DIVALPREX ER
GADOR
Antiepiléptico y preventivo de la migraña. Antimaníaco.
Composición.
Cada comprimido de liberación extendida de DIVALPREX® ER 250 contiene: Divalproato de sodio (equivalente a 250 mg de Acido valproico) 269,05 mg, Hidroxipropilmetilcelulosa K 15 M CR 227,5 mg, Lactosa monohidrato 70,965 mg, Celulosa microcristalina PH 101 62,985 mg, Silica coloidal anhidra 19,5 mg, Hidroxipropilmetilcelulosa 3cP1) 1,4586 mg, Hidroxipropilmetilcelulosa 6cP1) 1,4586 mg, Dióxido de titanio1) 1,50722 mg, Polietilenglicol 4001) 0,38896 mg, Polisorbato 801) 0,04862 mg, Etanol2) 0,08556 mg, Goma laca2) 0,03588 mg, Cera blanca2) 0,00828 mg, Cera carnauba2)0,00828 mg. 1) Componentes que corresponden a 4,862 mg de Opadry YS 1 7003 blanco. 2) Componentes que corresponden a 0,138 mg de Opaglos GS 2 0700. Cada comprimido de liberación extendida de DIVALPREX® ER 500 contiene: Divalproato de sodio (equivalente a 500 mg de Acido valproico).. 538,1 mg Hidroxipropilmetilcelulosa K 15 M CR 350 mg, Lactosa monohidrato 36,9 mg, Celulosa microcristalina PH 101 45 mg, Silica coloidal anhidra 30 mg, Hidroxipropilmetilcelulosa 3cP1) 5,4201 mg, Hidroxipropilmetilcelulosa 6cP1) 5,4201 mg, Dióxido de titanio1) 5,60077 mg, Polietilenglicol 4001) 1,44536 mg, Polisorbato 801) 0,18067 mg, Óxido de hierro amarillo 10 1,549 mg, Etanol2) 0,23808 mg, Goma laca2) 0,09984 mg, Cera blanca2) 0,02304 mg, Cera carnauba2) 0,02304 mg. 1) Componentes que corresponden a 18,067 mg de Opadry YS 1 7003 blanco. 2) Componentes que corresponden a 0,384 mg de Opaglos GS 2 0700.
Farmacología.
Mecanismo de acción: El Divalproato de sodio se disocia a ión Valproato en el tracto gastrointestinal. Aún no se ha establecido el mecanismo por el cual el Valproato ejerce su acción terapéutica. Se ha sugerido que su acción antiepiléptica se debe al aumento de las concentraciones de Ácido gama-aminobutírico (GABA) en el cerebro. Farmacodinamia: La relación entre concentración plasmática y respuesta clínica no está bien documentada. Un factor contribuyente es la unión a las proteínas no lineal dependiente de la concentración del Valproato, lo que afecta el clearance de la droga. Por lo tanto, el monitoreo de la concentración sérica de Valproato total no constituye un índice confiable de los compuestos bioactivos del Valproato. Por ejemplo, debido a que la unión del Valproato a las proteínas plasmáticas depende de la concentración, la fracción libre aumenta aproximadamente entre un 10% con concentraciones de 40 mcg/ml y un 18,5% con concentraciones de 130 mcg/ml. En ancianos, en pacientes con hiperlipemia y en aquéllos con enfermedades hepáticas o renales, la fracción libre es más elevada que la esperada. Manía: En un estudio clínico controlado contra placebo en manía aguda, los pacientes fueron dosificados hasta lograr la respuesta clínica, con concentraciones plasmáticas valle de entre 85 y 125 mcg/ml (ver Posología y forma de administración). Epilepsia: Comúnmente, se considera que el rango terapéutico en la epilepsia es de 50 a 100 mcg/ml de Valproato total, a pesar de que algunos pacientes pueden ser controlados con concentraciones plasmáticas menores o mayores. Farmacocinética: Absorción/Biodisponibilidad: La biodisponibilidad absoluta de los comprimidos de Divalproato de sodio ER administradas como una dosis única después de las comidas, fue aproximadamente del 90% en relación a la infusión intravenosa. Cuando se administra en iguales dosis totales diarias, la biodisponibilidad de Divalproato de sodio ER es menor que la del Divalproato de sodio (comprimidos recubiertos gastrorresistentes). En estudios de dosis múltiples en sujetos sanos (N=82) y en pacientes con epilepsia (N=86), cuando se administró en ayuno y sin ayuno Divalproato de sodio ER una vez al día, produjo un promedio de biodisponibilidad del 89% en relación a una dosis diaria total e igual de Divalproato de sodio administrado 2, 3 ó 4 veces al día. El tiempo medio para alcanzar las concentraciones máximas de Valproato (Cmáx) después de la administración de Divalproato de sodio ER, variaron de 4 a 17 hs. Después de la administración de varias dosis únicas diarias de Divalproato de sodio ER, la fluctuación entre las concentraciones plasmáticas máximas y mínimas de Valproato, fue de 10-20% inferior a la de Divalproato de sodio convencional administrado 2,3 ó 4 veces por día. Conversión de Divalproato de sodio a Divalproato de sodio ER: Cuando el Divalproato de sodio ER es dado en dosis de 8 a 20% mayores a la dosis diaria total de Divalproato de sodio, ambas formulaciones son bioequivalentes. En 2 estudios cruzados, aleatorizados, fueron comparadas dosis diarias múltiples de Divalproato de sodio con dosis diarias únicas de un 8 a 20% mayores de Divalproato de sodio ER. En dichos estudios los regímenes de Divalproato de sodio ER y Divalproato de sodio fueron equivalentes en relación al área bajo la curva. Adicionalmente, la Cmáx de Valproato fue menor y la Cmín fue mayor o igual para Divalproato de sodio ER en relación a los regímenes de Divalproato de sodio (ver tabla).
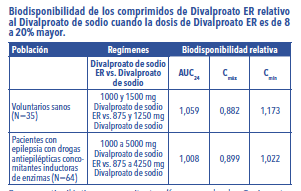
Drogas antiepilépticas concomitantes (fueron evaluadas Topiramato, Fenobarbital, Carbamazepina, Fenitoína y Lamotrigina), que inducen el sistema de la citocromo P450, no alteraron significativamente la biodisponibilidad de Valproato cuando se realizó la conversión de Divalproato de sodio a Divalproato de sodio ER. Distribución: Unión a proteínas: La unión del Valproato a proteínas plasmáticas depende de la concentración y la fracción libre aumenta aproximadamente entre un 10% con concentraciones de 40 mcg/ml y un 18,5% con concentraciones de 130 mcg/ml. La unión del Valproato a las proteínas disminuye en los ancianos, en pacientes con hepatopatías crónicas, pacientes con deterioro renal y en presencia de otras drogas (por ejemplo, Aspirina). Por el contrario, el Valproato puede desplazar a ciertas drogas que se unen a las proteínas (por ejemplo la Fenitoína, Carbamazepina, Warfarina y Tolbutamida) (ver Precauciones, Interacciones medicamentosas). Distribución en el SNC: Las concentraciones de Valproato en el líquido cefalorraquídeo (LCR) se aproximan a las concentraciones libres en el plasma (alrededor del 10% de la concentración total). Metabolismo: El Valproato es metabolizado casi en su totalidad por el hígado. En pacientes adultos que reciben monoterapia, el 30-50% de una dosis administrada aparece en la orina como conjugado glucurónico, La otra vía metabólica principal es la Beta-oxidación en las mitocondrias, lo que generalmente representa más del 40% de la dosis. Normalmente, menos del 15-20% de la dosis se elimina por otros mecanismos oxidativos. Menos del 3% de la dosis administrada se excreta intacta en la orina. La relación entre la dosis y la concentración de Valproato total no es lineal; la concentración no aumenta en forma proporcional a la dosis, sino que aumenta en menor grado debido a la saturación de los sitios de unión a las proteínas plasmáticas. La cinética de la droga libre es lineal. Eliminación: El clearance plasmático medio y el volumen de distribución del Valproato total son de 0,56 litro/h/1,73 m2 y 11 litros/1,73 m2, respectivamente. El clearance plasmático medio y el volumen de distribución del Valproato libre son de 4,6 litros/h/1,73 m2 y 92 litros/1,73 m2. La vida media terminal promedio del Valproato como monoterapia osciló entre 9 a 16 horas después de la administración oral de 250 mg a 1000 mg. Los valores citados se aplican principalmente a pacientes que no reciben drogas que afectan los sistemas enzimáticos del metabolismo hepático. Por ejemplo, los pacientes que reciben agentes antiepilépticos inductores de enzimas (Carbamazepina, Fenitoína y Fenobarbital) depurarán el Valproato en forma más rápida. Debido a estos cambios en el clearance del Valproato, deberá intensificarse el monitoreo de las concentraciones antiepilépticas cada vez que se agreguen o se suspendan fármacos antiepilépticos concomitantes. Poblaciones especiales: Niños: El perfil farmacocinético de valproato seguido de la administración de divalproato de sodio ER fue caracterizado en un estudio mulicéntrico, abierto, sin ayunar y de dosis múltiple en niños y adolescentes. Las dosis de divalproato de sodio ER una vez al día oscilaron entre 250-1750 mg. La administración de divalproato de sodio ER una vez al día en pacientes pediátricos (10 a 17 años) produjo perfiles de tiempo-concentración plasmáticos de Valproato similar a aquellos que han sido observados en adultos. Los pacientes pediátricos (entre 3 meses y 10 años) poseen un 50% más de clearance, expresado en el peso (es decir, ml/min/kg), que los adultos. Los niños mayores de 10 años presentan parámetros farmacocinéticos similares a los de los adultos. Ancianos: Se ha demostrado que la capacidad de los pacientes de edad avanzada (rango etario: 68 a 89 años) para eliminar Valproato es reducida en comparación con la de los pacientes adultos jóvenes (entre 22 y 26 años). El clearance intrínseco se reduce en un 39% y la fracción libre aumenta en un 44%. Por consiguiente, deberá reducirse la dosis inicial en los ancianos (ver Posología y forma de administración). Sexo: No existen diferencias en el clearance de la fracción libre, corregido por área de superficie corporal, entre hombres y mujeres (4,8±0,17 y 4,7±0,07 litros/h por 1,73 m2, respectivamente). Raza: No se han evaluado los efectos de la raza sobre la cinética del Valproato. Enfermedades hepáticas: Las enfermedades hepáticas alteran la capacidad de eliminar Valproato. En un estudio, el clearance del Valproato libre disminuyó un 50% en 7 pacientes con cirrosis y un 16% en 4 pacientes con hepatitis aguda, en comparación con 6 pacientes sanos. En dicho estudio, la vida media del Valproato aumentó de 12 a 18 horas. Las enfermedades hepáticas también están asociadas con menores concentraciones de albúmina y mayores fracciones libres (incremento de 2 a 2,6 veces) de Valproato. Por consiguiente, el monitoreo de las concentraciones totales puede dar lugar a interpretaciones falsas, ya que en pacientes con hepatopatías las concentraciones libres pueden ser muy elevadas, mientras que las concentraciones totales pueden parecer normales (ver Advertencias, Contraindicaciones). Enfermedades renales: Se ha informado de una ligera reducción (27%) en el clearance del Valproato libre en pacientes con insuficiencia renal (clearance de creatinina < 10 ml/min); sin embargo, la hemodiálisis generalmente reduce las concentraciones de Valproato en alrededor de un 20%. Por lo tanto, no sería necesario ajustar la dosis en pacientes con insuficiencia renal. En estos pacientes, la unión a las proteínas se ve considerablemente reducida, por lo que el monitoreo de las concentraciones totales puede llevar a interpretaciones erróneas.
Indicaciones.
Manía: Los comprimidos de Divalproato de sodio ER están indicados para el tratamiento de los episodios maníacos agudos o mixtos asociados con el trastorno bipolar, con o sin signos psicóticos. Un episodio maniaco es un período caracterizado por humor irritable, expansivo o anormal y persistentemente elevado. Los síntomas típicos de manía incluyen verborragia, hiperactividad motora, escasa necesidad de sueño, fuga de ideas, delirio de grandeza, juicio pobre, agresividad y posible hostilidad. Un episodio mixto está caracterizado por los criterios de un episodio maníaco junto con aquellos de un episodio de depresión mayor (humor deprimido, pérdida de interés o placer en casi todas las actividades). La eficacia de Divalproato de sodio ER está basada en parte en estudios llevados a cabo en esta indicación y fue confirmada en un estudio de tres semanas de duración en pacientes que reunían los criterios del DSM-IV TR para el trastorno bipolar I, de tipo maníaco o mixto, quienes fueron internados por manía aguda. La efectividad de Divalproato de sodio ER para el uso a largo plazo en manía, es decir, más de 3 semanas, no ha sido demostrada en estudios clínicos controlados. Por lo tanto, los médicos que decidan la utilización de Divalproato de sodio ER por períodos prolongados deberán reevaluar continuamente los riesgos y beneficios a largo plazo de la droga en cada caso en particular. Epilepsia: Divalproato de sodio ER está indicado como monoterapia y como terapia adjunta en tratamiento de adultos y niños de 10 años o más con crisis parciales complejas, ya sean aisladas o bien asociadas con otros tipos de crisis. Los comprimidos de Divalproato de sodio de liberación extendida, también están indicados para el uso como terapia única o adjunta en el tratamiento de crisis de ausencia simple y compleja en adultos y niños de 10 años o más y adicionalmente, en adultos y niños de 10 años o más con múltiples tipos de crisis que incluyan las crisis de ausencia. Migraña: Divalproato de sodio ER está indicado para la prevención de la migraña en adultos. No existe evidencia que avale la utilidad de Divalproato de sodio ER en el tratamiento del episodio agudo de este tipo de cefalea.
Dosificación.
DIVALPREX® ER es un producto de liberación extendida para administración oral una vez al día. Los comprimidos de DIVALPREX® ER deben ingerirse enteros, sin masticar. Manía: La dosis Inicial recomendada de DIVALPREX® ER es de 25 mg/kg/día administrada una vez al día. Esta dosis deberá aumentarse tan rápidamente como sea posible hasta alcanzar la menor dosis terapéutica que produzca el efecto clínico deseado o el rango de concentraciones plasmáticas deseadas. En un estudio clínico controlado contra placebo en manía aguda o tipo mixto, los pacientes se dosificaron hasta llegar a una respuesta clínica, con concentraciones plasmáticas valle de entre 85 y 125 mcg/ml. La dosis máxima recomendada es de 60 mg/kg/día. No hay evidencias disponibles de estudios controlados que orienten al médico en el manejo a más largo plazo de un paciente que mejore durante el tratamiento con Divalproato de sodio ER de un episodio maníaco agudo. Si bien existe consenso de que el tratamiento farmacológico más allá de una respuesta aguda resulta deseable, tanto para el mantenimiento de la dosis inicial como para la prevención de nuevos episodios maníacos, no hay datos que avalen los beneficios de Divalproato de sodio ER en tales tratamientos durante períodos más largos (es decir, más allá de 3 semanas). Epilepsia: DIVALPREX® ER Como la dosificación de Divalproato de sodio se titula en forma ascendente, las concentraciones de Fenobarbital, Carbamazepina, Fenitoína, Clonazepam, Diazepam, Etosuximida, Lamotrigina y/o Tolbutamida pueden verse afectadas (ver Precauciones, Interacciones medicamentosas). Crisis Parciales Complejas (CPC): Para adultos y niños de 10 o más años de edad. Monoterapia (terapia inicial): El Divalproato de sodio no ha sido sistemáticamente estudiado como terapia inicial. El tratamiento debe iniciarse con dosis de 10 a 15 mg/kg/día, con incrementos de 5 a 10 mg/kg/semana hasta alcanzar una respuesta clínica óptima; generalmente dicha respuesta se alcanza con dosis diarias inferiores a 60 mg/kg/día. Si no se logra una respuesta óptima, deberán medirse los niveles plasmáticos para determinar si están o no dentro de los niveles aceptados en general (50 a 100 mcg/ml). No pueden hacerse recomendaciones con respecto a la seguridad de Valproato en dosis superiores a 60 mg/kg/día. La probabilidad de trombocitopenia aumenta significativamente con concentraciones plasmáticas totales mínimas de Valproato superiores a 110 mcg/ml en mujeres y 135 mcg/ml en hombres. El beneficio de un mejor control de las crisis con mayores dosis debe ser evaluado en contraposición a la posibilidad de una mayor incidencia de reacciones adversas. Cambio a monoterapia: Los pacientes deberán iniciar el tratamiento con dosis de 10-15 mg/kg/día. La dosis deberá ser aumentada de 5 a 10 mg/kg/semana para alcanzar una respuesta clínica óptima. Habitualmente ésta se alcanza con dosis diarias inferiores a 60 mg/kg/día. Si dicha respuesta no se alcanza, deberán medirse los niveles plasmáticos para determinar si están dentro del rango terapéutico usualmente aceptado (50-100 mcg/ml). No pueden hacerse recomendaciones con respecto a la seguridad del Valproato para su uso en dosis mayores a 60 mg/kg/día. La dosificación de las drogas antiepilépticas concomitantes (AED) puede reducirse habitualmente en aproximadamente un 25% cada dos semanas. Esta reducción puede iniciarse al comienzo del tratamiento con Divalproato sódico o demorarse por una a dos semanas si se sospecha la ocurrencia de crisis como consecuencia de la misma. La velocidad y duración de la suspensión de la droga antiepiléptica concomitante puede ser altamente variable y los pacientes serán rigurosamente monitoreados durante este período debido al posible aumento de frecuencia de las crisis. Terapia adjunta: El Divalproato de sodio puede agregarse al régimen en dosis de 10 a 15 mg/ kg/día. La dosificación puede aumentarse de 5 a 10 mg/kg/semana hasta alcanzar una respuesta clínica óptima. Habitualmente dicha respuesta se logra con dosis diarias inferiores a 60 mg/kg/día. Si el resultado no se alcanzara, deberán medirse los niveles plasmáticos para determinar si están o no dentro del rango usualmente aceptado (50 a 100 mcg/ml). No puede hacerse ninguna recomendación acerca de la seguridad del Valproato en dosis mayores a 60 mg/kg/día. En un estudio de tratamiento adjunto para CPC, en el cual los pacientes recibían Carbamazepina o Fenitoína agregados al Valproato, no fue necesario realizar ajustes de la dosis de Carbamazepina o Fenitoína. Sin embargo, dado que el valproato puede interactuar con estas u otras drogas antiepilépticas administradas concomitantemente, así como con otras drogas (ver Precauciones, Interacciones medicamentosas), se recomienda determinar periódicamente las concentraciones plasmáticas de las drogas antiepilépticas concomitantes durante la primera fase del tratamiento (ver Precauciones, Interacciones medicamentosas). Crisis de ausencia simple y compleja: La dosis inicial recomendada es de 15 mg/kg/día, aumentando a intervalos de una semana entre 5 y 10 mg/kg/día hasta que puedan controlarse las crisis o los efectos adversos impidan posteriores aumentos. La dosis máxima recomendada es de 60 mg/kg/día. No se ha establecido una correlación válida entre la dosis diaria, las concentraciones séricas y el efecto terapéutico. Sin embargo, se considera que la concentración sérica terapéutica de Valproato para la mayoría de los pacientes con crisis de ausencia oscila entre 50 y 100 mcg/ml. Algunos pacientes pueden ser controlados con concentraciones séricas menores o mayores (ver Acción farmacológica - Farmacodinamia). Como la dosificación de Divalproato de sodio es titulada en forma ascendente, las concentraciones sanguíneas de Fenobarbital y/o Fenitoína pueden verse afectadas (ver Precauciones, Interacciones). Las medicaciones antiepilépticas no deberán suspenderse en forma abrupta en pacientes que reciben la droga para prevenir crisis más graves, debido a la posibilidad de precipitar un estado epiléptico con hipoxia y riesgo de muerte consiguientes (ver Advertencias). Profilaxis de la migraña: La dosis inicial recomendada es de 500 mg una vez por día durante 1 semana, aumentándola luego a 1 g una vez al día. Si bien sólo se han evaluado dosis de 1 g de Divalproato de sodio ER una vez al día en pacientes con migraña, el rango posológico efectivo de DIVALPREX® ER en dichos pacientes es de 500 mg a 1000 mg por día. Al igual que con otros productos a base de Valproato, las dosis de DIVALPREX® ER deberán ser individualizadas y podrán requerirse ajustes posológicos. Conversión de Divalproato de sodio a Divalproato de sodio de liberación extendida: En pacientes adultos y niños de 10 años o más con epilepsia, que recibían previamente Divalproato de sodio, el Divalproato de sodio de liberación extendida deberá ser administrado una vez por día utilizando 8 a 20% más que la dosis diaria total de Divalproato de sodio (ver Tabla de conversión de dosis). Para los pacientes a quienes la dosis diaria total de Divalproato de sodio no pueda ser directamente convertida al Divalproato de sodio de liberación extendida, se deberá considerar la opinión del médico para aumentar la dosis diaria total de Divalproato de sodio del paciente a la dosis mayor próxima antes de convertir a la dosis diaria total. No hay datos suficientes para permitir una recomendación de un factor de conversión para pacientes con dosis de Divalproato de sodio mayor a 3125 mg/día.
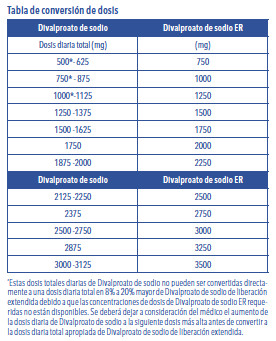
Las concentraciones plasmáticas Cmín de Divalproato de sodio de liberación extendida son equivalentes al Divalproato de sodio pero pueden variar con los pacientes luego de la conversión. Si no se lograra una respuesta clínica satisfactoria se deberán medir niveles plasmáticos para determinar si estos se encuentran o no dentro del rango terapéutico usualmente aceptado (50 a 100 mcg/ml). Recomendaciones posológicas generales: Posología en pacientes anciano: Debido a una disminución en el clearance de la fracción libre de Valproato y a una posible mayor susceptibilidad a la somnolencia, las dosis iniciales en pacientes ancianos deberán reducirse. La dosificación deberá ir aumentándose en forma más paulatina, con monitoreo periódico de la ingesta de alimentos y de agua, deshidratación, somnolencia y otros episodios adversos. Deberá considerarse la reducción de la dosis o la suspensión de Valproato en pacientes con ingesta deficiente de líquidos o alimentos y en pacientes con excesiva somnolencia. La dosis terapéutica definitiva deberá alcanzarse en base a la respuesta clínica y tolerabilidad del paciente (ver Advertencias). Reacciones adversas relacionadas a la dosis: La incidencia de efectos adversos (particularmente elevación de las enzimas hepáticas y trombocitopenia) puede estar relacionada con la dosis. La probabilidad de trombocitopenia aumenta significativamente con concentraciones totales de Valproato ≥110 mcg/ml en mujeres ó ≥135 mcg/ ml en hombres (ver Precauciones). Deberá evaluarse el beneficio de un mayor efecto terapéutico con dosis más altas frente a la posibilidad de una mayor incidencia de las reacciones adversas. Irritación gastrointestinal: Los pacientes que sufren de irritación gastrointestinal podrán beneficiarse con la administración de la medicación con las comidas o con una dosis inicial más baja. Cumplimiento: El paciente deberá tomar Divalproato de sodio ER todos los días según la indicación del médico. Si se olvidara de tomar un comprimido, deberá hacerlo tan pronto como le sea posible, salvo que sea casi la hora de la próxima dosis. Si se salteara una dosis, no deberá duplicar la siguiente. Dosificación en pacientes tomando Rufinamida: Pacientes estabilizados con rufinamida antes de recibir la prescripción de valproato deberían comenzar el tratamiento con valproato a dosis bajas y titular a una dosis clínicamente efectiva (ver Precauciones, Interacciones medicamentosas). Niñas y mujeres con capacidad de gestación: Valproato se debe iniciar y supervisar por un especialista con experiencia en el manejo de la epilepsia, trastorno bipolar o migraña. Valproato no se debe utilizar en niñas, ni en mujeres con capacidad de gestación, a menos que otros tratamientos no sean efectivos o tolerados. Valproato se prescribe y dispensa de acuerdo al Plan de Prevención de Embarazo (ver recuadro en Advertencias). Valproato se debe prescribir preferiblemente como monoterapia y a la menor dosis efectiva, si fuera posible como formulaciones de liberación prolongada. La dosis diaria debe ser dividida en al menos dos dosis individuales.
Contraindicaciones.
DIVALPREX® ER no deberá administrarse en las siguientes situaciones: Tratamiento de la epilepsia: en el embarazo, a menos que no exista otro tratamiento alternativo adecuado. En mujeres con capacidad de gestación, a menos que se cumplan las condiciones del Plan de Prevención de Embarazo. Tratamiento del trastorno bipolar y tratamiento profiláctico de la migraña: en el embarazo. En mujeres con capacidad de gestación, a menos que se cumplan las condiciones del Plan de Prevención de Embarazo. Pacientes con enfermedad hepática o disfunción hepática significativa. Pacientes con conocidos trastornos del ciclo de la urea (ver Advertencias). Pacientes que tienen enfermedades mitocondriales causadas por mutaciones en el ADN de la polimerasa gamma (POLG; por ejemplo, Síndrome de Alpers-Huttenlocher) y en niños menores de dos años de edad en los que se sospecha que tienen un trastorno relacionado con la POLG (ver Advertencias - Hepatotoxicidad). Pacientes con hipersensibilidad a la droga.
Reacciones adversas.
Manía: Los eventos adversos informados en estudios en manía aguda, en los que la incidencia en el grupo tratado con Divalproato de sodio ER fue superior al 5% y mayor a la del placebo, fueron: Somnolencia, dispepsia, náuseas, vómitos, diarrea, mareos, dolor, dolor abdominal, astenia, faringitis, lesiones accidentales. Se presentó cefalea con una incidencia Igual o mayor en el grupo placebo que en el de Divalproato de sodio ER. Los siguientes eventos adversos adicionales fueron informados en más del 1% pero en menos del 5% de los pacientes tratados con Divalproato de sodio ER en estudios clínicos controlados: Generales: dolor de espalda, escalofríos, escalofríos y fiebre, nivel del fármaco incrementado, síndrome gripal, infección, infección fúngica, rigidez de cuello. Sistema cardiovascular: arritmia, hipertensión, hipotensión, hipotensión postural. Sistema digestivo: constipación, boca seca, disfagia, incontinencia fecal, flatulencia, gastroenteritis, glositis, hemorragia de las encías, ulceración bucal. Sistema hemolinfático: anemia, tiempo de sangrado incrementado, equimosis, leucopenia. Trastornos metabólicos y nutricionales: hipoproteinemia, edema periférico. Sistema musculoesquelético: artrosis, mialgia. Sistema nervioso: marcha anormal, agitación, reacción catatónica, disartria, alucinaciones, hipertonía, hipocinesia, psicosis, reflejos incrementados, trastornos del sueño, hipoquinesia, temblor. Sistema respiratorio: hipo, rinitis. Piel: lupus eritematoso discoide, Eritema nodoso, forunculosis, erupción maculopapular, prurito, erupción, seborrea, sudoración, erupción vesiculobullosa. Sentidos especiales: conjuntivitis, ojos secos, trastornos del ojo, dolor de ojo, fotofobia, perversión del gusto. Sistema urogenital: cistitis, infección urinaria, trastorno menstrual, vaginitis. Epilepsia: Crisis Parciales Complejas (CPC): Basado en un estudio controlado con placebo de terapéutica adjunta de crisis parciales complejas, el Divalproato de sodio fue generalmente bien tolerado, siendo los efectos adversos de leves a moderados en gravedad. La intolerancia fue la razón primaria de discontinuación en pacientes tratados con Divalproato de sodio (6%) comparado con un 1% de los pacientes tratados con placebo. Los efectos adversos emergentes del tratamiento registrado en ≥5% de pacientes tratados con Divalproato de sodio y para los que la incidencia fue mayor que en el grupo placebo, en estudio controlado con placebo de terapia adjunta para el tratamiento de CPC, fueron los siguientes: Generales: cefalea, astenia, fiebre. Sistema gastrointestinal: náuseas, vómitos, dolor abdominal, diarrea, anorexia, dispepsia, constipación. Sistema nervioso: somnolencia, temblores, mareos, diplopía, ambiopía/ visión borrosa, ataxia, nistagmo, labilidad emocional, pensamiento anormal, amnesia. Sistema respiratorio: síndrome gripal, infección, bronquitis, rinitis. Otros: alopecia, pérdida de peso. Dado que los pacientes también estaban recibiendo otras drogas antiepilépticas, no es posible determinar en la mayoría de los casos si los efectos adversos enumerados anteriormente pueden ser atribuidos al Divalproato de sodio solamente o a la combinación con otras drogas antiepilépticas. Los efectos adversos emergentes del tratamiento registrados en ≥5% de los pacientes en el grupo de Divalproato en altas dosis y para los cuales la incidencia fue mayor que en el grupo de dosis bajas, en un estudio controlado de monoterapia con Divalproato de sodio para CPC, fueron los siguientes: Generales: astenia. Sistema digestivo: náuseas, diarrea, vómitos, dolor abdominal, anorexia, dispepsia. Sistema hemolinfático: trombocitopenia, equimosis. Metabólico-nutricional: aumento de peso, edema periférico. Sistema nervioso: temblor, somnolencia, mareos, insomnio, nerviosismo, amnesia, nistagmus, depresión. Sistema respiratorio: infección, faringitis, disnea. Piel: alopecia. Sentidos especiales: ambliopía/visión borrosa, tinnitus. La cefalea fue el único evento adverso que ocurrió sólo en ≥5% de los pacientes del grupo de altas dosis con igual o mayor incidencia que en el grupo de bajas dosis. Dado que a los pacientes se les estaba discontinuando otra droga antiepiléptica durante la primera fase del estudio, no es posible determinar si los siguientes efectos adversos pueden ser atribuidos sólo al Divalproato de sodio o a la combinación con otras drogas antiepilépticas. Los efectos adversos adicionales siguientes fueron informados en más del 1% pero en menos del 5% de los pacientes tratados con valproato en los estudios controlados de CPC: Generales: dorsalgia, dolor de pecho, malestar. Sistema cardiovascular: taquicardia, hipertensión, palpitaciones. Sistema digestivo: aumento del apetito, flatulencia, hematemesis, eructos, pancreatitis, absceso periodontal. Sistema hemolinfático: petequias. Trastornos metabólicos/nutricionales: aumento de TGO y TGP. Sistema musculoesquelético: mialgia, espasmos, artralgia, calambres en las piernas, miastenia. Sistema nervioso: ansiedad, confusión, marcha anormal, parestesia, hipertonía, incoordinación, sueños anormales, trastorno de la personalidad. Sistema respiratorio: sinusitis, tos incrementada, neumonía, epistaxis. Piel: erupción, prurito, piel seca. Sentidos: perversión del gusto, visión anormal, sordera, otitis media. Sistema urogenital: Incontinencia urinaria, vaginitis, dismenorrea, amenorrea, frecuencia urinaria. Migraña: De acuerdo con los resultados de dos estudios clínicos controlados con placebo y respectiva extensión a largo plazo, valproato fue en general bien tolerado; la mayoría de las reacciones adversas fueron consideradas leves a moderadas. Incluyendo los datos del estudio de extensión a largo plazo, las reacciones adversas registradas en ≥1% de 248 pacientes tratados con valproato como la razón principal de suspensión del tratamiento fueron: alopecia (6%), náuseas y/o vómitos (5%), aumento de peso (2%), temblor (2%), somnolencia (1%), TGO y/o TGP elevada (1%) y depresión (1%). Abajo se describen las reacciones adversas registradas por los pacientes tratados con Divalproato de sodio ER en un estudio controlado con placebo con una incidencia mayor del 5% y mayor que en el grupo de placebo: Sistema gastrointestinal: náuseas, dispepsia, diarrea, vómitos, dolor abdominal. Sistema nervioso: somnolencia. Otros: infección. Las reacciones adversas astenia y síndrome gripal, se produjeron en más del 5% de los pacientes tratados con Divalproato de sodio ER y con mayor incidencia en los pacientes que recibieron placebo que en los que recibieron Divalproato de sodio ER Otras reacciones adversas reportadas en más del 1% pero no más del 5% de los pacientes tratados con Divalproato de sodio ER en un estudio clínico controlado con placebo para el tratamiento preventivo de la migraña y con mayor incidencia que en el grupo placebo, fueron: Generales: lesión accidental, Infección viral. Sistema digestivo: aumento del apetito, trastornos dentales. Trastornos metabólicos/nutricionales: edema, aumento de peso. Sistema nervioso: marcha anormal, mareos, hipertonía, insomnio, nerviosismo, temblor, vértigo. Sistema respiratorio: faringitis, rinitis. Piel: erupción. Sentidos: tinnitus. Las reacciones adversas reportadas en los estudios controlados con placebo con una incidencia mayor del 5% y mayor que en el grupo de placebo en los pacientes tratados con valproato, fueron: Sistema gastrointestinal: náusea, dispepsia, diarrea, vómitos, dolor abdominal, aumento del apetito. Sistema nervioso: astenia, somnolencia, mareos, temblor. Las reacciones adversas síndrome gripal y faringitis, se produjeron en más del 5% de los pacientes tratados con Divalproato de sodio y con mayor incidencia en los pacientes que recibieron placebo que en los que recibieron Divalproato de sodio. Otras reacciones adversas no incluidas más arriba y registradas en más del 1% pero no más del 5% de los pacientes tratados con valproato fueron: Generales: dolor en el pecho. Sistema cardiovascular: vasodilatación. Sistema digestivo: constipación, boca seca, flatulencia, estomatitis. Sistema hemolinfático: equimosis. Trastornos metabólicos/nutricionales: edema periférico. Sistema musculoesquelético: calambres en las piernas. Sistema nervioso: sueños anormales, confusión, parestesia, trastornos del habla, alteraciones del pensamiento. Sistema respiratorio: disnea, sinusitis. Piel: prurito. Sistema urogenital: metrorragia. Experiencia post-marketing: Se han identificado las siguientes reacciones adversas luego de la aprobación de uso de Divalproato de sodio. Debido a que estas reacciones son reportadas voluntariamente por una población de tamaño incierto, no siempre es posible realizar una estimación confiable de su frecuencia o establecer una relación causal con la exposición al fármaco. Dermatológicos: caída transitoria del cabello, cambio de color de cabello, fotosensibilidad, necrólisis epidermal tóxica, trastornos de las uñas y lecho ungueal, eritema multiforme y síndrome de Stevens-Johnson. Psiquiátricos: alteración emocional, psicosis, agresividad, hiperactividad psicomotora, hostilidad, alteración en la atención, trastornos en el aprendizaje y deterioro del comportamiento. Neurológicos: se han reportado varios casos de deterioro cognitivo agudo o subagudo y cambios en el comportamiento (apatía o irritabilidad) con seudoatrofia cerebral en imágenes asociada con el tratamiento con Valproato. Ambos, los cambios cognitivos y de comportamiento y seudoatrofia cerebral revirtieron parcialmente o totalmente luego de la discontinuación del valproato. Musculoesqueléticos: fracturas, disminución de la densidad mineral ósea, osteopenia, osteoporosis y debilidad. Hematológicos: linfocitosis relativa, macrocitosis, leucopenia, supresión de la médula ósea, pancitopenia, anemia Incluyendo la forma macrocítlca con o sin deficiencia de folatos, anemia aplástica, agranulocltosls y porfiria intermitente aguda. Endócrinos: menstruación irregular, amenorrea secundaria, hiperandrogenismo, hirsutismo, nivel de tetosterona elevado, aumento mamario, galactorrea, inflamación de la glándula paratiroidea, poliquistosis ovárica, disminución de las concentraciones de carnitina, hiponatremia, hiperglucemia y secreción inapropiada de HAD. Se han registrado casos aislados de síndrome de Fanconi, principalmente en niños. Metabolismo y nutrición: aumento de peso. Reproductivo: aspermia, azoospermia, disminución del conteo de espermatozoides, disminución de la motilidad de espermatozoides, infertilidad masculina y morfología anormal de espermatozoides. Genitourinarios: enuresis e infección del tracto urinario. Sentidos: pérdida de la audición. Otros: reacción alérgica, anafilaxia, dolor óseo, bradicardia, vasculitis cutánea, retraso en el desarrollo. Riesgo de malformaciones congénitas y trastornos del desarrollo: Valproato tiene efectos teratogénicos que pueden causar malformaciones congénitas y también puede ocasionar trastornos graves del desarrollo neurocognitivo en los hijos expuestos durante el embarazo. Notificación de Sospecha de Reacciones Adversas: Es importante notificar la sospecha de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia al siguiente al siguiente teléfono (+54-11) 4340-0866 o por la página web https://www.argentina.gob.ar/anmat/farmacovigilancia/notificanos/eventosadversos o al Departamento de Farmacovigilancia de GADOR S.A. vía email a farmacovigilancia@gador.com o telefónicamente al 0800-220-2273.
Precauciones.
Interacciones con antibióticos carbapenémicos: Los antibióticos carbapenémicos (por ej. Ertapenem, Imipenem, Meropenem) pueden reducir las concentraciones séricas del Ácido Valproico a niveles subterapéuticos y en consecuencia producir pérdida del control de las convulsiones. Se recomienda controlar frecuentemente las concentraciones séricas de Ácido Valproico después del inicio del tratamiento con Carbapenem. Deberá considerarse un tratamiento antibacteriano o anticonvulsivante alternativo si las concentraciones séricas de Ácido Valproico descienden significativamente o se deteriora el control de las convulsiones (ver Precauciones, Interacciones medicamentosas). Reacción del fármaco con eosinofilia y síntomas sistémicos (DRESS) / Reacciones de hipersensibilidad multiorgánica: Se ha reportado reacción del fármaco con eosinofilia y síntomas sistémicos (DRESS) en pacientes que toman valproato, también conocido como hipersensibilidad multiorgánica, DRESS puede ser fatal o poner en riesgo la vida. Típicamente, DRESS, aunque no exclusivamente, se presenta con fiebre, erupción, y/o linfadenopatía, en asociación con otro sistema de órganos involucrado, como ser hepatitis, nefritis, anormalidades hematológicas, miocarditis o miositis a veces similar a una infección viral aguda. Frecuentemente se presenta eosinofilia. Debido a que este trastorno es variable en su expresión, otros sistemas de órganos no mencionados aquí pueden estar implicados. Es importante mencionar que las manifestaciones tempranas de hipersensibilidad, como ser fiebre o linfadenopatía, pueden estar presentes aun cuando el erupción no es evidente. Si tales signos y síntomas están presentes, el paciente debe ser evaluado inmediatamente. Valproato debería ser discontinuado y no ser reanudado si no se puede establecer una alternativa etiológica para los signos y síntomas. Hiperamoniemia: La hiperamoniemia se ha reportado en asociación con la terapia con valproato y puede presentarse aún con tests de función hepática normales. En pacientes que desarrollen letargia inexplicable y vómitos o cambios en el estado mental, deberá considerarse la encefalopatía hiperamoniémica por lo que deberán medirse los niveles de amonio. Asimismo, deberá considerarse la hiperamoniemia en pacientes con hipotermia (ver Precauciones - Hipotermia). Si el amonio estuviera aumentado se deberá discontinuar el tratamiento con Valproato. Deberán iniciarse intervenciones apropiadas para el tratamiento de la hiperamoniemia y tales pacientes deberán someterse a una investigación para trastornos subyacentes del ciclo de la urea (ver Contraindicaciones y Advertencias - Trastornos del ciclo de la urea y Precauciones - Hiperamoniemia y encefalopatía asociadas con el uso concomitante de Topiramato). Las elevaciones asintomáticas del amonio son más frecuentes y cuando se presentan, requieren un estrecho monitoreo de los niveles plasmáticos de amonio. Si persiste la elevación, deberá considerarse la discontinuación del tratamiento con Valproato. Hiperamoniemia y encefalopatía asociadas con el uso concomitante de Topiramato: La administración concomitante de Topiramato y Ácido Valproico se ha visto asociada con hiperamoniemia con o sin encefalopatía en pacientes que habían tolerado cada una de las drogas por separado. L
os síntomas clínicos de la encefalopatía hiperamoniémica a menudo incluyen alteraciones agudas en los niveles de conciencia y/o función cognitiva con letargia o vómitos. La hipotermia también puede ser una manifestación de la hiperamoniemia (ver Precauciones - Hipotermia). En la mayoría de los casos, los síntomas desaparecen luego de la interrupción de cualquiera de las drogas. Este evento adverso no se debe a una interacción farmacocinética. Los pacientes con errores congénitos del metabolismo o actividad mitocondrial hepática reducida pueden presentar mayor riesgo de hiperamoniemia con o sin encefalopatía. Aunque no se ha estudiado, una interacción entre Topiramato y Ácido Valproico puede exacerbar defectos existentes o desenmascarar deficiencias en personas susceptibles. En pacientes que desarrollen letargia inexplicable y vómitos o cambios en el estado mental, deberá considerarse la encefalopatía hiperamoniémica por lo que deberán medirse los niveles de amonio. (Ver Contraindicaciones y Advertencias - Trastornos del ciclo de la urea y Precauciones - Hiperamoniemia). Hipotermia: Se ha reportado hipotermia, definida como el descenso no intencional de la temperatura corporal por debajo de 35°C, asociada con el tratamiento con Valproato junto con y en ausencia de hiperamoniemia. Esta reacción adversa también puede producirse en pacientes que reciben tratamiento concomitante de Topiramato con Valproato luego de iniciado el tratamiento con Topiramato o al aumentar la dosis diaria de Topiramato (ver Interacciones medicamentosas - Topiramato). Deberá considerarse la suspensión del tratamiento con Valproato en pacientes que desarrollen hipotermia, que puede manifestarse en una variedad de anormalidades clínicas tales como letargia, confusión, coma y alteraciones significativas en otros sistemas orgánicos importantes tales como el sistema cardiovascular y respiratorio. El tratamiento y la evaluación clínica deben incluir análisis de los niveles de amonio en sangre. Monitoreo de concentración plasmática de droga: Dado que Divalproato de sodio puede interactuar con agentes que actúan como inductores enzimáticos, administrados en forma concomitante, se recomienda la determinación periódica de los niveles plasmáticos de Valproato y de dichas drogas concomitantes durante el curso inicial de la terapia, según indicación clínica (ver Precauciones, Interacciones medicamentosas). Efecto en pruebas tiroideas y de cetonas: El Valproato es eliminado parcialmente en la orina como un cetometabolito, lo que puede llevar a una falsa interpretación de cetonuria. Se han registrado alteraciones en las pruebas de la función tiroidea asociadas con la administración de Valproato. Se desconoce su significado clínico. Efecto en HIV y replicación de virus CMV: Existen estudios in vitro que sugieren que el Valproato estimula la replicación de los virus HIV y CMV bajo ciertas condiciones experimentales. Se desconocen sus consecuencias clínicas, si es que existen. Además, estos hallazgos in vitro son de dudosa importancia para los pacientes que reciben tratamiento antirretroviral de máxima supresión. Sin embargo, estos datos deberán tenerse en cuenta al interpretar los resultados del control rutinario de la carga viral en pacientes con HIV que reciben Valproato, o durante el seguimiento clínico de pacientes con CMV. Medicación residual en heces: Se han notificado casos raros de residuos del medicamento en las heces, algunos de los cuales se produjeron en pacientes con trastornos gastrointestinales anatómicos (incluida Ileostomía o colostomía) o funcionales con tiempos de tránsito gastrointestinal cortos. En algunos Informes, los residuos del medicamento se produjeron en el contexto de la diarrea. Se recomienda controlar los niveles plasmáticos de Valproato en pacientes que presenten residuos del medicamento en las heces y el estado clínico de los pacientes. Si estuviera clínicamente indicado, podrá considerarse el tratamiento alternativo. Carcinogénesis, mutagénesis, daño a la fertilidad: Carcinogénesis: Se administró valproato oralmente a ratas y ratones en dosis de 80 y 170 mg/kg/día (menos que la dosis máxima recomendada en humanos sobre la base en mg/m2) por dos años. Los encuentros primarios fueron un incremento en la incidencia de fibrosarcoma subcutáneo en ratas macho que recibieron valproato a dosis altas y adenoma pulmonar benigno, relacionado a la dosis, en ratones macho que recibieron valproato. Se desconoce el significado de estos hallazgos en seres humanos. Mutagénesis: El Valproato no demostró ser mutagénico en un ensayo bacteriano in vitro (Test de Ames), no produjo efecto letal dominante en el ratón ni incremento de la frecuencia de la aberración cromosómica en un estudio citogenético in vivo en ratas. Se informó de un aumento en la frecuencia de intercambio de cromátidas hermanas en niños epilépticos tratados con Valproato, pero no se observó dicha asociación en otro estudio realizado en adultos. Existe evidencia de que dicho aumento podría estar asociado a la epilepsia, pero se desconoce su significado biológico. Fertilidad: Los estudios de toxicidad crónica de valproato en perros y ratas jóvenes y adultas demostraron reducción de la espermatogénesis y atrofia testicular a dosis de 400 mg/kg/día o mayor en ratas (aproximadamente equivalente o superior que la dosis máxima recomendada en humanos sobre la base en mg/m2) y 150 mg/kg/día o mayor en perros (aproximadamente 1,4 veces o más la dosis máxima recomendada en humanos sobre la base en mg/m2). Estudios de fertilidad en ratas no han demostrado efectos sobre la fertilidad a dosis de 350 mg/kg/día de valproato (aproximadamente igual a la dosis máxima recomendada en humanos sobre la base en mg/ m2) durante 60 días. Se desconoce el efecto del Valproato sobre el desarrollo testicular, los parámetros espermáticos y la fertilidad en seres humanos. Embarazo: Valproato está contraindicado como tratamiento para el trastorno bipolar y la migraña durante el embarazo. Valproato está contraindicado como tratamiento para la epilepsia durante el embarazo a menos que no haya una alternativa adecuada para tratar la epilepsia. Valproato está contraindicado para su uso en mujeres con capacidad de gestación a menos que se cumplan las condiciones del Plan de Prevención de Embarazo. Riesgo de malformaciones congénitas y trastornos del desarrollo: Valproato tiene efectos teratogénicos que pueden causar malformaciones congénitas y también puede ocasionar trastornos graves del desarrollo neurocognitivo en los hijos expuestos durante el embarazo. Malformaciones congénitas: Los datos derivados de dos metanálisis muestran que del 8% al 13% de los hijos de mujeres epilépticas expuestas a monoterapia con valproato durante el embarazo sufren malformaciones congénitas. Esto representa un riesgo más elevado de sufrir malformaciones graves respecto a la población general, para la cual el riesgo es de aproximadamente el 2-3%. Los datos disponibles indican que el riesgo es dosis-dependiente. El riesgo es mayor con dosis elevadas (más de 1g al día) y los datos disponibles no permiten establecer una dosis umbral por debajo de la cual no exista riesgo. Los tipos de malformaciones más frecuentes son los defectos del tubo neural, dismorfias faciales, fisuras de labio y paladar, craneosinostosis, defectos cardiacos, renales y urogenitales, defectos en las extremidades (incluida la aplasia bilateral del radio) y múltiples anomalías que afectan a varios sistemas. La suplementación con ácido fólico antes del embarazo y durante el primer trimestre puede reducir el riesgo de defectos del tubo neural, el cual puede ocurrir en cualquier embarazo, pero no previene las malformaciones congénitas asociadas al uso de valproato durante el embarazo. Trastornos del desarrollo neurocognitivo: La exposición a valproato en el útero puede conllevar efectos adversos en el desarrollo mental y físico de los niños expuestos. El riesgo es dosis-dependiente, pero los datos disponibles no permiten establecer una dosis umbral por debajo de la cual no existan riesgos. El período gestacional de mayor riesgo para estos efectos es incierto y no se puede excluir la posibilidad de riesgo en cualquier etapa del embarazo. Estudios en niños en edad preescolar con antecedentes de exposición a valproato en el útero muestran que hasta el 30-40% de los niños experimenta retrasos en las etapas iniciales de desarrollo, como por ejemplo hablar y caminar más tarde, capacidades intelectuales disminuidas, aptitudes lingüísticas deficientes (habla y comprensión) y problemas de memoria. El coeficiente intelectual (CI) medido en niños en edad escolar (6 años de edad) con antecedentes de exposición a valproato en el útero fue un promedio de 7-10 puntos menor que el de niños expuestos a otros antiepilépticos. Aunque no se puede excluir la influencia de otros factores, existe evidencia en los niños expuestos a valproato de que el riesgo de padecer deficiencias intelectuales es independiente del CI de la madre. Los datos son limitados en cuanto a otras posibles consecuencias a largo plazo. Estos niños presentan también un mayor riesgo de padecer trastornos del espectro autista (aproximadamente tres veces más) y autismo infantil (aproximadamente cinco veces más) que la población general estudiada y podrían presentar una mayor tendencia a desarrollar síntomas del trastorno por déficit de atención con hiperactividad (TDAH), aunque los datos sobre esto último son más limitados. Teratogenicidad y efectos sobre el desarrollo mental: Si una mujer planea un embarazo: Para la indicación de epilepsia, si una mujer planea quedar embarazada, un especialista con experiencia en el tratamiento de la epilepsia debe reevaluar el tratamiento con valproato y considerar otras posibles alternativas terapéuticas. Se debe hacer todo lo posible para cambiar a un tratamiento alternativo apropiado antes de la concepción y antes de interrumpir la anticoncepción. Si el cambio no es posible, la mujer debe recibir asesoramiento adicional sobre los riesgos del valproato sobre el feto para ayudar a su toma de decisiones informada con respecto a la planificación familiar. Para la(s) indicación(es) trastorno bipolar y migraña, sí una mujer planea quedar embarazada, se debe consultar a un especialista con experiencia en el tratamiento de trastorno bipolar o migraña y se debe interrumpir el tratamiento con valproato, y si fuera necesario cambiar a un tratamiento alternativo antes de la concepción y antes de interrumpir la anticoncepción. Mujeres embarazadas: El valproato como tratamiento para el trastorno bipolar y el tratamiento profiláctico de la migraña está contraindicado para su uso durante el embarazo. El valproato como tratamiento para la epilepsia está contraindicado en el embarazo, a menos que no exista otro tratamiento alternativo adecuado. Si una mujer en tratamiento con valproato queda embarazada, debe ser referida inmediatamente a un especialista para considerar otras posibles alternativas terapéuticas. Durante el embarazo, las convulsiones clónicas tónicas maternas y el estado epiléptico con hipoxia pueden conllevar un riesgo particular de muerte para la madre y el feto. Si a pesar de los riesgos conocidos del valproato en el embarazo y después de una cuidadosa consideración del tratamiento alternativo, en circunstancias excepcionales una mujer embarazada debe recibir valproato para la epilepsia, se recomienda: Utilizar la mínima dosis efectiva y dividir la dosis diaria de valproato en varias dosis menores para tomar a lo largo del día. El uso de formulaciones de liberación prolongada es preferible a otras formulaciones para evitar picos altos de concentraciones plasmáticas. Todas las pacientes con un embarazo expuesto a valproato y sus parejas deben ser referidas a un especialista con experiencia en Teratología para la evaluación y el asesoramiento con respecto al embarazo expuesto. Debe realizarse un control prenatal especializado para detectar la posible aparición de defectos del tubo neural u otras malformaciones. La administración de suplementos de folato antes del embarazo puede disminuir el riesgo de defectos del tubo neural que pueden ocurrir en cualquier embarazo en la población general. Sin embargo, la evidencia disponible no sugiere que prevenga los defectos congénitos o malformaciones debido a la exposición al valproato. Las mujeres embarazadas que reciben Valproato pueden desarrollar anormalidades en la coagulación, incluyendo trombocitopenia, hipofibrinogenemia y/o descenso de otros factores de la coagulación, que pueden resultar en complicaciones hemorrágicas en el neonato, incluyendo muerte. Los parámetros de coagulación deberán ser estrictamente controlados cuando se emplee Valproato durante el embarazo. Si los parámetros fueran anormales en la madre, éstos deberían ser monitoreados también en el neonato. Los pacientes que toman valproato pueden desarrollar insuficiencia hepática. También se reportaron casos fatales de insuficiencia hepática en niños expuestos a valproato en el útero, luego del uso de valproato por parte de la madre durante el embarazo. Se ha reportado hipoglucemia en neonatos cuyas madres han tomado valproato durante el embarazo. Para prevenir crisis mayores, Valproato no deberá discontinuarse en forma abrupta, debido a la posibilidad de precipitar un status epilepticus con hipoxia fetal y materna y amenaza de vida. Incluso las convulsiones menores pueden suponer un riesgo para el embrión o el feto en desarrollo. Sin embargo, puede considerarse la discontinuación de la droga previamente o durante el embarazo en casos individuales si la frecuencia y severidad de la crisis, no supone una amenaza seria para la paciente. Lactancia: El Valproato se excreta en la leche materna. Deberá considerarse la suspensión de la lactancia cuando se administre Divalproato sódico en mujeres en este período. Empleo en pediatría: La seguridad y eficacia de los comprimidos de Divalproato de sodio de liberación extendida para el tratamiento de crisis parciales complejas, crisis simples y complejas de ausencia y crisis de tipo múltiple que incluyen crisis de ausencia, no han sido establecidas en pacientes pediátricos de menos de 10 años de edad. La experiencia indica que los niños menores de 2 años están expuestos a un riesgo considerablemente mayor de hepatotoxicidad fatal, especialmente aquellos con patologías de particular riesgo. Cuando valproato es utilizado en este grupo de pacientes, debería ser utilizado con extrema precaución y en monoterapia. La experiencia en epilepsia ha demostrado que la incidencia de hepatotoxicidad fatal en niños mayores de 2 años decrece considerablemente a medida que aumenta la edad. Empleo en ancianos: No se ha establecido la seguridad y eficacia de Divalproato de sodio ER para la prevención de la migraña en pacientes mayores de 65 años. No participaron pacientes mayores de 65 años en los estudios clínicos prospectivos doble ciego de manía asociada con trastorno bipolar. Un estudio en pacientes geriátricos con demencia reveló somnolencia relacionada a la droga y suspensión del tratamiento debido a somnolencia (ver Advertencias - Somnolencia en ancianos). En estos pacientes deberá reducirse la dosis inicial y deberán considerarse reducciones en la dosificación o suspensión del tratamiento en pacientes con excesiva somnolencia (ver Posología y forma de administración). Empleo en enfermedad hepática La enfermedad hepática afecta la capacidad de eliminar el valproato. (Ver Contraindicaciones, Advertencias y Precauciones). Intolerancia a la lactosa: Este medicamento contiene lactosa. Pacientes con problemas raros hereditarios de intolerancia a la galactosa, deficiencia de Lapp lactasa o malabsorción de glucosa-galactosa no deben tomar este medicamento.
Advertencias.
Hepatotoxicidad: Información general sobre hepatotoxicidad: Se han producido casos fatales de insuficiencia hepática en pacientes que recibían Ácido Valproico. Por lo general, se registraron durante los primeros seis meses de tratamiento. La hepatotoxicidad severa o fatal puede estar precedida de síntomas inespecíficos, tales como malestar general, debilidad, letargia, edema facial, anorexia y vómitos. En pacientes con epilepsia, también podría suceder pérdida del control de las convulsiones. Los pacientes deberán ser estrictamente controlados para detectar la presencia de dichos síntomas. Deberán realizarse análisis de la función hepática antes de iniciar la terapia, y a intervalos regulares durante la misma, especialmente durante los primeros 6 meses de tratamiento. Sin embargo, los médicos no deberán confiar solamente en la bioquímica sérica, ya que los resultados de dichas pruebas pueden no ser anormales en todos los casos. También deberán considerarse los resultados de los exámenes físicos periódicos y la historia clínica de los pacientes. Deberá tenerse mucha precaución cuando se administre valproato en pacientes con antecedentes de enfermedad hepática. Los pacientes que reciben anticonvulsivantes múltiples, los niños, pacientes con trastornos metabólicos congénitos, con severos trastornos convulsivos acompañados de retraso mental y con enfermedad cerebral orgánica pueden constituir un grupo de particular riesgo. La experiencia indica que los niños menores de 2 años están expuestos a un riesgo considerablemente mayor de hepatotoxicidad fatal, especialmente si pertenecen a alguno de los grupos de particular riesgo antes mencionados. Cuando DIVALPREX® ER es utilizado en este grupo de pacientes, debería ser usado con extrema precaución y como único agente. Debería evaluarse el beneficio del tratamiento por sobre los riesgos. La experiencia en epilepsia ha demostrado que la incidencia de hepatotoxicidad fatal en pacientes mayores de 2 años decrece considerablemente a medida que aumenta la edad. Pacientes con enfermedad mitocondrial sospechada o conocida: DIVALPREX® ER está contraindicado en pacientes con enfermedad mitocondrial conocida causada por mutaciones POLG y niños menores a 2 años de edad sobre quienes se tiene sospecha clínica de tener un trastornos mitocondrial (ver Contraindicaciones). Han sido reportado casos de insuficiencia hepática aguda y muertes relacionadas con trastornos hepáticos inducidas por Valproato en pacientes con síndromes neurometabólicos hereditarios causados por mutaciones en el gen mitocondrial de la ADN polimerasa gamma (POLG) (por ejemplo, Síndrome de Alpers-Huttenlocher) en una tasa mayor que aquéllos sin estos síndromes, la mayoría de los casos reportados de insuficiencia hepática aguda en pacientes con estos síndromes han sido identificados en niños y adolescentes. Deben sospecharse trastornos relacionados a la POLG en pacientes con historia familiar o síntomas sugestivos de trastornos relacionados a la POLG, incluyendo pero no limitándose a encefalopatía inexplicable, epilepsia refractaria (focal, mioclónica), presentación de status epilepticus, retrasos en el desarrollo, retraso psicomotor, neuropatía axonal sensitivo motora, ataxia miopática cerebelosa, oftalmoplejía o migraña complicada con aura occipital. Las pruebas de mutación de la POLG deben ser realizadas de acuerdo con la práctica clínica habitual para la evaluación diagnóstica de dichos trastornos. Las mutaciones A467T y W748S están presentes en aproximadamente 2/3 de los pacientes con trastornos relacionados con POLG autosómicos recesivos. En pacientes de más de dos años de edad en los que se tiene sospechas de tener una enfermedad mitocondrial hereditaria, el Divalproato de sodio debe ser usado únicamente luego de que otros anticonvulsivantes fallaron. Este grupo de mayor edad debe ser monitoreado de forma cercana durante el tratamiento con Divalproato de sodio por el desarrollo de daño hepático agudo con evaluaciones clínicas regulares y pruebas de monitoreo de la función hepática. El medicamento debería ser discontinuado inmediatamente en la presencia de disfunción hepática significativa, sospechada o aparente. En algunos casos, la disfunción hepática ha avanzado a pesar de la discontinuación del medicamento. Pancreatitis: Se han informado casos de pancreatitis potencialmente mortales en niños y adultos tratados con Valproato. Algunos de los casos se describieron como pancreatitis hemorrágicas cuyos síntomas iniciales progresaron rápidamente hasta la muerte. Algunos casos se presentaron poco después de iniciado el tratamiento y otros después de varios años de terapia. En base a los casos registrados, el índice excede al esperado en la población general y hubo casos de pancreatitis con recidiva después de reiniciado el tratamiento con Valproato. En estudios clínicos llevados a cabo en 2416 pacientes se observaron dos casos de pancreatitis sin etiología alternativa, lo que representa una experiencia de 1044 pacientes/año. Los pacientes y/o tutores deberán tener en cuenta que el dolor abdominal, las náuseas, los vómitos y/o la anorexia pueden ser síntomas de pancreatitis que requieren evaluación clínica inmediata. Si se diagnostica pancreatitis, deberá suspenderse la terapéutica con DIVALPREX® ER e iniciar un tratamiento alternativo para la afección clínica subyacente, según indicación clínica. Somnolencia en ancianos: En los pacientes de edad avanzada, la dosificación deberá incrementarse más paulatinamente, con monitoreo periódico de la ingesta de líquidos y nutrientes, deshidratación, somnolencia y de otros episodios adversos. Deberá considerarse la reducción de la dosis o la suspensión de Valproato en pacientes con ingesta hídrica o alimentaria deficiente y en pacientes con somnolencia excesiva (ver Posología y forma de administración). Trastornos hematopoyéticos y sangrado: La trombocitopenia (ver Precauciones - Embarazo) puede estar relacionada a la dosis de valproato. La probabilidad de aparición de trombocitopenia incrementa significativamente a concentraciones totales de valproato de 110 mcg/mL (mujeres) o 135 mcg/mL (hombres). Deberá evaluarse el beneficio de un mayor efecto terapéutico con dosis más elevadas frente a la posibilidad de una mayor incidencia de los efectos adversos. El uso de Valproato ha sido asociado con la disminución de otros líneas celulares y mielodisplasia. Dado que se han comunicado casos de citopenias, inhibición de la fase secundaria de la agregación plaquetaria y anormalidades en los parámetros de coagulación (por ejemplo, fibrinógeno bajo, deficiencia de factores de coagulación, enfermedad adquirida de Von Willebrand), se recomienda realizar pruebas de coagulación y hemograma completo antes de iniciar el tratamiento y a intervalos regulares durante el mismo. En los pacientes tratados con DIVALPREX® ER, se recomienda monitorear parámetros de coagulación y hemograma completo antes de ser sometidos a procedimientos quirúrgicos y durante el embarazo. La presencia de hemorragias, hematomas o trastornos de la hemostasia/coagulación constituye una indicación para reducir la dosis o suspender el tratamiento. Trastornos del ciclo de la urea: Se han informado casos de encefalopatía hiperamoniémica, algunas veces fatales, luego de la iniciación del tratamiento con valproato en pacientes con trastornos del ciclo de la urea, un grupo de anomalías genéticas infrecuentes, particularmente la deficiencia de la ornitina transcarbamilasa. Antes de la iniciación del tratamiento con DIVALPREX® ER deberá considerarse la evaluación de trastornos del ciclo de la urea en: 1. Pacientes con antecedentes de encefalopatía inexplicada o coma, encefalopatía asociada con una carga proteica, encefalopatía relacionada con el embarazo o el post-parto, retraso mental inexplicado, o antecedentes de niveles plasmáticos elevados de amonio o glutamina; 2. Pacientes con vómitos cíclicos y letargia, irritabilidad episódica extrema, ataxia, nitrógeno ureico bajo o supresión proteica; 3. Pacientes con antecedentes familiares de trastornos del ciclo de la urea o antecedentes familiares de muertes infantiles inexplicadas (particularmente varones); 4. Pacientes con otros signos o síntomas de trastornos del ciclo de la urea. Los pacientes que desarrollen síntomas de encefalopatía hiperamoniémica inexplicada mientras reciben tratamiento con Valproato deberán recibir tratamiento inmediato (incluyendo discontinuación de la terapia con Valproato) y ser evaluados para descartar trastornos subyacentes del ciclo de la urea (ver Contraindicaciones). Conducta e ideas suicidas: Se ha reportado un incremento del riesgo de pensamientos o conducta suicida en pacientes tratados con antiepilépticos, incluyendo Divalproato de sodio ER, para cualquiera de las indicaciones. Este mayor riesgo de conducta o pensamientos suicidas con los antiepilépticos se observó ya en la primera semana después de iniciado el tratamiento y continuó durante el período de tratamiento evaluado. El riesgo relativo de pensamientos o conducta suicida fue más elevado en los estudios clínicos en epilepsia que en los estudios clínicos en trastornos psiquiátricos u otros trastornos, pero las diferencias en el riesgo absoluto fueron similares en las indicaciones para epilepsia y psiquiátricas. Los pacientes tratados con antiepilépticos para alguna de las indicaciones deben ser monitoreados por posible aparición o agravamiento de la depresión, conducta o pensamientos suicidas y/o cambios inusuales en el estado de ánimo o comportamiento. El médico que esté considerando recetar Divalproato de sodio u otro antiepiléptico deberá evaluar el riesgo de pensamientos o conducta suicida frente al riesgo de la enfermedad sin tratar. La epilepsia y muchos otros trastornos para los cuales se recetan antiepilépticos se encuentran de por sí asociados con morbilidad y mortalidad y mayor riesgo de pensamientos o conducta suicida. En caso de aparición de pensamientos o conducta suicida durante el tratamiento, el médico deberá considerar si la aparición de estos síntomas en un determinado paciente puede estar relacionada con la enfermedad que está siendo tratada. Se deberá informar a los pacientes, cuidadores y familiares que los antiepilépticos aumentan el riesgo de pensamientos o conducta suicida y advertirles de la necesidad de estar alertas ante la aparición o agravamiento de los signos y síntomas de depresión, cambios inusuales en el estado de ánimo o comportamiento, o la aparición de pensamientos o conducta suicida o daño hacia ellos mismos. Los comportamientos que generen preocupación deben ser informados inmediatamente al médico. Mujeres en edad fértil: Debido al riesgo para el feto de malformaciones congénitas mayores (incluyendo defectos del tubo neural), se considerará la utilización de Valproato de sodio en mujeres en edad fértil, únicamente después de haber conversado extensamente con la paciente y evaluado los riesgos versus los beneficios potenciales del tratamiento (ver Precauciones - Embarazo). Esto es especialmente importante cuando el uso del Valproato se considera para una condición que no suele asociarse a daño permanentemente o muerte (por ejemplo, migraña). Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces durante el uso de Valproato. Para prevenir crisis mayores, Valproato no deberá discontinuarse en forma abrupta, debido a la posibilidad de precipitar un status epilepticus con hipoxia fetal y materna y amenaza de vida. Hay evidencia que sugiere que la suplementación con ácido fólico previo a la concepción y durante el primer trimestre de embarazo, disminuye el riesgo de defectos congénitos del tubo neural en la población general. Se desconoce si también se reduce el riesgo en mujeres que reciben valproato y suplementación con ácido fólico. Se recomienda en pacientes que están tomando valproato, que reciban suplementación dietaria de ácido fólico previo a la concepción y durante el embarazo de manera rutinaria. Defectos de nacimiento: El valproato, cuando se administra a mujeres embarazadas, puede causar malformaciones fetales, tales como defecto del tubo neural y otras anormalidades estructurales (ej. defectos craneofacial, malformaciones cardiovasculares, hipospadias, malformaciones de los miembros). La tasa de malformaciones congénitas en niños de madres que utilizaron valproato durante en el embarazo es 4 veces más alta que los niños nacidos de madres epilépticas que utilizaron otro antiepiléptico en monoterapia. La evidencia sugiere que la suplementación con ácido fólico previo a la concepción y durante el primer trimestre de embarazo, disminuye el riesgo de defectos del tubo neural en la población general. Disminución del coeficiente intelectual (CI) seguida a la exposición uterina: El Valproato puede causar disminución de las puntuaciones del coeficiente intelectual luego de la exposición intrauterina. Los estudios epidemiológicos publicados han indicado que los niños expuestos al Valproato en el útero tienen, en las pruebas cognitivas, puntajes más bajos que los niños expuestos a cualquier otro medicamento antiepiléptico o sin fármacos antiepilépticos. El más grande de esos estudios es un estudio de cohorte, prospectivo realizado en los Estados Unidos y el Reino Unido, que encontró que los niños con exposición pre-natal al Valproato tuvieron un puntaje de CI más bajo a los 6 años que los chicos con exposición pre-natal a otro antiepiléptico en monoterapia (lamotrigina, carbamazepina y fenitoína). Valproato está contraindicado durante el embarazo para mujeres que estén siendo tratadas para la profilaxis de la migraña o Trastorno bipolar. Las mujeres con epilepsia que estén embarazadas o planeen estarlo, no deberían ser tratadas con valproato, a menos que otro tratamiento haya fallado en el control adecuado de los síntomas o sea inaceptable. Plan de Prevención de Embarazo: Valproato tiene un alto potencial teratogénico y los niños expuestos a valproato en el útero tienen un alto riesgo de malformaciones congénitas y trastornos del neurodesarrollo. DIVALPREX® ER está contraindicado en las siguientes situaciones: Tratamiento de la epilepsia: En el embarazo, a menos que no exista otro tratamiento alternativo adecuado. En mujeres con capacidad de gestación, a menos que se cumplan las condiciones del Plan de Prevención de Embarazo. Tratamiento del trastorno bipolar y tratamiento profiláctico de la migraña: En el embarazo. En mujeres con capacidad de gestación, a menos que se cumplan las condiciones del Plan de Prevención de Embarazo. Condiciones del Plan de Prevención de Embarazo: El médico prescriptor se debe asegurar que: Se deben evaluar las circunstancias individuales en cada caso, involucrar al paciente en la discusión, garantizar su compromiso, discutir las alternativas terapéuticas y asegurar el entendimiento de los riesgos y las medidas necesarias para minimizarlos. En todas las pacientes se debe valorar la posibilidad de embarazo. La paciente ha entendido y conoce los riesgos de malformaciones congénitas y trastornos del neurodesarrollo, incluyendo la magnitud de estos riesgos para los niños expuestos a valproato en el útero. La paciente entiende que necesita realizarse un test de embarazo antes de iniciar el tratamiento y durante el tratamiento, si fuera necesario. La paciente recibe asesoramiento sobre anticoncepción. La paciente es capaz de cumplir con la necesidad de utilizar un método anticonceptivo eficaz (ver detalles a continuación en Anticoncepción), sin interrupción durante todo el tratamiento con valproato. La paciente entiende la necesidad de una revisión regular del tratamiento (al menos anualmente) por un especialista con experiencia en el manejo de la epilepsia, del trastorno bipolar o de la migraña. La paciente entiende la necesidad de consultar con su médico tan pronto como esté planeando un embarazo, para asegurar una discusión a tiempo y evaluar el cambio a otras posibles alternativas de tratamiento, antes de la concepción y antes de que se interrumpa el tratamiento anticonceptivo. La paciente entiende la necesidad de consultar de forma urgente con su médico en caso de embarazo La paciente ha reconocido que entiende los riesgos y precauciones necesarias asociadas al uso de valproato. Estas condiciones también afectan a mujeres no activas sexualmente en la actualidad, a menos que el prescriptor considere que existen razones convincentes que indican que no hay riesgo de embarazo. Niñas: Los prescriptores se deben asegurar que los padres/cuidadores de las niñas entienden la necesidad de contactar con un especialista cuando la niña en tratamiento con valproato tenga la menarca. El prescriptor se debe asegurar que se les ha facilitado a los padres/cuidadores de las niñas que han tenido la menarca, información completa sobre los riesgos de malformaciones congénitas y trastornos del neurodesarrollo, incluyendo la magnitud de estos riesgos para los niños expuestos a valproato en el útero. En las pacientes que tuvieron la menarca, el especialista prescriptor debe reevaluar la terapia con valproato anualmente y considerar las posibles alternativas de tratamiento. Si valproato es el único tratamiento apropiado, se debe evaluar la necesidad de utilizar un método anticonceptivo eficaz y las demás condiciones del Plan de Prevención de Embarazo. El especialista debe hacer todos los esfuerzos posibles para cambiar a las niñas a un tratamiento alternativo antes de llegar a la edad adulta. Test de embarazo: Se debe excluir la condición de embarazo antes de empezar el tratamiento con valproato. No se debe iniciar el tratamiento con· valproato en mujeres con capacidad de gestación sin un resultado negativo en el test de embarazo (test de embarazo en plasma), confirmado por un médico, para descartar el uso involuntario de valproato durante el embarazo. Anticoncepción: Las mujeres con capacidad de gestación a las que se les prescriba valproato deben usar métodos anticonceptivos efectivos, sin interrupción, durante toda la duración del tratamiento con valproato. A estas pacientes se les debe proporcionar información completa sobre la prevención del embarazo y se les debe aconsejar sobre anticoncepción, si no están usando métodos anticonceptivos efectivos. Se debe utilizar al menos un método anticonceptivo eficaz (preferiblemente una forma independiente del usuario, como un dispositivo intrauterino o un implante) o dos formas complementarias de anticoncepción, que incluya un método de barrera. Se deben evaluar las circunstancias individuales en cada caso. Al elegir el método anticonceptivo se debe involucrar a la paciente en la discusión, para garantizar su compromiso y el cumplimiento con las medidas elegidas. Incluso si tiene amenorrea, debe seguir todos los consejos sobre anticoncepción eficaz. Revisiones anuales del tratamiento por un especialista: El especialista debe revisar al menos una vez al año si valproato es el tratamiento más apropiado para la paciente. Planificación del embarazo: Para la indicación de epilepsia, si una mujer planea quedar embarazada, un especialista con experiencia en el tratamiento de la epilepsia debe volver a evaluar el tratamiento con valproato y considerar las posibles alternativas de tratamiento. Se deben hacer todos los esfuerzos posibles para cambiar a un tratamiento alternativo apropiado antes de la concepción y antes de que se interrumpa la anticoncepción. Si el cambio no es posible, la mujer debe recibir asesoramiento adicional sobre los riesgos del valproato sobre el feto para apoyar a su toma de decisión informada con respecto a la planificación familiar. Para la(s) indicación(es) trastorno bipolar y migraña, si una mujer planea quedar embarazada, se debe consultar a un especialista con experiencia en el tratamiento de trastorno bipolar o migraña y se debe interrumpir el tratamiento con valproato y si fuera necesario cambiar a un tratamiento alternativo antes de la concepción, y antes de que se suspenda la anticoncepción. En caso de embarazo: Si una mujer en tratamiento con valproato quedara embarazada, debe ser referida inmediatamente a un especialista para volver a evaluar el tratamiento con valproato y considerar las opciones alternativas. Además, las pacientes con un embarazo expuesto a valproato y sus parejas deben ser derivadas a un especialista con experiencia en Teratología para la evaluación y el asesoramiento del embarazo expuesto. El farmacéutico se debe asegurar que: Se aconseja a las pacientes que no interrumpan el tratamiento con valproato y que se contacten inmediatamente con un especialista en caso de embarazo planificado o sospecha de embarazo.
Interacciones.
Efectos de las drogas coadministradas sobre el clearance del valproato: Las drogas que afectan el nivel de expresión de las enzimas hepáticas, particularmente aquellas que elevan los niveles de las glucuronil-transferasas (por ejemplo Ritonavir), pueden aumentar el clearance del Valproato. Por ejemplo, la Fenitoína, la Carbamazepina y el Fenobarbital (o la Primidona) pueden duplicar el clearance del Valproato. Por lo tanto, los pacientes bajo monoterapia generalmente presentan vidas medias más prolongadas y concentraciones más elevadas que los pacientes bajo politerapia con drogas antiepilépticas. En cambio, los inhibidores de las isozimas del citocromo P450 (por ejemplo, los antidepresivos) ejercen poco efecto sobre el clearance del Valproato debido a que la oxidación mediada por los microsomas del citocromo P450 es una vía metabólica secundaria de relativamente poca importancia en comparación con la glucuronización y la Beta-oxidación. Debido a estas variaciones en el clearance del Valproato, siempre que se agreguen o se suspendan agentes inductores enzimáticos, deberá intensificarse el monitoreo de las concentraciones de Valproato y drogas concomitantes. La siguiente lista proporciona información respecto del potencial de influencia de varias medicaciones comúnmente recetadas sobre la farmacocinética del Valproato. Esta lista no está completa y no podría estarlo nunca ya que continuamente se está informando de nuevas interacciones. Drogas con las que se ha observado una interacción potencialmente significativa: Aspirina: Un estudio que comprendió la coadministración de Aspirina en dosis antipiréticas (11 a 16 mg/kg) y Valproato en niños (n=6) reveló una menor unión a las proteínas y una inhibición del metabolismo del Valproato. La fracción libre de Valproato se cuadruplicó en presencia de Aspirina en comparación con el Valproato solo. La vía de la Beta-oxidación que comprende el 2-E-Ácido Valproico, 3-OH-Acido Valproico y 3-ceto Ácido Valproico disminuyó del 25% de los metabolitos totales excretados con Valproato solo a 8,3% en presencia de la Aspirina. No se ha determinado si la interacción observada en este estudio se aplica también a los adultos, pero se recomienda precaución al coadministrar Valproato y Aspirina. Felbamato: Un estudio que comprendió la coadministración de 1200 mg/día de Felbamato y Valproato a pacientes con epilepsia (n=10) reveló un aumento del 35% (de 86 a 115 mcg/ml) en la concentración máxima media del Valproato en comparación con el Valproato solo. El incremento de la dosis de Felbamato a 2400 mg/día aumentó la concentración máxima media del Valproato a 133 mcg/ml (aumento adicional del 16%). Podrá ser necesario disminuir la dosis del Valproato cuando se inicie la administración de Felbamato. Antibióticos carbapenémicos: Se ha informado de una reducción clínicamente significativa de la concentración sérica del Ácido Valproico en pacientes tratados con antibióticos carbapenémicos (por ej. Ertapenem, Imipenem, Meropenem) que puede producir pérdida del control de las crisis. No se ha dilucidado el mecanismo de esta interacción. Se recomienda controlar frecuentemente las concentraciones séricas de Ácido Valproico después del inicio del tratamiento con Carbapenem. Deberá considerarse un tratamiento antibacteriano o anticonvulsivante alternativo si las concentraciones séricas de Ácido Valp