ECALTA
PFIZER
Agente antimicótico de uso sistémico.
Composición.
Cada frasco ampolla con polvo liofilizado contiene: Anidulafungina 100 mg. Fructosa 100 mg. Manitol 500 mg. Polisorbato 80: 250 mg. Acido tartárico 11,2 mg. Hidróxido de sodio y/o ácido clorhídrico para ajustar el pH.
Farmacología.
Descripción: ECALTA (anidulafungina) es un lipopéptido semisintético sintetizado a partir de un producto de la fermentación del Aspergillus nidulans. La anidulafungina es una equinocandina, una clase de antifúngico que inhibe la síntesis del 1,3-b-D-glucano, un componente esencial de la pared celular de los hongos. ECALTA (anidulafungina) es 1-[(4R,5R)-4,5-dihidroxi-N2-[[4"-(pentiloxi)[1,1':4',1"-terfenil]-4-il]carbonil]Lornitina] equinocandina B. La anidulafungina es un polvo blanco a blancuzco prácticamente insoluble en agua y levemente soluble en etanol. La fórmula empírica de la anidulafungina es C58H73N7O17 y el peso molecular es 1140,3. Su fórmula estructural es la siguiente: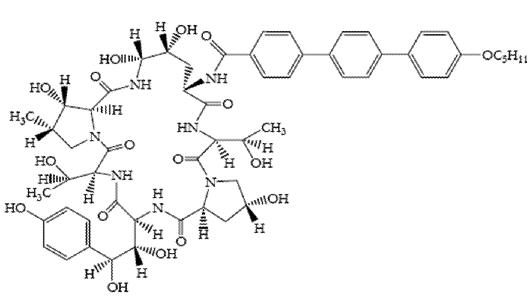
Antes de la administración de ECALTA Inyectable, es necesario reconstituir el producto con agua para inyectable y posteriormente diluirlo en dextrosa al 5% para inyectable o cloruro de sodio al 0,9% para inyectable (solución salina normal). No diluir con otras soluciones ni perfundir el producto junto con otros medicamentos o electrólitos (ver Dosificación). Mecanismo de acción: La anidulafungina es una equinocandina semisintética con actividad antifúngica. La anidulafungina inhibe la glucano sintetasa, una enzima que se encuentra presente en las células fúngicas, pero no en las células de los mamíferos. Esto produce la inhibición de la formación de 1,3-b-D-glucano, un componente esencial de la pared celular de los hongos. Actividad in vitro: La anidulafungina es activa in vitro contra Candida albicans, C. glabrata, C. parapsilosis y C. tropicalis (ver Indicaciones y Estudios clínicos). Las CIM se determinaron según el método de referencia estándar M27 aprobado por el Instituto para Estándares Clínicos y de Laboratorio (CLSI, Clinical and Laboratory Standards Institute) para la prueba de susceptibilidad para las levaduras. Sin embargo, aún no se ha establecido la correlación entre la actividad in vitro (CIM) según lo determinó este método y el resultado clínico. Actividad in vivo: La anidulafungina administrada por vía parenteral fue eficaz contra la Candida albicans en ratones y conejos inmunocompetentes e inmunosuprimidos con infección diseminada según la medición de la prolongación de la sobrevida y la reducción de la carga micológica. La anidulafungina también redujo la carga micológica de C. albicans resistente al fluconazol en un modelo de infección orofaríngea/esofágica de conejos inmunosuprimidos. Resistencia a la droga: Ha habido informes aislados de Candida con susceptibilidad reducida a la anidulafungina, lo que sugiere un potencial para el desarrollo de resistencia a la droga. La importancia clínica de esta observación es desconocida. Propiedades farmacocinéticas: Se ha caracterizado la farmacocinética de la anidulafungina tras la administración I.V. a voluntarios sanos, poblaciones especiales y pacientes. Las exposiciones sistémicas a la anidulafungina son proporcionales a la dosis y tienen una baja variabilidad de un intersujeto a otro (coeficiente de variación: < 25%), según se puede observar en la Tabla 1. El estado de equilibrio se alcanzó el primer día, después de la administración de una dosis de ataque (el doble de la dosis diaria de mantenimiento) y el factor de acumulación plasmática estimado en condiciones de equilibrio es aproximadamente 2.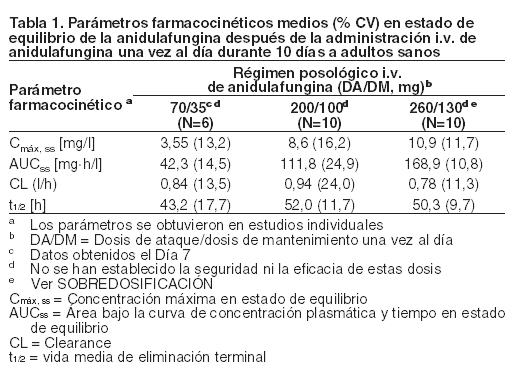
El clearance de la anidulafungina es de aproximadamente 1 l/h y la anidulafungina tiene una vida media de eliminación terminal de 40-50 horas. Distribución: La farmacocinética de la anidulafungina tras la administración por vía I.V. se caracteriza por una vida media de distribución breve (0,5 a 1 hora) y un volumen de distribución de 30-50 litros, que es similar al volumen total de líquidos corporales. La anidulafungina se une ampliamente a las proteínas plasmáticas humanas ( > 99%). Metabolismo: No se ha observado que la anidulafungina tenga un metabolismo hepático. La anidulafungina no es un sustrato, inductor o inhibidor clínicamente relevante de las isoenzimas del citocromo P450 (CYP450). Es improbable que la anidulafungina tenga efectos clínicamente relevantes sobre el metabolismo de los fármacos que se metabolizan a través de las isoenzimas del CYP450. La anidulafungina sufre una lenta degradación química, a temperatura y un pH fisiológicos, en un péptido de anillo abierto que carece de actividad antifúngica. La vida media de la degradación in vitro de la anidulafungina en condiciones fisiológicas es de 24 horas aproximadamente. In vivo, el producto de anillo abierto se convierte posteriormente en degradantes peptídicos y se elimina. Excreción: En un estudio clínico con una dosis única, se administró anidulafungina marcada con 14C a voluntarios sanos. Aproximadamente el 30% de la dosis radiactiva administrada se eliminó en las heces a lo largo de 9 días, menos del 10% de la cual fue en forma de droga intacta. Menos del 1% de la dosis radiactiva se excretó en la orina. Las concentraciones de anidulafungina estuvieron debajo del límite de cuantificación 6 días después de la administración de la dosis. Se recuperaron cantidades despreciables de radiactividad derivada del fármaco en la sangre, la orina y las heces, 8 semanas después de la administración de la dosis. Poblaciones especiales: Pacientes con infecciones fúngicas Los análisis farmacocinéticos de la población realizados a partir de cuatro estudios clínicos de fase 2/3 en los que se incluyen 107 pacientes de sexo masculino y 118 de sexo femenino con infecciones fúngicas demostraron que los parámetros farmacocinéticos de la anidulafungina no se ven afectados por la edad, la raza o la presencia de medicamentos concomitantes que son conocidos sustratos, inhibidores o inductores metabólicos. La farmacocinética de la anidulafungina en pacientes con infecciones fúngicas es similar a la que se observó en pacientes sanos. En la Tabla 2 se presentan los parámetros farmacocinéticos de la anidulafungina calculados utilizando una modelación farmacocinética poblacional después de la administración I.V. de una dosis de mantenimiento de 50 mg/día o 100 mg/día (posterior a la administración de una dosis de ataque).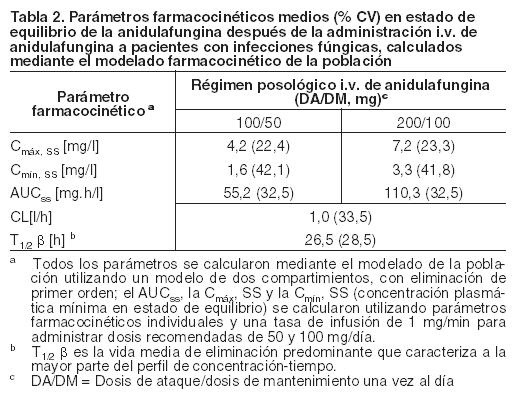
Sexo: No es necesario realizar ajustes de la dosis en función del sexo. Las concentraciones plasmáticas de anidulafungina en hombres y mujeres sanos fueron similares. En estudios realizados en pacientes a quienes se les administraron dosis múltiples, el clearance de la droga fue levemente más rápido (aproximadamente 22%) en los hombres. Población geriátrica: No es necesario realizar ajustes de la dosis para los pacientes de edad avanzada. Los análisis farmacocinéticos de la población demostraron que la mediana del clearance es levemente diferente entre el grupo de pacientes de edad avanzada (pacientes ≥ 65, mediana del CL = 1,07 l/h) y el grupo de pacientes de menor edad (pacientes < 65, mediana del CL = 1,22 l/h) y el rango de clearance fue similar. Raza: No es necesario realizar ajustes de la dosis en función de la raza. La farmacocinética de la anidulafungina fue similar entre los grupos de raza blanca, negra, asiática e hispánica. Infección por VIH: No es necesario realizar ajustes de la dosis en función de la infección por el VIH, independientemente de la terapia antirretroviral concomitante. Insuficiencia hepática: No es necesario realizar ajustes de la dosis en función de la insuficiencia hepática leve, moderada o grave. La anidulafungina no se metaboliza por vía hepática. Se examinó la farmacocinética de la anidulafungina en pacientes con insuficiencia hepática clase A, B o C de la escala de Child-Pugh. No se produjeron aumentos de las concentraciones de anidulafungina en los pacientes con algún grado de insuficiencia hepática. Aunque se observó una leve disminución del AUC en pacientes con insuficiencia hepática clase C de la escala de Child- Pugh, se mantuvo dentro del rango de las estimaciones para la población de voluntarios sanos. Insuficiencia renal: No es necesario realizar ajustes de la dosis para los pacientes con algún grado de insuficiencia renal, incluidos los pacientes que deben realizarse hemodiálisis. La anidulafungina tiene un clearance renal despreciable. En un estudio clínico realizado en pacientes con insuficiencia renal leve, moderada, grave o terminal (dependiente de diálisis), la farmacocinética de la anidulafungina fue similar a la que se observó en los pacientes con función renal normal. La anidulafungina no es dializable y puede ser administrada independientemente del horario de la hemodiálisis. Pacientes pediátricos: Se investigó la farmacocinética de la anidulafungina después de la administración de dosis diarias a pacientes pediátricos (2 a 11 años) y adolescentes (12 a 17 años) inmunocomprometidos con neutropenia. El estado de equilibrio se alcanzó el primer día, después de la administración de una dosis de ataque (el doble de la dosis de mantenimiento), y la Cmáx y el AUCss aumentaron de manera proporcional a la dosis. Las concentraciones y las exposiciones después de la administración de la dosis de mantenimiento de 0,75 y 1,5 mg/kg/día en esta población fueron similares a las que se observaron en adultos después de la administración de dosis de mantenimiento de 50 y 100 mg/día, respectivamente (según se muestra en la Tabla 3) (ver Advertencias, Uso en pacientes pediátricos).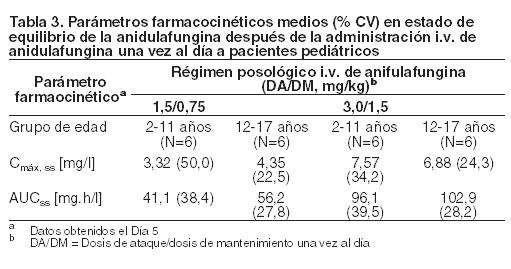
Estudios sobre interacciones medicamentosas: Estudios in vitro demostraron que la anidulafungina no se metaboliza a través del citocromo P450 humano ni de los hepatocitos humanos aislados; tampoco inhibe significativamente la actividad de las isoformas CYP humanas clínicamente importantes (1A2, 2C9, 2D6, 3A4). No se observaron interacciones medicamentosas clínicamente relevantes con los fármacos que probablemente se administren junto con la anidulafungina. Ciclosporina (sustrato del CYP3A4): En un estudio en el que 12 pacientes adultos sanos recibieron una dosis de mantenimiento de 100 mg/día de anidulafungina después de la administración de una dosis de ataque de 200 mg (Días 1 a 8) y en combinación con 1,25 mg/kg de ciclosporina administrada por vía oral dos veces al día (Días 5 a 8), la Cmáx en estado de equilibrio de anidulafungina no fue afectada significativamente por la ciclosporina; el AUC en estado de equilibrio de la anidulafungina aumentó un 22%. Un estudio in vitro separado demostró que la anidulafungina no influyó sobre el metabolismo de la ciclosporina. No se recomienda ajustar la dosis de ninguno de los fármacos cuando se administran de manera concomitante. Voriconazol (inhibidor y sustrato del CYP2C19, CYP2C9, CYP3A4): En un estudio en el que 17 pacientes sanos recibieron una dosis de mantenimiento de 100 mg/día de anidulafungina después de la administración de una dosis de ataque de 200 mg, 200 mg de voriconazol por vía oral dos veces al día (después de la administración de dos dosis de ataque de 400 mg) y ambas en combinación, la administración concomitante no alteró significativamente la Cmáx ni el AUC en estado de equilibrio de anidulafungina y voriconazol. No se recomienda ajustar la dosis de ninguno de los fármacos cuando se administran de manera concomitante. Tacrolimus (sustrato del CYP3A4): En un estudio en el que 35 pacientes sanos recibieron una dosis única de 5 mg de tacrolimus administrada por vía oral (el Día 1), una dosis de mantenimiento de 100 mg/día de anidulafungina después de la administración de una dosis de ataque de 200 mg (Días 4 a 12) y ambas en combinación (Día 13), la administración concomitante no alteró significativamente la Cmáx ni el AUC en estado de equilibrio de anidulafungina y tacrolimus. No se recomienda ajustar la dosis de ninguno de los fármacos cuando se administran de manera concomitante. AmBisome® (anfotericina B liposomal): Se examinó la farmacocinética de la anidulafungina en 27 pacientes que recibieron anfotericina B liposómica de manera concomitante. Los análisis farmacocinéticos de la población demostraron que, al realizar una comparación con los datos obtenidos de los pacientes que no recibieron anfotericina B, la administración concomitante con anfotericina B no alteró significativamente la farmacocinética de la anidulafungina. No se recomienda ajustar la dosis de anidulafungina. Rifampina (potente inductor de CYP450): Se examinó la farmacocinética de la anidulafungina en 27 pacientes que recibieron anidulafungina y rifampina de manera concomitante. Los análisis farmacocinéticos de la población demostraron que, al realizar una comparación con los datos obtenidos de los pacientes que no recibieron rifampina, la administración concomitante con rifampina no alteró significativamente la farmacocinética de la anidulafungina. No se recomienda ajustar la dosis de anidulafungina. Estudios clínicos: Candidemia y otras infecciones por Candida (absceso intraabdominal y peritonitis). Se evaluaron la seguridad y la eficacia de ECALTA en un estudio de fase 3, randomizado, doble ciego realizado en pacientes con candidemia y/u otras formas de candidiasis invasiva. Los pacientes fueron randomizados para recibir ECALTA por vía I.V. una vez al día (dosis de ataque de 200 mg seguida por una dosis de mantenimiento de 100 mg) o fluconazol por vía I.V. (dosis de ataque de 800 mg seguida por una dosis de mantenimiento de 400 mg). Los pacientes fueron estratificados en función del puntaje APACHE II (≤ 20 y > 20) y de la presencia o la ausencia de neutropenia. Se excluyeron del estudio a los pacientes con endocarditis, osteomielitis o meningitis por Candida, o a aquellos con una infección causada por C. krusei. El tratamiento se administró durante un mínimo de 14 días y un máximo de 42 días. Se permitió que los pacientes de ambos grupos de estudio pasaran al tratamiento con fluconazol por vía oral después de un mínimo de 10 días de tratamiento intravenoso, siempre que pudieran tolerar el medicamento oral, no presentaran fiebre durante un mínimo de 24 horas y que los resultados de los últimos cultivos sanguíneos fueran negativos para las especies de Candida. Los pacientes que recibieron al menos una dosis del medicamento de estudio y que presentaron un resultado positivo para las especies de Candida en el cultivo obtenido de una zona normalmente estéril antes del ingreso al estudio (población según intención de tratar modificada [MITT]) fueron incluidos en el análisis primario de la respuesta global al final del tratamiento intravenoso. Una respuesta global exitosa requirió cura o mejoría clínica (resolución significativa, aunque incompleta, de los signos y síntomas de la infección por Candida, sin tratamiento antifúngico adicional), y erradicación microbiológica documentada o supuesta. En esta población, los pacientes que obtuvieron un resultado indeterminado se analizaron como fracasos. Se randomizaron doscientos cincuenta y seis pacientes, a quienes se les administró al menos una dosis del medicamento de estudio. La mediana de la duración del tratamiento I.V. fue de 14 y 11 días en los grupos que recibieron ECALTA y el fluconazol, respectivamente. En el caso de los pacientes que recibieron fluconazol por vía oral, la mediana de la duración del tratamiento oral fue de 7 días para el grupo que recibió ECALTA y de 5 días para el grupo que recibió el fluconazol. En la Tabla 4 se presenta la distribución de los pacientes.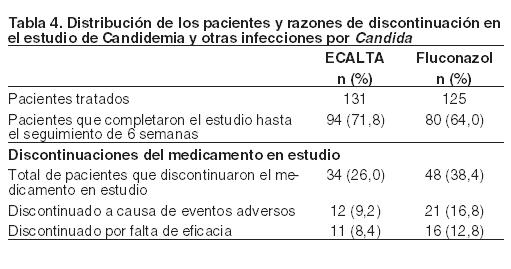
Doscientos cuarenta y cinco pacientes (127 recibieron ECALTA, 118 recibieron fluconazol) cumplieron con los criterios de inclusión en la población MITT. De estos 245 pacientes, 219 (116 recibieron ECALTA, 103 recibieron fluconazol) presentaron candidemia únicamente. En este estudio, los factores de riesgo para candidemia entre los pacientes de ambos grupos de tratamiento fueron: presencia de un catéter venoso central (78%), tratamiento con antibióticos de amplio espectro (69%), cirugía reciente (42%), hiperalimentación reciente (25%) y malignidad de base (22%). La especie aislada más frecuente en el nivel basal fue C. albicans (61,6%), seguida por C. glabrata (20,4%), C. parapsilosis (11,8%) y C. tropicalis (10,6%). La mayoría (97%) de los pacientes fueron no neutropénicos (RAN > 500) y el 81% presentó puntajes de APACHE II inferiores o iguales a 20. En la Tabla 5 se resumen las tasas de éxito global en pacientes con candidemia y otras infecciones por Candida.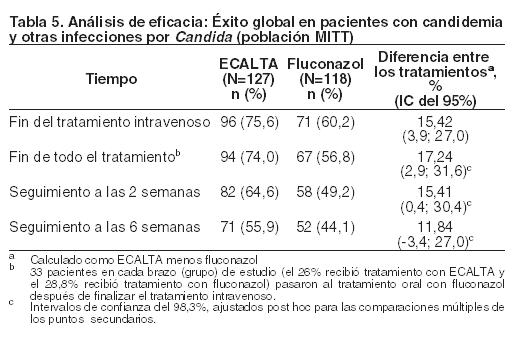
La Tabla 6 presenta los datos sobre los resultados y la mortalidad para la población MITT.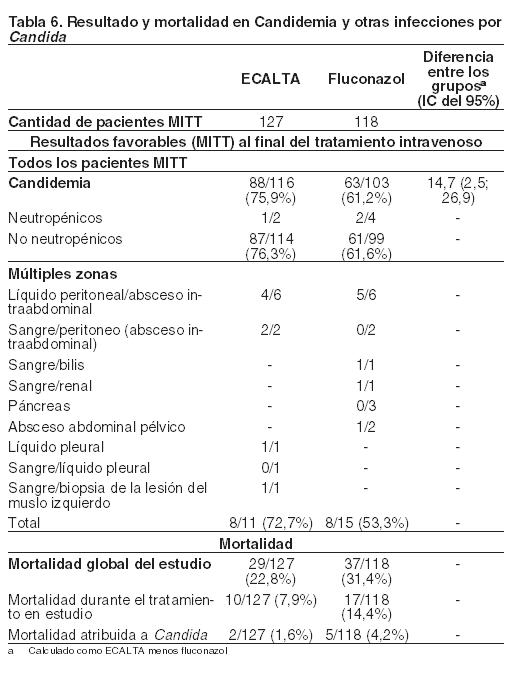
Indicaciones.
ECALTA está indicado para el tratamiento de las siguientes infecciones fúngicas: Candidemia y otras formas de infecciones por Candida (absceso intraabdominal y peritonitis) (ver Estudios clínicos y Farmacología). No se han realizado estudios con ECALTA en el tratamiento de endocarditis, osteomielitis y meningitis causadas por Candida y no se han realizado estudios en cantidades suficientes de pacientes neutropénicos que permitan determinar la eficacia en este grupo. Se deben obtener las muestras para cultivo fúngico y otros estudios de laboratorio relevantes (incluida la histopatología) antes del tratamiento a fin de poder aislar e identificar el/los organismo(s) causante(s). Se puede iniciar el tratamiento antes de conocer los resultados de los cultivos y de otros estudios de laboratorio. Sin embargo, una vez que estos resultados estén disponibles, se debe ajustar el tratamiento antifúngico en función de estos.
Dosificación.
Candidemia y otras infecciones por Candida (absceso intraabdominal y peritonitis): La dosis recomendada es una dosis de ataque única de 200 mg de ECALTA el día 1, seguida por una dosis diaria de 100 mg a partir de entonces. La duración del tratamiento debe basarse en la respuesta clínica del paciente. En general, el tratamiento antifúngico debe continuar durante un mínimo de 14 días posteriores al último cultivo con resultado positivo. En base a la evidencia actual, no parece necesario realizar un ajuste de la dosis para los pacientes con algún grado de insuficiencia renal o hepática, los pacientes que utilizan medicamentos concomitantes o aquellos que pertenecen a otras poblaciones especiales (ver Farmacología - Poblaciones especiales y Estudios sobre interacciones medicamentosas). Preparación de ECALTA para la administración: ECALTA inyectable se debe reconstituir con agua estéril para inyectable y posteriormente diluir sólo con dextrosa para inyectable al 5% o cloruro de sodio para inyectable al 0,9% (solución salina). Aún no se ha establecido la compatibilidad de ECALTA reconstituido con sustancias, aditivos o medicamentos intravenosos que no sean dextrosa para inyectable al 5% o cloruro de sodio para inyectable al 0,9% (solución salina). Reconstitución de los frascos ampolla de 100 mg. Reconstituir de manera aséptica cada frasco ampolla de 100 mg con 30 ml de agua estéril para inyectable para obtener una concentración de 3,33 mg/ml. La solución reconstituida puede conservarse por un máximo de una hora en heladera entre 2°C - 8°C antes de la disolución en la solución para perfusión. No congelar. Dilución y perfusión: Transferir de manera aséptica el contenido de el/los frasco(s) ampolla reconstituido(s) a una bolsa I.V. (o botella) de tamaño adecuado que contenga dextrosa para inyectable al 5% o cloruro de sodio para inyectable al 0,9% (solución salina). La siguiente tabla presenta las instrucciones de disolución y perfusión de cada dosis.
La tasa de perfusión no debe superar 1,1 mg/minuto (equivalente a 1,4 ml/minuto u 84 ml/hora cuando se reconstituye y diluye según las instrucciones). ECALTA no debe administrarse en forma de bolo. Si la solución para la perfusión no se utiliza inmediatamente, se debe conservar en heladera entre 2°C-8°C. No congelar. La solución para perfusión debe administrarse dentro de las 24 horas de la preparación. Se deben inspeccionar visualmente los productos medicinales parenterales para detectar cualquier partícula o decoloración antes de la administración, siempre que la solución y el envase lo permitan. Si se identifica alguna partícula o decoloración, desechar la solución. Cada envase de ECALTA sirve para una sola administración, descartar el remanente.
Contraindicaciones.
ECALTA está contraindicado en personas con hipersensibilidad conocida a la anidulafungina, a cualquier componente de ECALTA o a otras equinocandinas.
Reacciones adversas.
Generales: Se han informado posibles síntomas mediados por la histamina con la administración de ECALTA, incluidos erupción cutánea, urticaria, rubor, prurito, broncoespasmo, disnea e hipotensión. Estos eventos son poco frecuentes cuando la tasa de perfusión de ECALTA no supera 1,1 mg/minuto. Experiencia general sobre la seguridad de ECALTA: Se evaluó la seguridad de ECALTA inyectable en 929 individuos, incluidos 672 pacientes en estudios clínicos y 257 individuos en estudios de fase 1. Un total de 633 pacientes recibió dosis diarias de ECALTA de 50 o 100 mg. Un total de 481 pacientes recibió ECALTA durante ≥ 14 días. Tres estudios (uno comparativo con fluconazol, dos no comparativos) evaluaron la eficacia y la seguridad de ECALTA (100 mg) en pacientes con candidemia y otras infecciones por Candida. La Tabla 7 presenta los eventos adversos relacionados con el tratamiento que se informaron en ≥ 2,0% de pacientes que recibieron tratamiento con ECALTA o fluconazol en el estudio comparativo de candidemia.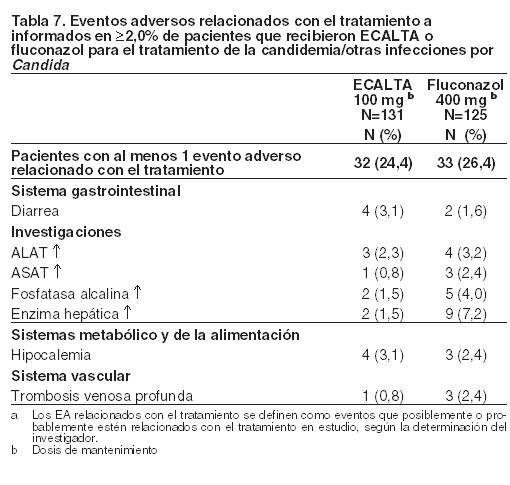
Un estudio único, de fase 3, randomizado, doble ciego comparó la eficacia y la seguridad de ECALTA con la del fluconazol en pacientes con candidiasis esofágica. La Tabla 8 presenta los eventos adversos relacionados con el tratamiento que fueron informados en ≥ 1,0% de pacientes que recibieron tratamiento con ECALTA. (No se informaron eventos adversos con una frecuencia del 2% o mayor en pacientes con candidiasis esofágica).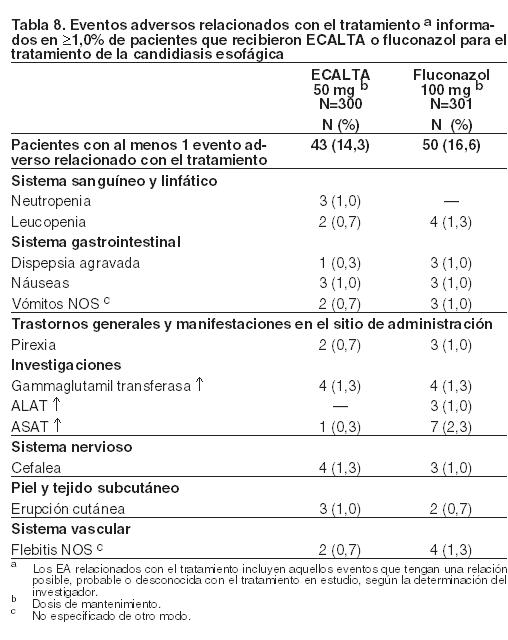
Los siguientes eventos ocurrieron en < 2% de los pacientes tratados por candidemia/otras infecciones por Candida o en < 1% de los pacientes tratados por candidiasis esofágica y, según el criterio del investigador, estaban al menos posiblemente relacionados con ECALTA: Sanguíneos y linfáticos: coagulopatía, trombocitopenia. Cardíacos: fibrilación auricular, bloqueo de rama (derecha), arritmia sinusal, extrasístoles ventriculares. Oculares: dolor ocular, visión borrosa, alteración de la visión. Gastrointestinales: dolor abdominal superior, constipación, diarrea no especificada de otro modo, dispepsia, incontinencia fecal, náuseas, vómitos. Generales y en el sitio de administración: reacción relacionada con la perfusión, edema periférico, rigidez en el sitio de administración. Hepatobiliares: pruebas de la función hepática anormal no especificadas de otro modo, colestasis, necrosis hepática. Infecciones: candidiasis, infección por Clostridium, fungemia, candidiasis oral. Investigaciones: amilasa ↑, bilirrubina ↑, CPK ↑, creatinina ↑, QT prolongado en el electrocardiograma, gammaglutamil transferasa ↑, transición temprana en el electrocardiograma, lipasa ↑, magnesio ↓, recuento plaquetario ↑, recuento plaquetario ↓, potasio ↓, tiempo de protrombina prolongado, urea ↑. Del metabolismo y la alimentación: hipercalcemia, hiperglucemia, hipercalemia, hipernatremia, hipomagnesemia. Musculoesqueléticos y del tejido conectivo: dolor de espalda. Sistema nervioso: convulsiones, mareos, cefaleas. Respiratorio, torácico y del mediastino: tos. Cutáneos y del tejido conectivo: edema angioneurótico, eritema, prurito, prurito generalizado, aumento de la sudoración, urticaria, urticaria no especificada de otra forma. Vasculares: rubor, sofocos de calor, hipertensión, hipotensión, tromboflebitis superficial.
Advertencias.
Efectos hepáticos: Se han observado anormalidades de laboratorio en las pruebas de la función hepática en voluntarios sanos y en pacientes tratados con ECALTA. En algunos pacientes con afecciones serias de base, quienes recibían múltiples medicaciones concomitantes junto con ECALTA, ocurrieron anormalidades hepáticas clínicamente significativas. Se han informado casos aislados de disfunción hepática significativa, hepatitis o insuficiencia hepática en pacientes; no se ha establecido una relación causal con ECALTA. Los pacientes que desarrollen pruebas de la función hepática anormales durante el tratamiento con ECALTA deben ser controlados para detectar cualquier evidencia de empeoramiento de la función hepática y se deben evaluar los riesgos/beneficios de continuar con el tratamiento con ECALTA. Interacciones medicamentosas: Los estudios clínicos y pre-clínicos in vitro e in vivo demostraron que la anidulafungina no es un sustrato, inductor o inhibidor de las isoenzimas del citocromo P450 clínicamente relevantes. La anidulafungina presenta un clearance renal despreciable. Se esperan interacciones mínimas con los medicamentos concomitantes (ver Farmacología - Estudios sobre interacciones medicamentosas). Se llevaron a cabo estudios de interacción medicamentosa de la anidulafungina con otros fármacos que posiblemente se administren de manera concomitante. Cuando se utiliza en dosis terapéuticas, no se recomienda realizar un ajuste de la dosis de ninguno de los fármacos cuando la anidulafungina se administra concomitantemente con voriconazol o tacrolimus y no se recomienda realizar un ajuste de la dosis de la anidulafungina cuando se administre concomitantemente con anfotericina B o rifampina (ver Farmacología - Estudios sobre interacciones medicamentosas). La administración concomitante con ciclosporina aumentó levemente el AUC en estado de equilibrio de la anidulafungina en un 22%. Un estudio separado in vitro demostró que la anidulafungina no influyó sobre el metabolismo de la ciclosporina. Los eventos adversos observados en el estudio fueron consistentes con los eventos adversos observados en otros estudios con la administración de anidulafungina únicamente. No se recomienda ajustar la dosis de ninguno de los fármacos para los pacientes que reciben ciclosporina concomitantemente (ver Farmacología - Estudios sobre interacciones medicamentosas). Farmacología y toxicología en animales: En estudios de 3 meses de duración, se observó toxicidad hepática, incluidas necrosis hepatocelular de célula única, hipertrofia hepatocelular y aumento de los pesos hepáticos, en monos y ratas que recibieron dosis equivalentes a 5-6 veces la exposición en seres humanos. Para ambas especies, la hipertrofia hepatocelular aún se observó un mes después de finalizar la administración de la dosis. Carcinogénesis, mutagénesis, trastornos de la fertilidad: Aún no se han llevado a cabo estudios de carcinogenicidad a largo plazo en animales con la administración de anidulafungina. La anidulafungina no fue genotóxica en los siguientes estudios in vitro: ensayos de mutación inversa bacteriana, un ensayo de aberración cromosómica con células de ovario de hámster chino y un ensayo de mutación genética directa con células de linfoma de ratón. La anidulafungina no fue genotóxica en ratones en el ensayo de micronúcleo in vivo. La anidulafungina no produjo eventos adversos sobre la fertilidad en ratas macho o hembra con la administración intravenosa de dosis de 20 mg/kg/día (equivalente al doble de la dosis de mantenimiento terapéutica propuesta de 100 mg/día en base al área de superficie corporal relativa). Embarazo: Embarazo categoría C: Se llevaron a cabo estudios sobre el desarrollo embrionario-fetal con la administración de dosis máximas de 20 mg/kg/día en ratas y conejos (equivalente a 2 y 4 veces, respectivamente, la dosis de mantenimiento terapéutica propuesta de 100 mg/día en base al área de superficie corporal relativa). La administración de anidulafungina provocó cambios esqueléticos en los fetos de ratas incluida la osificación incompleta de diversos huesos y costillas onduladas, desalineadas o deformes. Estos cambios no estuvieron relacionados con la dosis y se mantuvieron dentro del rango de la base de datos de control histórico del laboratorio. Los efectos sobre el desarrollo observados en conejos (leve disminución de los pesos fetales) ocurrieron en el grupo que recibió dosis altas; esta dosis también produjo toxicidad maternal. La anidulafungina atravesó la barrera placentaria en ratas y se detectó en el plasma del feto. No se han realizado estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios sobre reproducción realizados en animales no siempre predicen la respuesta en seres humanos, ECALTA debe utilizarse durante el embarazo sólo si el beneficio potencial justifica el riesgo para el feto. Lactancia: ECALTA debe administrarse a madres en período de lactancia sólo si el beneficio potencial justifica el riesgo. Se detectó anidulafungina en la leche de las ratas en período de lactancia. Se desconoce si la anidulafungina se excreta en la leche humana. Uso en pacientes pediátricos: Aún no se han establecido la seguridad ni la efectividad de la anidulafungina en pacientes pediátricos (ver Farmacología - Poblaciones especiales/Pacientes pediátricos).
Conservación.
Producto sin reconstituir: Los frascos ampolla de ECALTA Inyectable sin reconstituir deben conservarse en heladera entre 2°C y 8°C. No congelar. Producto reconstituido: ECALTA inyectable reconstituido puede conservarse en heladera entre 2°C y 8°C durante un máximo de 1 hora. No congelar. La estabilidad química y física durante el uso de la solución reconstituida ha sido demostrada durante 1 hora a 5°C. Solución para perfusión: La solución para perfusión de ECALTA puede conservarse en heladera entre 2°C-8°C, pero debe administrarse dentro de las 24 horas. No congelar. La estabilidad química y física durante el uso de la solución reconstituida ha sido demostrada durante 24 horas a 5°C.
Sobredosificación.
Durante los estudios clínicos, se administró una dosis única de ECALTA de 400 mg inadvertidamente como dosis de ataque. No se informaron eventos adversos clínicos. En un estudio realizado en 10 pacientes sanos se administró una dosis de ataque de 260 mg seguida por 130 mg diarios; por lo general, ECALTA fue bien tolerado: 3 de 10 pacientes experimentaron aumentos de la transaminasa transitorios y asintomáticos (≤ 3 x LSN). La anidulafungina no es dializable. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse a los Centros de Toxicología: Hospital de Pediatría Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital A. Posadas: (011) 4658-7777 / 4654-6648.
Presentación.
ECALTA 100 mg: Envases que contienen 1 frasco ampolla con polvo liofilizado estéril.
Revisión.
Diciembre 2011. LPD: 04/Nov/2010.