 103 mg. Excipientes.
Farmacología.
Descripción: ENTRESTO™ (Sacubitrilo y Valsartán) es una combinación de un inhibidor de la neprilisina y un bloqueante del receptor de angiotensina II. ENTRESTO™ contiene un complejo compuesto de formas aniónicas de sacubitrilo y valsartán, cationes de sodio y moléculas de agua en un coeficiente molar de 1:1:3:2,5; respectivamente. Luego de la administración oral, el complejo se disocia en sacubitrilo (que se sigue metabolizando hasta convertirse en LBQ657) y valsartán. La estructura química del complejo se describe como Octadecasodiohexakis(4-{[(1S,3R)-1-([1,1´bifenil]-4-ilmetil)-4-etoxi-3-metil-4-oxobutil]amino}-4oxobutanoato)hexakis(N-pentanoilN-{[2´-(1H-tetrazol-1-id-5-il)[1,1´-bifenil]-4-il]metil}-L-valinato)-agua (1/15). Su fórmula empírica (hemipentahidrato) es C48H55N6O8Na3 2,5 H2O. Su masa molecular es 957,99 y su fórmula estructural esquemática es: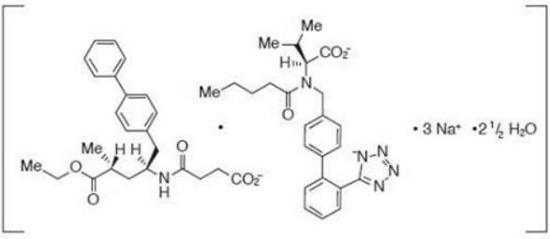
Mecanismo de Acción: ENTRESTO™ contiene un inhibidor de la neprilisina, sacubitrilo, y un bloqueante del receptor de angiotensina II, valsartán. ENTRESTO™ inhibe la neprilisina (endopeptidasa neutra; NEP) por medio de LBQ657, el metabolito activo del profármaco sacubitrilo, y bloquea el receptor tipo 1 (AT1) de la angiotensina II por medio de valsartán. Los efectos cardiovasculares y renales de ENTRESTO en los pacientes con insuficiencia cardiaca se atribuyen a los niveles elevados de péptidos que son degradados por la neprilisina, tales como los péptidos natriuréticos, por LBQ657, y la inhibición simultánea de los efectos de la angiotensina II por medio de valsartán. Valsartán inhibe los efectos de la angiotensina II al bloquear selectivamente el receptor AT1 y también inhibe la liberación de la aldosterona que depende de la angiotensina II. Propiedades Farmacodinámicas: Se evaluaron los efectos farmacodinámicos de ENTRESTO luego de administrar dosis únicas y múltiples en sujetos sanos y en pacientes con insuficiencia cardiaca, y resultaron consistentes con la inhibición simultánea de la neprilisina y el bloqueo del sistema renina angiotensina. En un estudio controlado con valsartán de 7 días de duración conducido en pacientes con Fracción de Eyección Reducida (HFrEF), la administración de ENTRESTO™ produjo un incremento significativo no sostenido de la natriuresis, Cgmp urinario elevado, y una disminución en los niveles plasmáticos de MR-proANP y NTproBNP en comparación con valsartán. En un estudio de 21 días de duración conducido en pacientes con HFrEF, ENTRESTO incrementó significativamente el ANP y cGMP urinarios y el cGMP plasmático, y disminuyó los niveles plasmáticos de NT-proBNP, de aldosterona y de endotelina-1. ENTRESTO™ también bloqueó el receptor AT1 evidenciado por la mayor actividad de la renina plasmática y de las concentraciones de renina plasmática. En el estudio PARADIGM-HF, ENTRESTO™ redujo el NT-proBNT plasmático (que no es un sustrato de la neprilisina) e incrementó el BNP plasmático (un sustrato de la neprilisina) y el cGMP urinario en comparación con enalapril. Prolongación QT: En un estudio clínico minucioso de QTc conducido en sujetos masculinos sanos, las dosis únicas de ENTRESTO 194 mg de sacubitrilo/206 mg de valsartán y 583 mg de sacubitrilo/617 mg de valsartán no tuvieron efecto alguno sobre la repolarización cardiaca. Beta-amiloide: La neprilisina es una de las múltiples enzimas que están involucradas en la depuración del beta-amiloide (Ab) del cerebro y del Líquido Cefalorraquídeo (LCR). La administración de ENTRESTO 194 mg de sacubitrilo/206 mg de valsartán una vez por día durante 2 semanas a sujetos sanos se asoció con un incremento en el LCR Ab1-38 en comparación con placebo. No se observaron cambios en las concentraciones de LCR Ab1-40 ni de LCR Ab1-42. Se desconoce la relevancia clínica de este hallazgo (Ver datos de toxicidad preclínica). Presión Arterial: El agregado de una dosis única de 50 mg de sildenafilo a ENTRESTO en estado estable (194 mg de sacubitrilo/206 mg de valsartán una vez por día durante 5 días) en pacientes con hipertensión se asoció con una reducción adicional en la presión arterial (~5/4 mmHg, presion arterial sistólica/diastólica) en comparación con la administración de ENTRESTO™ solo. La co-administración de ENTRESTO no alteró de manera significativa el efecto sobre la presión arterial de la nitroglicerina intravenosa. Propiedades Farmacocinéticas: Absorción: Luego de la administración oral, ENTRESTO se disocia en sacubitrilo y valsartán. Sacubitrilo se sigue metabolizando hasta convertirse en LBQ657. Las concentraciones plasmáticas pico de sacubitrilo, LBQ657 y valsartán se alcanzan a las 0,5 horas, 2 horas y 1,5 horas, respectivamente. La biodisponibilidad absoluta oral de sacubitrilo se estima que es > 60%. El valsartán que contiene ENTRESTO tiene mayor biodisponibilidad que el valsartán contenido en otras formulaciones de comprimidos comercializados. El contenido de 26 mg, 51 mg y 103 mg de valsartán en ENTRESTO™ es equivalente a 40 mg, 80 mg y 160 mg de valsartán en otras formulaciones de comprimidos comercializados, respectivamente. Luego de administrar ENTRESTO™ dos veces por día, los niveles estables de sacubitrilo, LBQ657 y valsartán se alcanzaron a los 3 días. En estado estable, sacubitrilo y valsartán no se acumularon en forma significativa, en cambio LBQ657 se acumuló en alrededor de 1,6 veces. La administración de ENTRESTO con alimentos no tuvo ningún efecto clínicamente significativo sobre las exposiciones sistémicas de sacubitrilo, LBQ657 o valsartán. A pesar de que hay una disminución en la exposición a valsartán cuando se administra ENTRESTO™ con alimentos, esta disminución no está acompañada de una reducción clínicamente significativa en el efecto terapéutico. Por lo tanto, se puede administrar ENTRESTO con o sin alimentos. Distribución: Sacubitrilo, LBQ657 y valsartán tienen una gran capacidad para unirse a las proteínas plasmáticas (entre 94% y 97%). Sobre la base de la comparación del plasma y de las exposiciones del LCR, LBQ657 cruza la barrera hematoencefálica en forma limitada (0,28%). Los volúmenes aparentes promedio de distribución de valsartán y sacubitrilo son 75 y 103 L, respectivamente. Metabolismo: Sacubitrilo se convierte fácilmente en LBQ657 por medio de esterasas. LBQ657 no se sigue metabolizando de manera significativa. Valsartán se metaboliza muy poco. Solamente cerca del 20% de la dosis se recupera en la forma de metabolitos. Se ha identificado un metabolito hidroxilado en el plasma en concentraciones bajas ( < 10%). Eliminación: Luego de la administración oral, entre el 52% y 68% de sacubitrilo (principalmente como LBQ657) y ~13% de valsartán y sus metabolitos se excretan en la orina. Entre 37% y 48% de sacubitrilo (principalmente como LBQ657), y 86% de valsartán y sus metabolitos se excretan en las heces. Sacubitrilo, LBQ657 y valsartán se eliminan del plasma con una vida media de eliminación media (T1/2) de aproximadamente 1,4 horas, 11,5 horas y 9,9 horas, respectivamente. Linealidad/No Linealidad: La farmacocinética de sacubitrilo, LBQ657 y valsartán fue lineal sobre el rango de dosis de ENTRESTO de 24 mg de sacubitrilo/26 mg de valsartán a 194 mg de sacubitrilo/206 mg de valsartán. Interacciones Medicamentosas: Efecto de los fármacos co-administrados sobre ENTRESTO: Dado que el metabolismo de sacubitrilo y valsartán mediado por la enzima CYP450 es mínimo, no se espera que la co-administración con fármacos que afectan las enzimas CYP450 afecte la farmacocinética de ENTRESTO. Los estudios dedicados a la interacción medicamentosa demostraron que la co-administración de furosemida, warfarina, digoxina, carvedilol, combinación de levonorgestrel/etinilestradiol, amlodipina, omeprazol, hidroclorotiazida, metformina, atorvastatina y sildenafilo no alteró la exposición sistémica a sacubitrilo, LBQ657 ni a valsartán. Efecto de ENTRESTO™ sobre los fármacos co-administrados: Los datos in vitro indican que sacubitrilo inhibe los transportadores OATP1B1 y OATP1B3 Los efectos de ENTRESTO™ sobre la farmacocinética de fármacos coadministrados se resumen en la Figura 1.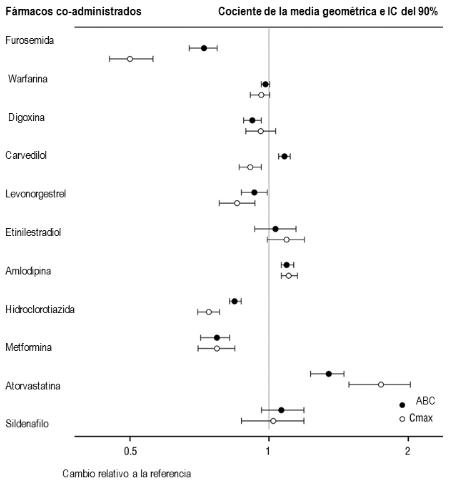
Poblaciones Específicas: El efecto de las poblaciones específicas sobre la farmacocinética de LBQ657 y valsartán se muestra en la Figura 2.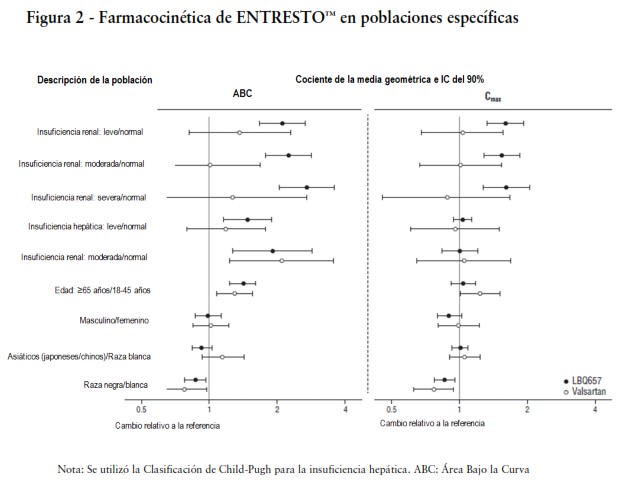
Ensayos clínicos: La dosificación en los estudios clínicos se basó en la cantidad total de ambos componentes de ENTRESTO es decir, 24/26 mg, 49/51 mg y 97/103 mg se refirieron como 50 mg, 100 mg y 200 mg, respectivamente. PARADIGM-HF: PARADIGM-HF fue un estudio doble ciego, aleatorizado, multicéntrico que comparó ENTRESTO con enalapril en 8442 pacientes adultos con insuficiencia cardiaca crónica sintomática (clase II-IV de la NYHA) y disfunción sistólica (fracción de eyección ventricular izquierda ≤ 40%). Los pacientes debían haber recibido un IECA o ARA II durante al menos cuatro semanas y dosis máximas toleradas de beta-bloqueantes. Se excluyeron los pacientes con presión arterial sistólica de < 100 mmHg en la selección. El objetivo primario del estudio PARADIGM-HF fue determinar si ENTRESTO, una combinación de sacubitrilo y un inhibidor del Sistema Renina Angiotensina Aldosterona (SRAA) (valsartán), fue superior al inhibidor del SRAA (enalapril) administrado sólo al reducir el riesgo del criterio de valoración combinado de muerte cardiovascular (CV) u hospitalización por Insuficiencia Cardiaca (IC). Después de interrumpir el tratamiento existente con un IECA o ARA II, los pacientes ingresaron a períodos de prueba simple ciegos secuenciales durante los cuales recibieron enalapril 10 mg dos veces por día, seguido de ENTRESTO 100 mg dos veces por día, que se incrementó a 200 mg dos veces por día. Los pacientes que completaron exitosamente los períodos de prueba secuenciales fueron aleatorizados para recibir ENTRESTO™ 200 mg (N=4209) dos veces por día o enalapril 10 mg (N=4233) dos veces por día. El criterio de valoración primario fue el primer evento en la combinación de muerte CV u hospitalización por IC. La duración mediana del seguimiento fue de 27 meses y se trató a los pacientes por hasta 4,3 años. La población estaba compuesta por un 66% de pacientes caucásicos, un 18 % de pacientes asiáticos y un 5% de pacientes de población negra. La edad media era de 64 años y el 78% eran hombres. En la aleatorización, el 70% de los pacientes era Clase II de la NYHA, 24% era Clase III de la NYHA y 0,7% era Clase IV de la NYHA. La fracción media de eyección ventricular izquierda fue de 29%. La causa subyacente de la insuficiencia cardiaca fue la enfermedad coronaria en el 60% de los pacientes, el 71% tenía antecedentes de hipertensión, el 43% tenía antecedentes de infarto de miocardio, el 37% tenía una tasa de filtrado glomerular (eGFR) < 60 mL/min/1,73m, y el 35% tenía diabetes mellitus. La mayoría de los pacientes estaban tomando beta-bloqueantes (94%), antagonistas mineralocorticoides (58%) y diuréticos (82%). Pocos pacientes tenían un desfibrilador cardioversor implantado o un desfibrilador para el tratamiento de resincronización cardiaca (15%). El estudio PARADIGM-HF demostró que ENTRESTO, una combinación de sacubitrilo y un inhibidor del SRAA (valsartán), fue superior al inhibidor del SRAA (enalapril), al reducir el riesgo del criterio de valoración combinado de muerte cardiovascular (CV) u hospitalización por insuficiencia cardiaca (IC), sobre la base del análisis del tiempo transcurrido hasta la aparición del evento (cociente de riesgo [HR]: 0,80, intervalo de confianza [IC] del 95%, 0,73; 0,87, p < 0,0001). El efecto del tratamiento reflejó una reducción tanto en la muerte cardiovascular como en la hospitalización por insuficiencia cardiaca. Ver Tabla 1 y Figura 3. La muerte súbita representó el 45% de las muertes cardiovasculares, seguidas de la falla de bombeo, que representó el 26%. ENTRESTO™ también mejoró la supervivencia global (HR 0,84; IC del 95% [0,76; 0,93], p = 0,0009) (Tabla 1). Este hallazgo fue determinado en su totalidad por una menor incidencia de la mortalidad cardiovascular con ENTRESTO™.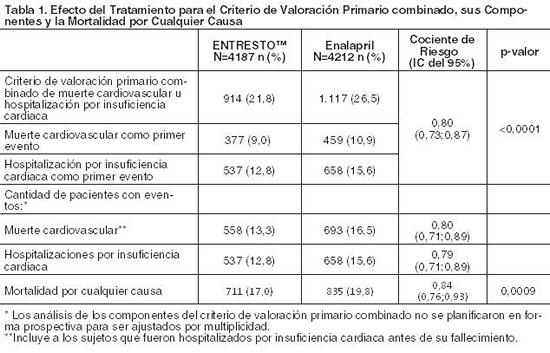
Las curvas de Kaplan-Meier que se presentan a continuación (Figura 3) muestran el tiempo transcurrido hasta la primera aparición del criterio de valoración combinado (3A), y el tiempo transcurrido hasta el acaecimiento de la muerte cardiovascular en cualquier momento (3B) y la primera hospitalización por insuficiencia renal (3C).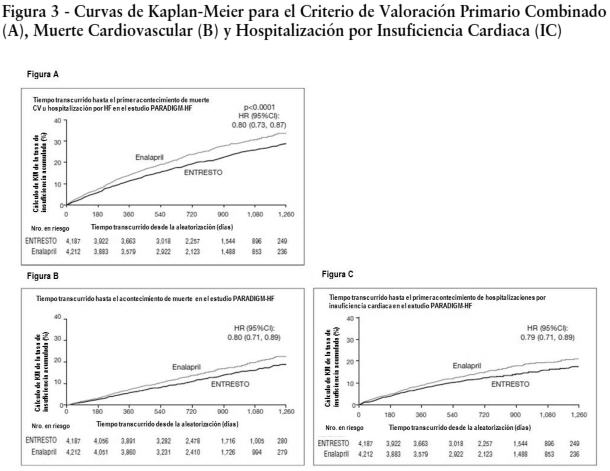
Se examinó una amplia variedad de características demográficas, características de la enfermedad basal y medicamentos concomitantes basales para determinar su influencia sobre los resultados. Los resultados del criterio de valoración primario combinado fueron consistentes en todos los subgrupos examinados (Figura 4).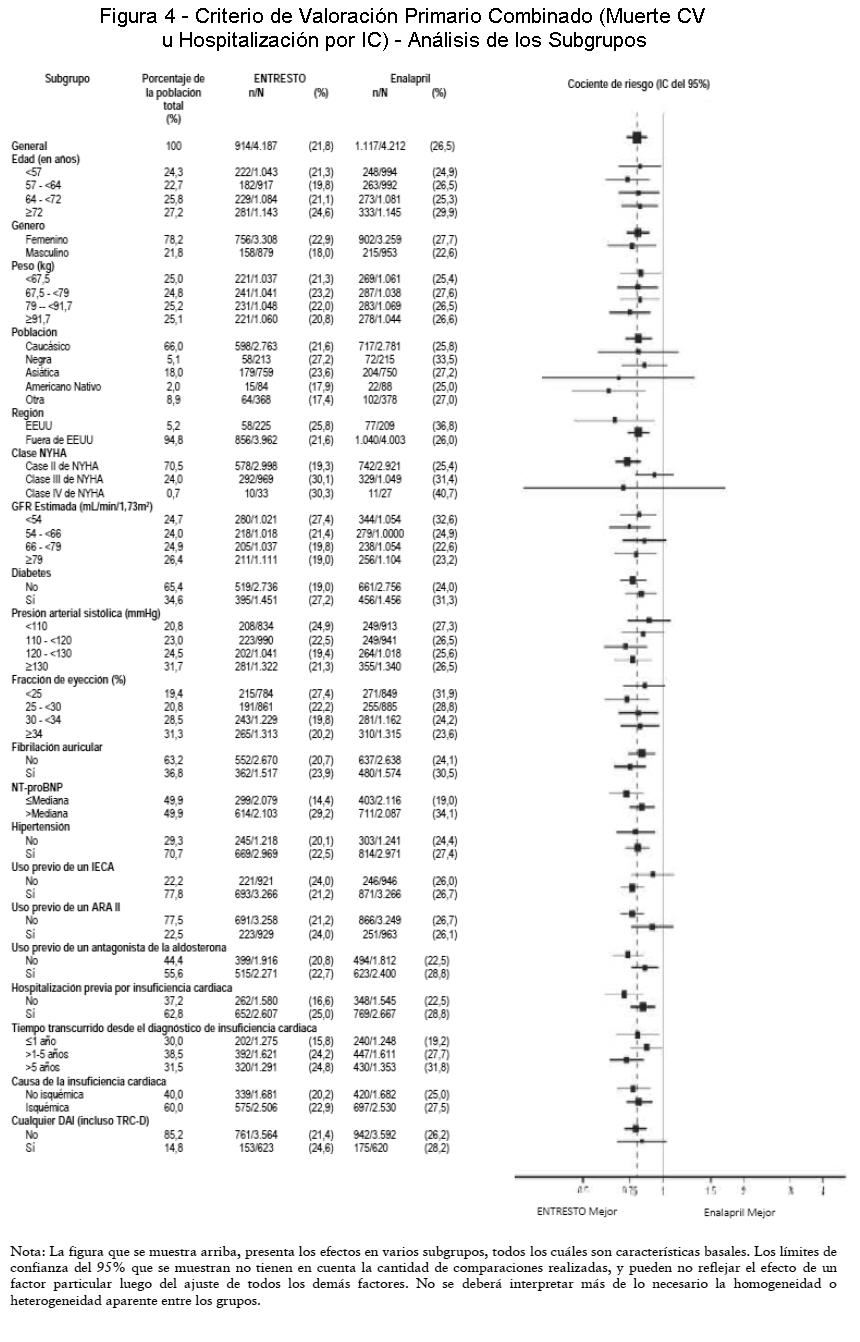
Datos de toxicidad preclínica: Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad: Carcinogénesis y Mutagénesis: Los estudios de carcinogénesis conducidos en ratones y ratas con sacubitrilo y valsartán no identificaron ningún potencial carcinogénico para ENTRESTO™. La Cmáx de LBQ657 con la dosis alta de 1200 mg/Kg/día en los ratones macho y hembra fue, respectivamente, de 14 y 16 veces aquella observada en los seres humanos con la Máxima Dosis Recomendada para Humanos (MDRH). La Cmáx de LBQ657 en las ratas macho y hembra con la HD de 400 mg/Kg/día fue, respectivamente, de 1,7 y 3,5 veces aquélla observada con la MDRH. Las dosis de valsartán estudiadas (dosis alta de 160 y 200 mg/Kg/día en ratones y ratas, respectivamente) fueron alrededor de 4 y 10 veces, respectivamente, la MDRH dependiendo de los mg/m2. Los estudios de mutagenicidad y clastogenicidad conducidos con ENTRESTO, sacubitrilo y valsartán no revelaron ningún efecto a nivel genético ni cromosómico. Deterioro de la Fertilidad: ENTRESTO no demostró ningún efecto sobre la fertilidad en las ratas hasta la dosis de 73 mg de sacubitrilo/77 mg de valsartán/Kg/día (≤1,0 vez y ≤0,18 veces la MDRH sobre la base de las ABC de valsartán y LBQ657, respectivamente). Toxicología y/o Farmacología Animal: Se evaluaron los efectos de ENTRESTO sobre las concentraciones de beta-amiloide en el LCR y en el tejido cerebral en monos cynomolgus jóvenes (de 2 a 4 años) tratados con ENTRESTO™ (24 mg de sacubitrilo/26 mg de valsartán/Kg/día) durante 2 semanas. En este estudio, ENTRESTO afectó la depuración de Ab del LCR, aumentando los niveles de LCR Ab1-40, 1-42 y 1-38 en el LCR. No hubo ningún aumento correlativo en los niveles de Ab en el cerebro. Además, en el estudio de toxicología conducido en los monos cynomolgus tratados con ENTRESTO con la dosis de 146 mg de sacubitrilo/154 mg de valsartán/Kg/día durante 39 semanas, no hubo acumulación de beta-amiloide en el cerebro.
Indicaciones.
ENTRESTO™ está indicado para reducir el riesgo de muerte cardiovascular y hospitalización por insuficiencia cardiaca en pacientes con insuficiencia cardiaca crónica (Clase II-IV de la New York Heart Association (NYHA)), y fracción de eyección reducida. ENTRESTO™ se administra generalmente en combinación con otros tratamientos para la insuficiencia cardiaca, en lugar de un Inhibidor de la Enzima Convertidora de Angiotensina (IECA) o de otro Bloqueante de Receptor de Angiotensina (ARA II).
Dosificación.
El uso concomitante de ENTRESTO™ con un Inhibidor de la Enzima Convertidora de Angiotensina (IECA) está contraindicado. Si se cambia de un IECA a ENTRESTO™, se deberá dejar transcurrir un período de lavado de 36 horas entre la administración de los dos fármacos (Ver Contraindicaciones e Interacciones Medicamentosas). La dosis inicial recomendada es ENTRESTO 100 mg (49/51 mg) dos veces por día. Duplicar la dosis de ENTRESTO™ luego de 2 a 4 semanas hasta alcanzar la dosis de mantenimiento objetivo de ENTRESTO™ 200 mg (97/103 mg) dos veces por día, según la tolerancia del paciente. Ajuste de las Dosis para los Pacientes que no reciben ningún IECA ni ARA II o que recibieron dosis bajas de estos agentes previamente: Se recomienda una dosis inicial de ENTRESTO™ 50 mg (24/26 mg) dos veces por día para los pacientes que no están recibiendo actualmente ningún inhibidor de la ACE ni ningún bloqueante del receptor de angiotensina II (ARA II) y para los pacientes que recibieron dosis bajas de estos agentes previamente. Duplicar la dosis de ENTRESTO™ cada 2 a 4 semanas hasta alcanzar la dosis de mantenimiento objetivo de ENTRESTO™ 200 mg (97/103 mg) dos veces por día, según la tolerancia del paciente. Ajuste de las dosis por insuficiencia renal severa: Se recomienda una dosis inicial de ENTRESTO™ 50 mg (24/26 mg) dos veces por día para los pacientes con insuficiencia renal severa (eGFR < 30 mL/min/1,73 m). Duplicar la dosis de ENTRESTO™ cada 2 a 4 semanas hasta alcanzar la dosis de mantenimiento objetivo de ENTRESTO™ 200 mg (97/103 mg) dos veces por día, según la tolerancia del paciente. No se necesita realizar ningún ajuste de la dosis inicial por insuficiencia renal leve a moderada. Ajuste de las Dosis por Insuficiencia Hepática: Se recomienda una dosis inicial de ENTRESTO™ 50 mg (24/26 mg) dos veces por día para los pacientes con insuficiencia hepática moderada (clasificación Child-Pugh B). Duplicar la dosis de ENTRESTO™ cada 2 a 4 semanas hasta alcanzar la dosis de mantenimiento objetivo de ENTRESTO™ 200 mg (97/103 mg) dos veces por día, según la tolerancia del paciente. No se necesita realizar ningún ajuste de la dosis inicial por insuficiencia hepática leve. No se recomienda su uso en pacientes con insuficiencia hepática severa.
Contraindicaciones.
ENTRESTO™ está contraindicado: En pacientes con hipersensibilidad a cualquiera de los componentes. En pacientes con antecedentes de angioedema relacionado con tratamientos previos con un IECA o ARA II (Ver Advertencias). Uso concomitante con IECAs. No administrar dentro de las 36 horas posteriores al cambio de o a un IECA (Ver Precauciones - Interacciones Medicamentosas). Con el uso concomitante con aliskireno en pacientes con diabetes (Ver Precauciones - Interacciones Medicamentosas).
Reacciones adversas.
Las reacciones adversas clínicamente significativas que se mencionan en otras secciones del prospecto incluyen: Angioedema (Ver Advertencias); Hipotensión (Ver Advertencias); Deterioro de la Función Renal (Ver Advertencias); Hiperkalemia (Ver Advertencias). Experiencia en Estudios Clínicos: Debido a que los estudios clínicos se llevan a cabo bajo condiciones ampliamente variables, las tasas de reacciones adversas observadas en los estudios clínicos de un fármaco no pueden compararse directamente con las tasas de los estudios clínicos de otro fármaco por lo que pueden no reflejar las tasas observadas en la práctica. En el estudio PARADIGM-HF, los sujetos completaron períodos de prueba secuenciales con enalapril y ENTRESTO™ de (mediana) 15 y 29 días, respectivamente, antes de ingresar al período doble ciego aleatorizado que comparó ENTRESTO™ con enalapril. Durante el período de prueba de enalapril, se retiraron 1102 pacientes (10,5%) del estudio en forma permanente, 5,6% debido a un evento adverso, que fueron con mayor frecuencia la disfunción renal (1,7%), hiperkalemia (1,7%) e hipotensión (1,4%). Durante el período de prueba de ENTRESTO™, se retiró un 10,4% adicional de pacientes del estudio en forma permanente, 5,9% debido a un evento adverso, que fueron con mayor frecuencia la disfunción renal (1,8%), hipotensión (1,7%) e hiperkalemia (1,3%). Debido a este diseño de prueba, los porcentajes de las reacciones adversas que se describen a continuación son menores que los esperados en la práctica. En el período doble ciego, se evaluó la seguridad en 4203 pacientes tratados con ENTRESTO™ y en 4229 pacientes tratados con enalapril. En el estudio PARADIGM-HF, los pacientes aleatorizados a la rama de tratamiento con ENTRESTO™ recibieron el tratamiento por hasta 4,3 años, con una duración mediana de la exposición de 24 meses; 3271 pacientes fueron tratados durante más de un año. La interrupción del tratamiento debido a la aparición de un evento adverso durante el período doble ciego ocurrió en 450 (10,7%) de los pacientes tratados con ENTRESTO™ y en 516 (12,2%) de los pacientes tratados con enalapril. Las reacciones adversas que ocurrieron con una incidencia de ≥5% en los pacientes que fueron tratados con ENTRESTO™ en el período doble ciego se muestran en la Tabla 2.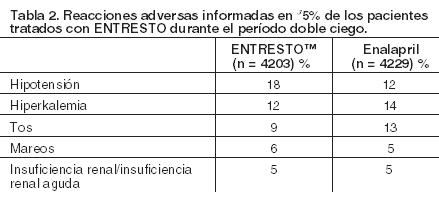
En el estudio PARADIGM-HF, la incidencia de angioedema fue de 0,1% tanto en el período de prueba de enalapril como en el de ENTRESTO. Durante el período doble ciego, la incidencia de angioedema fue superior en los pacientes tratados con ENTRESTO™ que en los pacientes tratados con enalapril (0,5% y 0,2%, respectivamente). La incidencia de angioedema en los pacientes de población negra fue de 2,4% con ENTRESTO y de 0,5% con enalapril (Ver Advertencias). Se informó la presencia de ortostatismo en el 2,1% de los pacientes tratados con ENTRESTO™ en comparación con el 1,1% de los pacientes tratados con enalapril durante el período doble ciego del estudio PARADIGM-HF. Se informaron caídas en el 1,9% de los pacientes tratados con ENTRESTO™ en comparación con el 1,3% de los pacientes tratados con enalapril. Anormalidades de Laboratorio: Hemoglobina y Hematocritos: Se observaron disminuciones en la hemoglobina/hematocritos de > 20% en aproximadamente el 5% de los pacientes tratados tanto con ENTRESTO como con enalapril durante el período doble ciego del estudio PARADIGM-HF. Creatinina Sérica: Se observaron incrementos en la creatinina sérica de > 50% en 1,4% de los pacientes en el período de prueba de enalapril y en el 2,2% de los pacientes en el período de prueba de ENTRESTO™. Durante el período doble ciego, aproximadamente 16% de los pacientes tratados tanto con ENTRESTO™ como con enalapril experimentaron incrementos en la creatinina sérica de > 50%. Potasio Sérico: Se observaron concentraciones de potasio de > 5,5 mEq/L en aproximadamente el 4% de los pacientes tanto en el período de prueba de enalapril como en el de ENTRESTO™. Durante el período doble ciego, aproximadamente el 16% de los pacientes tratados tanto con ENTRESTO™ como con enalapril experimentaron incrementos en las concentraciones de potasio de > 5,5 mEq/L. Información para Profesionales Médicos: El producto ENTRESTO™ cuenta con un Plan de Gestión de Riesgos cuya finalidad es garantizar la seguridad y protección de los pacientes, promoviendo el uso del producto de acuerdo a las recomendaciones de Novartis.
Precauciones.
Interacciones Medicamentosas: Bloqueo Dual del Sistema Renina-Angiotensina-Aldosterona (SRAA): El uso concomitante de ENTRESTO™ con un IECA está contraindicado debido a que existe un mayor riesgo de producir angioedema (Ver Contraindicaciones). Se deberá evitar el uso de ENTRESTO™ con un ARA II debido a que ENTRESTO™ contiene valsartán, un bloqueante del receptor de angiotensina II. El uso concomitante de ENTRESTO™ con aliskireno está contraindicado en pacientes con diabetes (Ver Contraindicaciones) Se deberá evitar el uso de aliskireno en pacientes con insuficiencia renal (eGFR < 60 mL/min/1,73 m2). Diuréticos Ahorradores de Potasio: Al igual que otros fármacos que bloquean la angiotensina II o sus efectos, el uso concomitante de diuréticos ahorradores de potasio (por ejemplo, espironolactona, triamtereno, amilorida), suplementos de potasio, o sustitutos de la sal que contienen potasio puede producir incrementos en el potasio sérico (Ver Advertencias). Fármacos Antiinflamatorios No Esteroides (AINEs) incluyendo a los Inhibidores Selectivos de la Ciclooxigenasa 2 (Inhibidores COX-2): En los pacientes que son adultos mayores, con depleción del volumen (incluso aquellos en tratamiento con diuréticos), o con la función renal comprometida, el uso concomitante de AINEs, que incluyen los inhibidores COX-2, con ENTRESTO™ puede resultar en el empeoramiento de la función renal, incluso en una posible insuficiencia renal aguda. Estos efectos son generalmente reversibles. Se deberá monitorear la función renal en forma periódica. Litio: Se han informado incrementos en las concentraciones de litio sérico y toxicidad por litio durante la administración concomitante de litio con antagonistas del receptor de angiotensina II. Se deberán monitorear los niveles de litio sérico durante el uso concomitante con ENTRESTO™. Uso en Poblaciones Específicas: Embarazo: Resumen de los Riesgos: ENTRESTO™ puede causar daño fetal cuando se lo administra a mujeres embarazadas. El uso de fármacos que actúan sobre el sistema renina-angiotensina durante el segundo y tercer trimestre del embarazo reduce la función renal fetal y aumenta la morbilidad y muerte fetal y neonatal. La mayoría de los estudios epidemiológicos que evalúan las anormalidades fetales luego de la exposición al uso de antihipertensivos durante el primer trimestre no han hecho ninguna distinción entre los fármacos que afectan el sistema renina-angiotensina y otros agentes antihipertensivos. En los estudios de reproducción animal, el tratamiento con ENTRESTO durante la organogénesis produjo una mayor mortalidad embriofetal en las ratas y en los conejos y teratogenicidad en los conejos. Cuando se detecta un embarazo, se deberá considerar un tratamiento con un fármaco alternativo e interrumpir ENTRESTO™. Sin embargo, si no existe ninguna alternativa adecuada al tratamiento con fármacos que afectan el sistema renina-angiotensina, y si se considera que el fármaco salva la vida de la madre, se deberá informar a la mujer embarazada sobre el riesgo potencial para el feto. Se desconoce el riesgo de base estimado de defectos congénitos mayores y aborto espontáneo para la población indicada. En la población general de los Estados Unidos, el riesgo de base estimado de defectos congénitos mayores y aborto espontáneo en embarazos reconocidos clínicamente es de 2-4% y de 15-20%, respectivamente. Consideraciones Clínicas: Reacciones Adversas Fetales/Neonatales: La oligohidramnios en las mujeres embarazadas que utilizan fármacos que afectan el sistema renina-angiotensina en el segundo y tercer trimestre del embarazo puede resultar en lo siguiente: función renal fetal reducida que derive en anuria e insuficiencia renal, hipoplasia pulmonar fetal, deformaciones esqueléticas, incluso hipoplasia craneal, hipotensión y muerte. Se deberán realizar ecografías seriadas para evaluar el entorno intraamniótico. Las pruebas fetales pueden ser adecuadas, dependiendo de la semana de gestación. Sin embargo, los pacientes y los médicos deberán tener presente que es posible que la oligohidramnios no se manifieste sino hasta después de que el feto hubiera sufrido un daño irreversible sostenido. Si se observa la presencia de oligohidramnios, se deberá considerar un tratamiento con un fármaco alternativo. Se deberá observar de cerca a los neonatos con antecedentes de exposición intrauterina a ENTRESTO para detectar la presencia de hipotensión, oliguria e hiperkalemia. En los neonatos con antecedentes de exposición intrauterina a ENTRESTO, si se produce oliguria o hipotensión, se deberá evaluar la presión arterial y la perfusión renal. Puede que sea necesario realizar exsanguinotransfusiones o diálisis como medio para revertir la hipotensión y reemplazar la función renal. Datos: Datos en Animales: El tratamiento con ENTRESTO™ durante la organogénesis produjo una mayor mortalidad embriofetal en las ratas con dosis ≥ 49 mg de sacubitrilo/51 mg de valsartán/Kg/día (≤ 0,14 [LBQ657, el metabolito activo] y 1,5 [valsartán] veces la dosis recomendada para seres humanos [MDRH] de 97/103 mg dos veces por día sobre la base del área bajo la curva de concentración plasmática del fármaco - tiempo [ABC]) y en los conejos con dosis ≥ 5 mg de sacubitrilo/5 mg de valsartán/Kg/día (4 veces y 0,06 veces la MDRH sobre la base de la ABC de valsartán y LBQ657, respectivamente). ENTRESTO™ es teratogénico sobre la base de una baja incidencia de hidrocefalia fetal, asociada con dosis tóxicas maternales, que se observó en los conejos tratados con dosis de ENTRESTO de ≥ 5 mg de sacubitrilo/5 mg de valsartán/Kg/día. Los efectos embriofetales adversos de ENTRESTO™ se atribuyen a la actividad del antagonista del receptor de angiotensina. Los estudios de desarrollo pre y posnatal en las ratas tratadas con dosis de sacubitrilo de hasta 750 mg/Kg/día (4,5 veces la MDRH sobre la base de la ABC del LBQ657) y con dosis de valsartán de hasta 600 mg/Kg/día (0,86 veces la MDRH sobre la base de la ABC) indican que el tratamiento con ENTRESTO™ durante la organogénesis, gestación y lactancia puede afectar el desarrollo y supervivencia de las crías. Lactancia: Resumen de los Riesgos: No existe información alguna referida a la presencia de sacubitrilo/valsartán en la leche humana, a los efectos sobre los lactantes amamantados ni a los efectos sobre la producción de leche. Sacubitrilo/valsartán está presente en la leche de las ratas. Debido al potencial de reacciones adversas serias en lactantes amamantados a partir de la exposición a sacubitrilo/valsartán, se deberá informar a las mujeres que amamantan que no se recomienda la lactancia durante el tratamiento con ENTRESTO™. Datos: Luego de administrar una dosis oral (15 mg de sacubitrilo/15 mg de valsartán/Kg) de [14C] ENTRESTO™ a las ratas lactantes, se observó el pasaje de LBQ657 a la leche. Con posterioridad a la administración de una dosis única oral de 3 mg/Kg de [C] valsartán a las ratas lactantes, se observó el pasaje de valsartán a la leche. Uso Pediátrico: No se estableció la seguridad ni efectividad de ENTRESTO™ en pacientes pediátricos. Uso en pacientes de edad avanzada: No se observaron diferencias farmacocinéticas relevantes en pacientes adultos mayores (≥65 años) o muy mayores (≥75 años) en comparación con la población general (Ver Farmacología/ Propiedades). Insuficiencia Hepática: No se requiere el ajuste de las dosis cuando se administra ENTRESTO™ a pacientes con insuficiencia hepática leve (clasificación A de Child-Pugh). La dosis inicial recomendada en pacientes con insuficiencia hepática moderada (clasificación B de Child Pugh) es de 24/26 mg dos veces por día. No se recomienda el uso de ENTRESTO™ en pacientes con insuficiencia hepática severa (clasificación C de Child-Pugh), debido a que no se realizaron estudios en estos pacientes. (Ver Dosificación). Insuficiencia Renal: No se requiere el ajuste de las dosis en pacientes con insuficiencia renal leve (eGFR 60 a 90 mL/min/1,73 m2) a moderada (eGFR 30 a 60 mL/min/1,73 m2). La dosis inicial recomendada en pacientes con insuficiencia renal severa (eGFR < 30 mL/min/1,73 m2) es de 24/26 mg dos veces por día. (Ver Dosificación y Advertencias).
Advertencias.
Angioedema: ENTRESTO puede causar angioedema. Durante el período doble ciego del estudio PARADIGM-HF, el 0,5% de los pacientes tratados con ENTRESTO™ y el 0,2% de los pacientes tratados con enalapril experimentaron angioedema (Ver Reacciones Adversas). Si se produce angioedema, se deberá interrumpir la administración de ENTRESTO inmediatamente, se deberá proporcionar un tratamiento adecuado y se deberá monitorear al paciente para evaluar un posible compromiso de las vías respiratorias. ENTRESTO no debe ser administrado nuevamente. En los casos de angioedema confirmado en los cuales la inflamación se limitó a la cara y a los labios, en general, la condición se resolvió sin tratamiento, a pesar de que los antihistamínicos han sido útiles para aliviar los síntomas. El angioedema asociado con el edema laríngeo puede ser mortal. Cuando existe compromiso de la lengua, de la glotis o de la laringe, que probablemente cause una obstrucción de las vías aéreas, se deberá administrar un tratamiento adecuado, por ejemplo, una solución de epinefrina/adrenalina en una proporción de 1:1000 (0,3 mL a 0,5 mL) por vía subcutánea y tomar las medidas necesarias para garantizar el mantenimiento de la permeabilidad de las vías aéreas. ENTRESTO ha sido asociado con una mayor tasa de incidencia de angioedema en pacientes de población negra que en otros pacientes. Los pacientes con antecedentes previos de angioedema pueden presentar un mayor riesgo de experimentar angioedema con ENTRESTO (Ver Reacciones Adversas). No se podrá utilizar ENTRESTO™ en pacientes con antecedentes conocidos de angioedema relacionado con tratamientos previos con un IECA o ARA II (Ver Contraindicaciones). Hipotensión: ENTRESTO disminuye la presión arterial y puede causar hipotensión sintomática. Los pacientes con un sistema renina-angiotensina activado, tales como los pacientes con depleción del volumen y/o de sal (por ejemplo, aquéllos tratados con dosis altas de diuréticos), presentan un mayor riesgo. Durante el período doble ciego del estudio PARADIGM-HF, el 18% de los pacientes tratados con ENTRESTO™ y el 12% de los pacientes tratados con enalapril informaron que experimentaron hipotensión como evento adverso (Ver Reacciones Adversas), habiéndose informado a la hipotensión como evento adverso serio en aproximadamente el 1.5% de los pacientes en ambas ramas de tratamiento. Se deberá corregir la depleción del volumen o de sal antes de administrar ENTRESTO™ o comenzar con una dosis menor. Si se produce hipotensión, se deberá considerar el ajuste de la dosis de diuréticos, de los fármacos antihipertensivos concomitantes y el tratamiento de otras causas de la hipotensión (por ejemplo, hipovolemia). Si la hipotensión persiste a pesar de dichas medidas, se deberá reducir la dosis o interrumpir ENTRESTO temporariamente. En general, no es necesario interrumpir el tratamiento. Deterioro de la Función Renal: Como consecuencia de la inhibición del sistema renina-angiotensina-aldosterona (SRAA), se pueden anticipar disminuciones en la función renal en individuos susceptibles tratados con ENTRESTO. Durante el período doble ciego del estudio PARADIGM-HF, el 5% de los pacientes, tanto en el grupo tratado con ENTRESTO™ como en el grupo tratado con enalapril, informaron que experimentaron insuficiencia renal como evento adverso (Ver Reacciones Adversas). En los pacientes cuya función renal depende de la actividad del sistema renina-angiotensina-aldosterona (por ejemplo, pacientes con insuficiencia cardiaca congestiva severa), el tratamiento con IECAs y antagonistas del receptor de angiotensina fue asociado con oliguria, azoemia progresiva y, rara vez, con insuficiencia renal aguda y muerte. Se deberá monitorear de cerca la creatinina sérica, y reducir la dosis o interrumpir ENTRESTO™ en pacientes que desarrollan una disminución en la función renal de relevancia clínica. Al igual que todos los fármacos que afectan el SRAA, ENTRESTO™ puede incrementar los niveles de urea en sangre y de la creatinina sérica en pacientes con estenosis arterial renal unilateral o bilateral. En los pacientes con estenosis arterial renal, se deberá monitorear la función renal. Hiperkalemia: A través de sus acciones sobre el SRAA, ENTRESTO™ puede producir hiperkalemia. Durante el período doble ciego del estudio PARADIGM-HF, el 12% de los pacientes tratados con ENTRESTO™ y el 14% de los pacientes tratados con enalapril informaron que experimentaron hiperkalemia como evento adverso (Ver Reacciones Adversas). Se deberá monitorear el potasio sérico periódicamente y tratar adecuadamente, en especial en pacientes con factores de riesgo por hiperkalemia tales como insuficiencia renal severa, diabetes, hipoaldosteronismo, o una dieta rica en potasio. Puede que sea necesario reducir las dosis o interrumpir ENTRESTO™ (Ver Dosificación). Toxicidad Fetal: ENTRESTO™ puede causar daño fetal cuando se lo administra a mujeres embarazadas. El uso de fármacos que actúan sobre el sistema renina-angiotensina durante el segundo y tercer trimestre del embarazo reduce la función renal fetal y aumenta la morbilidad y muerte fetal y neonatal. Cuando se detecta un embarazo, se deberá considerar un tratamiento con un fármaco alternativo e interrumpir ENTRESTO™. Sin embargo, si no existe ninguna alternativa adecuada al tratamiento con fármacos que afectan el sistema renina-angiotensina, y si se considera que el fármaco salva la vida de la madre, se deberá informar a la mujer embarazada sobre el riesgo potencial para el feto.
Conservación.
Conservar a menos de 30°C. Proteger de la humedad.
Sobredosificación.
Existen datos limitados disponibles referidos a la sobredosificación con ENTRESTO™ en los sujetos humanos. En los voluntarios sanos, se estudiaron las dosis únicas de ENTRESTO™ 583 mg de sacubitrilo/617 mg de valsartán, y las dosis múltiples de 437 mg de sacubitrilo/463 mg de valsartán (14 días), que fueron bien toleradas. La hipotensión es el resultado más probable de la sobredosificación debido a los efectos hipotensores de ENTRESTO™. Se deberá proporcionar un tratamiento sintomático. Es improbable que se pueda eliminar a ENTRESTO™ mediante hemodiálisis debido a su gran capacidad para unirse a las proteínas. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital A. Posadas: (011) 4654-6648/4658-7777.
Presentación.
ENTRESTO™ 50 mg: Envases conteniendo 30 y 60 comprimidos recubiertos. ENTRESTO™ 100 mg: Envases conteniendo 30 y 60 comprimidos recubiertos. ENTRESTO™ 200 mg: Envases conteniendo 30 y 60 comprimidos recubiertos.