EMECLAD
TUTEUR
Agentes antineoplásicos, antimetabolitos, análogos de la purina.
Composición.
Cada comprimido recubierto de EMECLAD® contiene: Cladribina 10 mg. Excipientes: Hidroxipropil-Beta-Ciclodextrina 143,76 mg, Sorbitol 64,04 mg, Estearato de magnesio 2,20 mg.
Farmacología.
Mecanismo de acción: Cladribina es un análogo nucleósido de la desoxiadenosina. Una sustitución con cloro en el anillo purínico protege a Cladribina de la degradación por la adenosina desaminasa, aumentando el tiempo de residencia intracelular del profármaco Cladribina. La subsiguiente fosforilación de Cladribina a su forma trifosfato activa, la 2-clorodesoxiadenosina trifosfato (Cd-ATP), se logra de una forma particularmente eficiente en los linfocitos debido a sus niveles constitucionalmente altos de desoxicitidina cinasa (DCK) y relativamente bajos de 5'-nucleotidasa (5'-NTasa). Un cociente DCK/5'-NTasa elevado favorece la acumulación del Cd-ATP, lo que hace a los linfocitos particularmente propensos a la muerte celular. Como consecuencia de un cociente DCK/5'-NTasa más bajo, otras células derivadas de la médula ósea se ven menos afectadas que los linfocitos. DCK es la enzima limitante de la velocidad de conversión del profármaco Cladribina a su forma trifosfato activa, lo que lleva a una depleción selectiva de las células T y B en proceso de división o no. El principal mecanismo de acción inductor de la apoptosis del Cd-ATP ejerce efectos directos e indirectos sobre la síntesis de DNA y la función mitocondrial. En las células en proceso de división, el Cd-ATP interfiere con la síntesis de DNA a través de la inhibición de la ribonucleótido reductasa y compite con la desoxiadenosina trifosfato por la incorporación al DNA mediante las DNA polimerasas. En las células en reposo, Cladribina causa rupturas monocatenarias del DNA, un rápido consumo del nicotinamida adenina dinucleótido, agotamiento del ATP y muerte celular. Existen datos indicativos de que Cladribina también puede causar apoptosis directa dependiente e independiente de la caspasa a través de la liberación del citocromo C y del factor inductor de la apoptosis en el citosol de las células que no se encuentran en proceso de división. La patogenia de la Esclerosis Múltiple implica una compleja cadena de eventos en los que distintos tipos de células inmunitarias, incluidas las células T y B autorreactivas, desempeñan un papel clave. El mecanismo mediante el que Cladribina ejerce sus efectos terapéuticos en Esclerosis Múltiple no está completamente esclarecido, pero se cree que su efecto predominante sobre los linfocitos B y T interrumpe la cascada de eventos inmunitarios centrales en la Esclerosis Múltiple. Las variaciones en los niveles de expresión de DCK y 5'-NTasa entre los subtipos de células inmunitarias pueden explicar las diferencias en la sensibilidad de dichas células a Cladribina. Debido a estos niveles de expresión, las células del sistema inmunitario innato se ven menos afectadas que las del sistema inmunitario adaptativo.
Farmacodinamia.
Se ha demostrado que Cladribina ejerce un efecto de larga duración mediante la acción dirigida preferencialmente sobre los linfocitos y los procesos autoinmunitarios que intervienen en la fisiopatología de la Esclerosis Múltiple. En los ensayos, la mayor proporción de pacientes con linfopenia de grado 3 o 4 ( < 500 a 200 células/mm³ o < 200 células/mm³) se observó dos meses después de la primera dosis de Cladribina de cada año, lo que indica un lapso entre las concentraciones plasmáticas de Cladribina y su máximo efecto hematológico. En los ensayos clínicos, los datos con la dosis acumulada propuesta de 3,5 mg/kg de peso corporal demuestran una mejoría gradual en la mediana de los recuentos de linfocitos hasta el rango de la normalidad en la semana 84 después de la primera dosis de Cladribina (aproximadamente 30 semanas después de la última dosis de Cladribina). Los recuentos de linfocitos de más del 75% de los pacientes retornaron al rango de la normalidad en la semana 144 después de la primera dosis de Cladribina (aproximadamente 90 semanas después de la última dosis de Cladribina). El tratamiento con Cladribina por vía oral genera una rápida disminución de las células T CD4+ y T CD8+ circulantes. Las células T CD8+ presentan una disminución menos pronunciada y una recuperación más rápida que las células T CD4+, lo que da lugar a una reducción temporal del cociente CD4 a CD8. Cladribina reduce las células B CD19+ y las células natural killers CD16+/CD56+, que también se recuperan más rápido que las células T CD4+.
Farmacocinética.
Cladribina es un profármaco que tiene que fosforilarse a nivel intracelular para hacerse biológicamente activo. Se estudiaron las propiedades farmacocinéticas de Cladribina después de su administración por vía oral e intravenosa en pacientes con Esclerosis Múltiple y en pacientes con neoplasias malignas, y en sistemas in vitro. Absorción: Después de la administración por vía oral de Cladribina, esta se absorbe rápidamente. La administración de 10 mg de Cladribina produjo una Cmax media de Cladribina dentro del intervalo de 22 a 29 ng/ml y un AUC medio correspondiente dentro del intervalo de 80 a 101 ng·h/ml (medias aritméticas de varios ensayos). Cuando se administró por vía oral Cladribina en ayunas, la mediana de la Tmax fue de 0,5 horas (rango de 0,5 a 1,5 horas). Cuando Cladribina se administró con una comida rica en grasas, su absorción se retrasó (mediana de la Tmax, 1,5 h, rango de 1 a 3 h) y la Cmax disminuyó un 29% (basado en la media geométrica), mientras que el AUC no se modificó. La biodisponibilidad de 10 mg de Cladribina por vía oral fue de aproximadamente un 40%. Distribución: El volumen de distribución es grande, lo que indica una amplia distribución tisular y captación intracelular. Los estudios mostraron un volumen medio de distribución de Cladribina en el rango de 480 a 490 l. La unión de Cladribina a las proteínas plasmáticas es del 20% y es independiente de la concentración plasmática. La distribución de Cladribina a través de las membranas biológicas se ve facilitada por diversas proteínas transportadoras, incluidas ENT1, CNT3 y BCRP. Los estudios in vitro indican que el fiujo de Cladribina solo está relacionado con la gp-P de forma mínima. No se prevén interacciones clínicamente relevantes con inhibidores de la gp-P. No se han estudiado formalmente las posibles consecuencias de la inducción de la gp-P sobre la biodisponibilidad de Cladribina. Los estudios in vitro mostraron una captación insignificante de Cladribina en los hepatocitos humanos mediada por transportadores. Cladribina puede penetrar la barrera hematoencefálica. Un pequeño estudio en pacientes con cáncer ha demostrado un cociente de concentraciones en el líquido cefalorraquídeo/plasma de aproximadamente 0,25. Cladribina y/o sus metabolitos fosforilados se acumulan y retienen de forma sustancial en los linfocitos humanos. In vitro, se constataron cocientes de acumulación intracelular frente a extracelular de aproximadamente 30 a 40 una hora después de la exposición a Cladribina. Metabolismo: Se estudió el metabolismo de Cladribina en pacientes con Esclerosis Múltiple después de la administración de un único comprimido de 10 mg y también después de una dosis única de 3 mg por vía intravenosa. Después de la administración tanto oral como intravenosa, el principal componente presente en el plasma y la orina fue el compuesto original Cladribina. El metabolito 2-cloroadenina fue un metabolito menor tanto en el plasma como en la orina, representando solo una tasa fi3% de la exposición plasmática al fármaco original tras la administración oral. Solo pudieron encontrarse trazas de otros metabolitos en el plasma y la orina. En sistemas hepáticos in vitro se observó un metabolismo insignificante de Cladribina (al menos el 90% fue Cladribina inalterada). Cladribina no es un sustrato relevante de las enzimas del citocromo P450 y no muestra un potencial significativo de actuar como inhibidor de CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 y CYP3A4. No se prevé que la inhibición de estas enzimas o polimorfismos genéticos (por ejemplo, CYP2D6, CYP2C9 o CYP2C19) dé lugar a efectos clínicamente significativos sobre las propiedades farmacocinéticas de Cladribina ni sobre la exposición al fármaco. Cladribina no tiene un efecto inductor clínicamente significativo sobre las enzimas CYP1A2, CYP2B6 y CYP3A4. Después de entrar en las células diana, Cladribina es fosforilada a monofosfato de Cladribina (Cd-AMP) por la DCK (y también por la desoxiguanosina cinasa en las mitocondrias). Posteriormente, el Cd-AMP es fosforilado a difosfato de Cladribina (Cd-ADP) y trifosfato de Cladribina (Cd-ATP). La desfosforilación y la desactivación del Cd-AMP son catalizadas por la 5´-NTasa citoplasmática. En un estudio de las propiedades farmacocinéticas intracelulares del CdAMP y el CdATP en pacientes con leucemia mieloide crónica, los niveles de Cd-ATP fueron aproximadamente la mitad de los de Cd-AMP. La semivida intracelular del Cd-AMP fue de 15 horas. La semivida intracelular del Cd-ATP fue de 10 horas. Eliminación: De acuerdo con los datos farmacocinéticos poblacionales agregados de diversos estudios, las medianas de los valores de eliminación fueron de 22,2 l/h para el clearance renal y de 23,4 l/h para el clearance no renal. El clearance renal superó a la tasa de filtración glomerular, lo que indica una secreción tubular renal activa de Cladribina. La porción no renal de la eliminación de la Cladribina (aproximadamente el 50%) consiste en un metabolismo hepático insignificante y en una amplia distribución intracelular y captación del principio activo de Cladribina (Cd-ATP) dentro del compartimento intracelular diana (es decir, los linfocitos) y la consiguiente eliminación del Cd-ATP intracelular según el ciclo vital y las vías de eliminación de estas células. La semivida terminal estimada para un paciente típico del análisis farmacocinético poblacional es de aproximadamente un día. Sin embargo, esto no genera una acumulación del fármaco tras la administración en dosis única diaria, ya que esta semivida solo representa una pequeña porción del AUC. Dependencia de la dosis y del tiempo: Después de la administración por vía oral de Cladribina en un intervalo de dosis de 3 a 20 mg, la Cmax y el AUC aumentaron de manera proporcional a la dosis, lo que sugiere que la absorción no se ve afectada por procesos limitados por la tasa de absorción o eliminación o limitados por la capacidad de las vías metabólicas, hasta una dosis de 20 mg por vía oral. No se ha observado una acumulación significativa de las concentraciones plasmáticas de Cladribina después de la administración repetida. No hay datos indicativos de que las propiedades farmacocinéticas de Cladribina puedan cambiar de manera dependiente del tiempo tras la administración repetida. Poblaciones especiales: No se ha realizado ningún estudio para evaluar las propiedades farmacocinéticas de Cladribina en pacientes con Esclerosis Múltiple de edades avanzadas o pediátricas, ni en pacientes con insuficiencia renal o hepática. Un análisis farmacocinético poblacional no mostró ningún efecto relacionado con la edad (rango de 18 a 65 años) ni el sexo sobre las propiedades farmacocinéticas de Cladribina. Insuficiencia renal Se ha observado que el clearance de Cladribina depende del clearance de creatinina (ClCr). A partir de un análisis farmacocinético de la población, en el que se incluyeron pacientes con función renal normal y con insuficiencia renal leve, se prevé que el clearance total en los pacientes con insuficiencia renal leve (ClCr: 60 ml/min) disminuya moderadamente, con un aumento resultante de la exposición del 25%. Insuficiencia hepática El papel de la función hepática para la eliminación de Cladribina se considera insignificante. Interacciones farmacocinéticas: En un estudio de interacción medicamentosa en pacientes con Esclerosis Múltiple se demostró que la biodisponibilidad de Cladribina con dosis de 10 mg por vía oral no se alteró cuando se administró simultáneamente con pantoprazol.
Indicaciones.
EMECLAD® está indicado para el tratamiento de pacientes adultos con Esclerosis Múltiple recurrente muy activa definida mediante características clínicas o de imagen.
Dosificación.
El tratamiento con EMECLAD® debe ser iniciado y supervisado por un médico con experiencia en Esclerosis Múltiple. Posología: La dosis acumulada recomendada de EMECLAD® es de 3,5 mg/kg de peso corporal a lo largo de dos años, administrados en forma de un curso de tratamiento de 1,75 mg/kg por año. Cada curso de tratamiento consiste en dos semanas de tratamiento, una al inicio del primer mes y otra al inicio del segundo mes del año de tratamiento respectivo. Cada semana de tratamiento consiste en cuatro o cinco días en los que el paciente recibe 10 mg o 20 mg (uno o dos comprimidos) como dosis diaria única, dependiendo del peso corporal. Ver detalles más abajo en las Tablas 1 y 2. Tras la finalización de los dos cursos de tratamiento, no es necesario un tratamiento ulterior con EMECLAD® en los años 3 y 4. No se ha estudiado el reinicio de la terapia después del año 4. Criterios para iniciar y continuar el tratamiento El recuento de linfocitos debe ser: normal antes de comenzar EMECLAD® en el año 1; de al menos 800 células/mm³ antes de comenzar EMECLAD® en el año 2. Si fuese necesario, el curso de tratamiento del año 2 puede retrasarse hasta un máximo de seis meses para permitir la recuperación de los linfocitos. Si esta recuperación tarda más de seis meses, el paciente no debe volver a tomar EMECLAD®. Distribución de la dosis: En la Tabla 1 se proporciona la distribución de la dosis total durante los dos años de tratamiento. En el caso de algunos intervalos de peso, el número de comprimidos puede variar de una semana de tratamiento a la siguiente. No se ha estudiado el uso de Cladribina por vía oral en los pacientes con un peso inferior a 40 kg.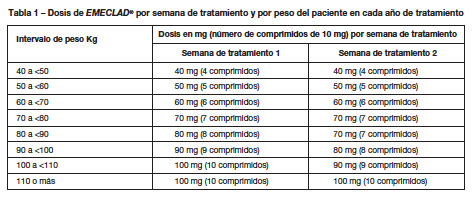
En la Tabla 2 se muestra cómo se distribuye el número total de comprimidos por semana de tratamiento en el transcurso de los días. Se recomienda que las dosis diarias de Cladribina de cada semana de tratamiento se tomen a intervalos de 24 horas, a aproximadamente la misma hora cada día. Si una dosis diaria consiste en dos comprimidos, ambos deben tomarse juntos como una dosis única.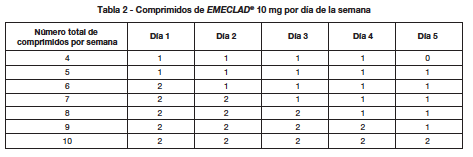
Una dosis olvidada debe tomarse en cuanto se recuerde, en el mismo día, de acuerdo con la pauta de tratamiento. Una dosis olvidada no debe tomarse junto con la siguiente dosis programada al día siguiente. En caso de una dosis olvidada, el paciente debe tomarla al día siguiente, y ampliar el número de días de esa semana de tratamiento. Si se olvidan dos dosis consecutivas, se aplica la misma regla y el número de días de la semana de tratamiento se amplía en dos días. Uso simultáneo de otros medicamentos por vía oral Se recomienda que la administración de cualquier otro medicamento por vía oral se separe de la de EMECLAD® por lo menos tres horas durante el número limitado de días de administración de Cladribina. Poblaciones especiales: Insuficiencia renal: No se han realizado estudios específicos en pacientes con insuficiencia renal. En los pacientes con insuficiencia renal leve (ClCr de 60 a 89 ml/min), no se considera necesario ajustar la dosis. No se ha establecido la seguridad y la eficacia en los pacientes con insuficiencia renal moderada o grave. Por lo tanto, EMECLAD® está contraindicado en estos pacientes. Insuficiencia hepática: No se han realizado estudios en pacientes con insuficiencia hepática. Aunque la importancia de la función hepática para la eliminación de Cladribina se considera insignificante, en ausencia de datos, no se recomienda el uso de EMECLAD® en los pacientes con insuficiencia hepática moderada o grave (puntuación de Child-Pugh > 6). Pacientes de edad avanzada: En los estudios clínicos con Cladribina por vía oral en Esclerosis Múltiple no se incluyeron pacientes de más de 65 años; por lo tanto, se desconoce si responden de manera diferente que los pacientes más jóvenes. Se recomienda precaución cuando se use EMECLAD® en pacientes de edad avanzada, teniendo en cuenta la posibilidad de que haya una mayor frecuencia de función hepática o renal reducida, enfermedades concomitantes y otros tratamientos medicamentosos. Población pediátrica: No se ha establecido la seguridad y la eficacia de EMECLAD® en los pacientes menores de 18 años. No se dispone de datos. Forma de administración: EMECLAD® se administra por vía oral. Los comprimidos se deben tomar con agua y tragarse sin masticar. Los comprimidos se pueden tomar independientemente de la ingesta de alimentos. Como los comprimidos no son recubiertos, deben tragarse inmediatamente una vez extraídos del blíster y no se deben dejar expuestos sobre superficies ni manipularse durante ningún período superior al requerido para la administración de la dosis. Si se deja un comprimido sobre una superficie o si se libera un comprimido roto o fragmentado del blíster, la zona debe limpiarse bien. Las manos del paciente deben estar secas al manipular los comprimidos y deben lavarse después.
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes. Infección por el virus de la inmunodeficiencia humana (VIH). Infección crónica activa (tuberculosis o hepatitis). Inicio del tratamiento con Cladribina en pacientes inmunocomprometidos, incluidos los que reciben actualmente tratamiento inmunosupresor o mielosupresor. Neoplasia maligna activa. Insuficiencia renal moderada o grave (ClCr < 60 ml/min). Embarazo y lactancia.
Reacciones adversas.
Resumen del perfil de seguridad: Las reacciones adversas de mayor relevancia clínica notificadas en los pacientes con Esclerosis Múltiple que recibieron Cladribina en la dosis acumulada recomendada de 3,5 mg/kg a lo largo de dos años en los ensayos clínicos fueron linfopenia y herpes zóster. La incidencia de herpes zóster fue mayor durante el periodo de linfopenia de grado 3 o 4 ( < 500 a 200 células/mm³ o < 200 células/mm³) que durante el tiempo en el que los pacientes no presentaron linfopenia de grado 3 o 4. Lista de reacciones adversas Las reacciones adversas que se describen en la siguiente lista derivan del conjunto de datos de los ensayos clínicos sobre Esclerosis Múltiple en los que se utilizó Cladribina por vía oral en monoterapia en una dosis acumulada de 3,5 mg/kg. La base de datos de seguridad de estos ensayos comprende 923 pacientes. Las siguientes definiciones se aplican a la terminología de frecuencia usada en adelante: Muy frecuentes (1/10), Frecuentes (1/100 a < 1/10), Poco frecuentes (1/1.000 a < 1/100), Raras (1/10.000 a < 1/1.000), Muy raras ( < 1/10.000) y Frecuencia no conocida (no puede estimarse a partir de los datos disponibles) Infecciones e infestaciones Frecuentes: Herpes oral, herpes zóster en dermatomas. Muy raras: Tuberculosis. Trastornos de la sangre y del sistema linfático Muy frecuentes: Linfopenia. Frecuentes: Disminución del recuento de neutrófilos. Trastornos de la piel y del tejido subcutáneo Frecuentes: Erupción cutánea, alopecia. Descripción de reacciones adversas seleccionadas: Linfopenia: En los ensayos clínicos, del 20 al 25% de los pacientes tratados con una dosis acumulada de Cladribina de 3,5 mg/kg a lo largo de dos años en monoterapia presentaron linfopenia transitoria de grado 3 o 4. La linfopenia de grado 4 se observó en menos del 1% de los pacientes. La mayor proporción de pacientes con linfopenia de grado 3 o 4 se observó dos meses después de la primera dosis de Cladribina de cada año (4,0% de los pacientes con linfopenia de grado 3 en el año 1 y 11,3% en el año 2,0% de los pacientes con linfopenia de grado 4 en el año 1 y 0,4% en el año 2). Se prevé que en la mayoría de los pacientes se produzca la recuperación a recuentos de linfocitos normales o a linfopenia de grado 1 en un plazo de nueve meses. Para reducir el riesgo de linfopenia grave, se deben realizar recuentos de linfocitos antes, durante y después del tratamiento con Cladribina, y seguir criterios estrictos para el inicio y la continuación del mismo. Neoplasias malignas: En los ensayos clínicos y en el seguimiento a largo plazo de los pacientes tratados con una dosis acumulada de 3,5 mg/kg de Cladribina oral, se observaron eventos correspondientes a neoplasias malignas con mayor frecuencia en los pacientes tratados con Cladribina [10 eventos en 3414 años-pacientes (0,29 eventos por 100 años-pacientes)] que en los que recibieron placebo [3 eventos en 2022 años-pacientes (0,15 eventos por 100 años-pacientes)]. Hipersensibilidad: En los ensayos clínicos con pacientes tratados con una dosis acumulada de 3,5 mg/kg de Cladribina oral, se observaron episodios de hipersensibilidad con mayor frecuencia en los pacientes tratados con Cladribina (11,8%) que en los pacientes que recibieron placebo (8,4%). Se observaron episodios graves de hipersensibilidad en el 0,3% de los pacientes tratados con Cladribina y en ninguno de los pacientes que recibieron placebo. Los episodios de hipersensibilidad dieron lugar a la interrupción del tratamiento en el 0,4% de los pacientes tratados con Cladribina y en el 0,3% de los pacientes que recibieron placebo. Lesión hepática: Durante la experiencia poscomercialización se notificaron eventos poco frecuentes de lesiones hepáticas en relación temporal con Cladribina, incluyendo casos graves y casos que provocaron la suspensión del tratamiento. Las elevaciones transitorias de las transaminasas séricas fueron normalmente superiores a 5 veces el LSN. Se observaron casos aislados de elevaciones transitorias de las transaminasas séricas de hasta 40 veces el LSN y/o hepatitis sintomática con elevación transitoria de la bilirrubina e ictericia. El tiempo hasta la aparición fue variable, presentándose la mayor parte de los casos dentro de las 8 semanas siguientes al primer ciclo de tratamiento.
Precauciones.
Interacción con otros medicamentos y otras formas de interacción: Cladribina contiene hidroxipropil-beta-ciclodextrina, que puede formar complejos con otros medicamentos, lo que puede causar un aumento de la biodisponibilidad de dichos medicamentos (especialmente los de baja solubilidad). Por lo tanto, se recomienda que la administración de cualquier otro medicamento por vía oral se separe de la de Cladribina por lo menos 3 horas durante el número limitado de días de administración de Cladribina.: Medicamentos inmunosupresores: El inicio del tratamiento con Cladribina está contraindicado en los pacientes inmunocomprometidos, incluidos los que reciben actualmente tratamiento inmunosupresor o mielosupresor con fármacos como metotrexate, ciclofosfamida, ciclosporina o azatioprina, o el uso crónico de corticoesteroides, a causa de un riesgo de efectos aditivos sobre el sistema inmunitario. Durante el tratamiento con Cladribina se puede administrar un tratamiento agudo y a corto plazo con corticoesteroides sistémicos. Otros medicamentos modificadores de la enfermedad: El uso de Cladribina con interferón beta provoca un aumento del riesgo de linfopenia. No se ha establecido la seguridad y la eficacia de Cladribina en combinación con otros tratamientos modificadores de la enfermedad para la Esclerosis Múltiple. No se recomienda el tratamiento concomitante. Medicamentos hematotóxicos: Debido a la disminución del recuento de linfocitos inducida por Cladribina, pueden esperarse reacciones adversas hematológicas aditivas si Cladribina se administra con anterioridad o simultáneamente con otras sustancias que afectan al perfil hematológico (por ejemplo, carbamacepina). En estos casos, se recomienda una vigilancia estrecha de los parámetros hematológicos. Vacunas vivas o vivas atenuadas: No se debe iniciar el tratamiento con Cladribina en las cuatro a seis semanas posteriores a la vacunación con vacunas vivas o vivas atenuadas, debido al riesgo de infección por la vacuna activa. Se debe evitar la vacunación con vacunas vivas o vivas atenuadas durante y después del tratamiento con Cladribina, mientras los recuentos de leucocitos del paciente no se encuentren dentro de los límites de la normalidad. Inhibidores potentes de los transportadores de ENT1, CNT3 y BCPR A nivel de la absorción de Cladribina, la única vía de interacción posible de importancia clínica sería la proteína de resistencia del cáncer de mama (BCRP o ABCG2). La inhibición de la BCRP en el tracto digestivo puede aumentar la biodisponibilidad oral y la exposición sistémica de Cladribina. Entre los inhibidores conocidos de la BCRP, que pueden alterar las propiedades farmacocinéticas de sustratos de la BCRP en un 20% in vivo, se encuentra eltrombopag. Los estudios in vitro indican que Cladribina es un sustrato de las proteínas de transporte del nucleósido equilibrativo (ENT1) y el nucleósido concentrativo (CNT3). Por consiguiente, los inhibidores potentes de los transportadores de ENT1 y CNT3, como dilazep, nifedipino, nimodipino, cilostazol, sulindaco o reserpina, pueden, en teoría, alterar la biodisponibilidad, la distribución intracelular y la eliminación renal de Cladribina. No obstante, los efectos netos en términos de posibles alteraciones de la exposición a Cladribina son difíciles de predecir. Aunque se desconoce la importancia clínica de estas interacciones, se recomienda evitar la administración simultánea de inhibidores potentes de ENT1, CNT3 o BCRP durante el tratamiento de 4-5 días con Cladribina. Si esto no fuera posible, debe plantearse la selección de otros medicamentos alternativos para su administración simultánea que carezcan de propiedades de inhibición de los transportadores de ENT1, CNT3 o BCRP, o en los que estas propiedades sean mínimas. Si esto no es posible, se recomienda la disminución de la dosis hasta la mínima dosis obligatoria de los medicamentos que contengan estos compuestos, la separación del momento de administración y la vigilancia estricta del paciente. Inductores potentes de los transportadores BCRP y P-gp No se han estudiado formalmente los efectos de los inductores potentes de los transportadores de fiujo BCRP y glucoproteína P (P-gp) sobre la biodisponibilidad y la eliminación de Cladribina. Se debe considerar una posible disminución de la exposición a Cladribina en caso de administración simultánea de inductores potentes de los transportadores BCRP (por ejemplo, corticoesteroides) o P-gp (por ejemplo, rifampicina, hierba de San Juan). Anticonceptivos hormonales: En la actualidad, se desconoce si Cladribina puede reducir la efectividad de los anticonceptivos hormonales de acción sistémica. Por lo tanto, las pacientes que utilizan anticonceptivos hormonales de acción sistémica deben añadir un método de barrera durante el tratamiento con Cladribina y al menos hasta 4 semanas después de la última dosis de cada año de tratamiento. Fertilidad, embarazo y lactancia: Anticoncepción en hombres y mujeres: Antes del inicio del tratamiento tanto en el año 1 como en el año 2, se debe asesorar a las mujeres en edad fértil y a los varones con parejas con capacidad de gestación respecto a los posibles riesgos graves para el feto y la necesidad de utilizar métodos anticonceptivos efectivos. En las mujeres en edad fértil, se debe descartar un embarazo antes del inicio de Cladribina en el año 1 y en el año 2, y evitar el embarazo mediante el uso de métodos anticonceptivos efectivos durante el tratamiento con Cladribina y por lo menos hasta seis meses después de la última dosis. Las usuarias de anticonceptivos hormonales de acción sistémica deben añadir un método de barrera durante el tratamiento con Cladribina y al menos hasta 4 semanas después de la última dosis de cada año de tratamiento. Las mujeres que queden embarazadas durante el tratamiento con Cladribina deben suspender el tratamiento. Dado que Cladribina interfiere con la síntesis del DNA, se esperaría encontrar efectos adversos sobre la gametogénesis humana. Por lo tanto, los pacientes varones deben tomar precauciones a fin de evitar el embarazo de su pareja durante el tratamiento con Cladribina y por lo menos hasta seis meses después de la última dosis. Embarazo: Teniendo en cuenta los datos derivados de la experiencia en humanos con otras sustancias inhibidoras de la síntesis de DNA, Cladribina podría causar malformaciones congénitas cuando se administra durante el embarazo. Los estudios realizados en animales han mostrado toxicidad para la reproducción. Cladribina está contraindicado en mujeres embarazadas. Lactancia: Se desconoce si Cladribina se excreta en la leche materna. Debido a la posibilidad de reacciones adversas graves en los niños alimentados con lactancia materna, durante el tratamiento con Cladribina y hasta una semana después de la última dosis la lactancia está contraindicada. Fertilidad: En los ratones, no hubo efectos sobre la fertilidad ni sobre las funciones reproductivas de las crías. Sin embargo, se observaron efectos testiculares en ratones y monos. Dado que Cladribina interfiere con la síntesis del DNA, son de prever efectos adversos sobre la gametogénesis humana. Por lo tanto, los pacientes varones deben tomar precauciones a fin de evitar el embarazo de su pareja durante el tratamiento con Cladribina y por lo menos hasta seis meses después de la última dosis. Efectos sobre la capacidad para conducir y utilizar máquinas: La infiuencia de Cladribina sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Datos preclínicos sobre seguridad: La evaluación farmacológica y toxicológica de la seguridad no clínica de Cladribina en modelos animales no reveló hallazgos significativos distintos de los esperados por el mecanismo farmacológico. Los principales órganos diana identificados en los estudios de toxicología con dosis repetidas por vías parenterales (intravenosa o subcutánea) de hasta 1 año de duración en ratones y monos fueron los sistemas linfático y hematopoyético. Otros órganos diana tras una administración más prolongada (14 ciclos) de Cladribina a monos por vía subcutánea fueron los riñones (cariomegalia del epitelio tubular renal), las glándulas suprarrenales (atrofia cortical y disminución de la vacuolización), el tracto gastrointestinal (atrofia de la mucosa) y los testículos. También se observaron efectos sobre los riñones en ratones. Mutagenicidad: Cladribina se incorpora a las cadenas del DNA e inhibe la síntesis y reparación de este. La Cladribina no indujo mutaciones genéticas en las bacterias ni en las células de mamífero, pero fue clastogénica, causando daños cromosómicos, en las células de mamífero in vitro en una concentración que era 17 veces superior a la Cmax clínica prevista. Se detectó clastogenicidad in vivo en ratones con una dosis de 10 mg/kg, la dosis más baja estudiada. Carcinogenicidad: El potencial carcinogénico de Cladribina fue evaluado en un ensayo a largo plazo de 22 meses de duración con administración subcutánea en ratones y en un ensayo a corto plazo de 26 semanas de duración por vía oral en ratones transgénicos. En el ensayo de carcinogenicidad a largo plazo en ratones, la dosis más alta utilizada fue 10 mg/kg que se constató genotóxica en el ensayo de micronúcleos de ratón (equivalente aproximadamente a 16 veces la exposición humana prevista en términos de AUC en los pacientes tratados con la dosis diaria máxima de 20 mg de Cladribina). No se observó una incidencia aumentada de trastornos linfoproliferativos ni de otros tipos de tumores (aparte de los de las glándulas de Harder, predominantemente adenomas) en los ratones. Los tumores de las glándulas de Harder no se consideran clínicamente relevantes, ya que los seres humanos carecen de estructuras anatómicas comparables. En el ensayo de carcinogenicidad a corto plazo en ratones Tg rasH2, no se observó un aumento relacionado con Cladribina en la incidencia de trastornos linfoproliferativos ni de otros tipos de tumores en ninguna de las dosis investigadas de hasta 30 mg/kg al día (equivalente aproximadamente a 25 veces la exposición humana prevista en términos de AUC en los pacientes tratados con la dosis diaria máxima de 20 mg de Cladribina). Cladribina también se evaluó en un ensayo de 1 año de duración en monos por vía subcutánea. En este ensayo, no se observó una incidencia aumentada de trastornos linfoproliferativos ni de tumores. Aunque Cladribina puede tener potencial genotóxico, los datos a largo plazo en ratones y monos no proporcionaron indicios de un incremento relevante del riesgo de carcinogenicidad en los seres humanos. Toxicidad para la reproducción: Si bien no se halló ningún efecto sobre la fertilidad de los ratones hembra, la función reproductiva ni las funciones generales de las crías, se demostró que Cladribina fue letal para el embrión cuando se administró a ratones hembra preñadas, y el compuesto fue teratógeno en ratones (también tras el tratamiento solamente de los machos) y conejos. Los efectos letales para el embrión y teratógenos observados son congruentes con los mecanismos farmacológicos de la Cladribina. En un estudio de fertilidad en ratones macho, se observaron fetos malformados con agenesia de porciones de uno o ambos apéndices distales del húmero y/o el fémur. La incidencia de fetos de ratón afectados en este estudio estuvo en el mismo intervalo de incidencia espontánea de amelia y focomelia en esta cepa de ratones. No obstante, teniendo en cuenta la genotoxicidad de Cladribina, no se pueden descartar efectos relacionados con una posible alteración genética de las células espermáticas en fase de diferenciación mediados por los machos. Cladribina no afectó la fertilidad de los ratones macho; sin embargo, los efectos testiculares observados fueron la disminución del peso de los testículos y el aumento de la cantidad de espermatozoides no móviles. En el mono también se observaron degeneración testicular y una disminución reversible de los espermatozoides con una motilidad progresiva rápida. Histológicamente, solo se observó degeneración testicular en un mono macho en un ensayo de toxicidad subcutánea de un año de duración.
Advertencias.
Control hematológico: El mecanismo de acción de Cladribina está ligado estrechamente a una disminución del recuento de linfocitos. El efecto sobre el recuento de linfocitos es dependiente de la dosis. En los ensayos clínicos se han observado también disminuciones del recuento de neutrófilos, del recuento de hematíes, del hematocrito, de la hemoglobina y del recuento de plaquetas, en comparación con los valores basales, aunque estos parámetros suelen mantenerse dentro de los límites de la normalidad. Pueden esperarse reacciones adversas hematológicas aditivas si Cladribina se administra con anterioridad o simultáneamente a otras sustancias que afectan al perfil hematológico. Se deben determinar los recuentos de linfocitos antes del inicio de Cladribina en el año 1, antes del inicio de Cladribina en el año 2, dos y seis meses después del inicio del tratamiento en cada año de tratamiento. Si el recuento de linfocitos es inferior a 500 células/mm³, se debe vigilar activamente hasta que los valores aumenten de nuevo. Para las decisiones terapéuticas basadas en los recuentos de linfocitos del paciente. Infecciones: Cladribina puede reducir las defensas inmunitarias del organismo y podría aumentar la probabilidad de desarrollar infecciones. Se debe descartar infección por el VIH, y tuberculosis y hepatitis activas antes del inicio del tratamiento con Cladribina. Las infecciones latentes pueden activarse, incluidas la tuberculosis o la hepatitis. Por lo tanto, se deben hacer pruebas de detección de infecciones latentes, en particular de tuberculosis y hepatitis B y C, antes del inicio del tratamiento en el año 1 y el año 2. El inicio del tratamiento con Cladribina debe retrasarse hasta que la infección haya sido adecuadamente tratada. También se debe considerar un retraso en el inicio del tratamiento con Cladribina en los pacientes con infección aguda hasta que ésta se haya controlado completamente. Se recomienda prestar una atención especial a los pacientes que no tienen antecedentes de exposición al virus de la varicela zóster. Se recomienda vacunar a los pacientes con anticuerpos negativos antes del inicio del tratamiento con Cladribina. El inicio del tratamiento con Cladribina debe posponerse durante cuatro a seis semanas para permitir que la vacunación sea efectiva. La incidencia de herpes zóster fue mayor en los pacientes en tratamiento con Cladribina. Si los recuentos de linfocitos descienden por debajo de 200 células/mm³, se debe considerar la administración de profilaxis contra el herpes de acuerdo con las prácticas locales habituales durante el tiempo que dure la linfopenia de grado 4. Se deben vigilar los signos y síntomas que sugieran infecciones, en particular herpes zóster, en los pacientes que presenten recuentos de linfocitos por debajo de 500 células/mm³. En caso de infección se debe iniciar el tratamiento antiinfeccioso según esté clínicamente indicado. Puede considerarse la interrupción o el retraso del tratamiento con Cladribina hasta la resolución de la infección. Se han notificado casos de leucoencefalopatía multifocal progresiva en pacientes tratados con Cladribina parenteral por tricoleucemia con una pauta de tratamiento diferente. En la base de datos de ensayos clínicos sobre Cladribina en Esclerosis Múltiple (1976 pacientes, 8650 años-paciente), no se notificó ningún caso de leucoencefalopatía multifocal progresiva. No obstante, se debe realizar una RM basal antes de iniciar Cladribina (habitualmente en un plazo de tres meses). Neoplasias malignas: En los estudios clínicos, se observaron casos de neoplasias malignas con mayor frecuencia en los pacientes tratados con Cladribina que en los que recibieron placebo. Cladribina está contraindicado en los pacientes con Esclerosis Múltiple que presentan neoplasias malignas activas. Se debe realizar una evaluación individual del beneficio-riesgo antes de iniciar el tratamiento con Cladribina en los pacientes con neoplasias malignas anteriores. Se debe aconsejar a los pacientes tratados con Cladribina que realicen una consulta con su médico oncólogo. Anticoncepción: Antes del inicio del tratamiento tanto en el año 1 como en el año 2, se debe asesorar a las mujeres en edad fértil y a los varones con parejas con capacidad de gestación respecto a la posibilidad de riesgos graves para el feto y la necesidad de utilizar métodos anticonceptivos efectivos. Las mujeres en edad fértil deben evitar el embarazo mediante el uso de métodos anticonceptivos efectivos durante el tratamiento con Cladribina y por lo menos hasta seis meses después de la última dosis. Los pacientes varones deben tomar precauciones para evitar el embarazo de su pareja durante el tratamiento con Cladribina y por lo menos hasta seis meses después de la última dosis. Transfusiones de sangre: En los pacientes que necesiten una transfusión de sangre, se recomienda la irradiación de los componentes hemáticos celulares antes de la administración, con el fin de evitar la enfermedad de injerto contra huésped relacionada con la transfusión. Se aconseja consultar con un hematólogo. Cambio de otro tratamiento a Cladribina o desde Cladribina a otro tratamiento: En los pacientes que han recibido tratamiento previo con medicamentos inmunomoduladores o inmunosupresores, se debe considerar el mecanismo de acción y la duración del efecto del otro medicamento antes del inicio de Cladribina. T