DEPAKENE
ABBOTT EPD
Anticonvulsivante.
Composición.
Depakene cápsulas - Lista N° 5681 - Cada cápsula contiene: Ácido Valproico 250 mg; aceite de maíz, 205,00 mg; glicerina, 72,00 mg; metilparabeno, 0,583 mg; Amarillo ocaso FD & C N° 6; 0,429 mg; dióxido de titanio, 3,282 mg; gelatina, 157,00 mg; propilparabeno, 0,146 mg; etilvainillina, 0,365 mg. Depakene jarabe - Lista N° 5682 - Cada 100 ml contiene: Ácido Valproico 5 g; metilparabeno, 0,100 g; propilparabeno, 0,020 g; azúcar, 60,000 g; vainillina, 0,010 g; amaranto, 0,004 g; sabor artificial a cerezas, 0,030 g; glicerina, 15,000 g; sorbitol, 15,000 g; agua destilada c.s.p.
Farmacología.
Mecanismo de Acción y Farmacodinamia: El Ácido Valproico se disocia en ión Valproato en el tracto intestinal. El mecanismo por el cual el Ácido Valproico ejerce sus efectos antiepilépticos no ha sido establecido. Se ha sugerido que su actividad está relacionada a niveles cerebrales aumentados de Ácido gama aminobutírico (GABA). Farmacocinética: Absorción/Biodisponibilidad: Dosis orales equivalentes de productos conteniendo Divalproato de sodio (Valcote) y cápsulas conteniendo Ácido Valproico (Depakene) administran sistémicamente cantidades equivalentes de ión Valproato. Si bien el índice de absorción del ión Valproato puede variar según la formulación administrada (líquida, sólida o Sprinkle), las condiciones de uso (por ejemplo en ayunas o posprandial) y el método de administración (por ejemplo sea que el contenido de la cápsula se espolvoree sobre la comida o se ingiera intacta), estas diferencias serán de escasa importancia clínica bajo las condiciones de estado de equilibrio alcanzadas con el uso crónico para el tratamiento de la epilepsia. Sin embargo, al iniciar el tratamiento, es posible que existan diferencias importantes en la Tmáx y Cmáx entre los diferentes fármacos que contienen Valproato. Por ejemplo, en estudios de dosis única, el efecto de la alimentación influyó más sobre el índice de absorción del comprimido (aumento en la Tmáx de 4 a 8 horas) que sobre el de las cápsulas Sprinkle (aumento en la Tmáx de 3.3 a 4.8 horas). Mientras que el índice de absorción desde el tracto gastrointestinal y la fluctuación en las concentraciones plasmáticas de Valproato varían según el régimen posológico y la formulación, es poco probable que se vea afectada la eficacia del Valproato como anticonvulsivante cuando se lo emplea en forma crónica. La experiencia con regímenes posológicos de 1 a 4 tomas diarias y los estudios en modelos de epilepsia en primates en los que se utilizó un ritmo de infusión constante, indican que la biodisponibilidad sistémica diaria total (grado de absorción) es el principal determinante del control de las convulsiones, y que las diferencias en las relaciones de las concentraciones plasmáticas máximas a mínimas entre las formulaciones de Valproato no son importantes desde el punto de vista clínico. La coadministración de productos orales conteniendo Valproato con las comidas y el reemplazo entre las distintas formulaciones de Divalproato de sodio y Ácido Valproico no deberán provocar problemas clínicos en el manejo de los pacientes epilépticos (Ver Dosificación). Sin embargo, cualquier variación en la posología o el agregado o interrupción de medicaciones concomitantes normalmente deberán ser acompañados por un estrecho monitoreo del estado clínico y de las concentraciones plasmáticas de Valproato. Distribución: Unión a las proteínas: La unión del Valproato a las proteínas plasmáticas depende de la concentración, y la fracción libre aumenta desde alrededor del 10% a 40 mcg/ml hasta el 18.5% a 130 mcg/ml. La unión del Valproato a las proteínas se ve reducida en los ancianos, en pacientes con hepatopatías crónicas, pacientes con insuficiencia renal y en presencia de otras drogas (por ejemplo Aspirina). Por el contrario, el Valproato puede desplazar a ciertas drogas que se unen a las proteínas (por ejemplo Fenitoína, Carbamazepina, Warfarina y Tolbutamida) (Ver Interacciones). Distribución en el SNC: Las concentraciones de Valproato en el líquido cefalorraquídeo se aproximan a las concentraciones libres en el plasma (alrededor del 10% de la concentración total). Metabolismo: El Valproato es metabolizado casi en su totalidad por el hígado. En los pacientes adultos que reciben monoterapia, el 30-50% de una dosis administrada aparece en la orina como conjugado glucurónico. La otra vía metabólica principal es la beta oxidación en las mitocondrias, lo que generalmente representa más del 40% de la dosis. Normalmente, menos del 15-20% de la dosis es eliminada por otros mecanismos oxidativos. Menos del 3% de una dosis administrada se excreta intacta en la orina. La relación entre la dosis y la concentración total de Valproato es no lineal, la concentración no aumenta en forma proporcional a la dosis, sino que aumenta en menor grado debido a la saturación de los sitios de unión a las proteínas plasmáticas. La cinética de la droga no unida es lineal. Eliminación: El clearance plasmático medio y volumen de distribución del Valproato total son de 0.56 l/h/1.73 m2 y de 11 litros/1.73 m2, respectivamente. El clearance plasmático medio y el volumen de distribución del Valproato libre son de 4.6 l/h/1.73 m2 y de 92 l/1.73 m2. La vida media terminal promedio del Valproato como monoterapia osciló entre 9 y 16 horas después de la administración oral de 250 mg a 1 g. Los estimados citados se aplican principalmente a pacientes que no reciben drogas que afectan los sistemas enzimáticos del metabolismo hepático. Por ejemplo, los pacientes que reciben drogas antiepilépticas enzimo-inductoras (Carbamazepina, Fenitoína y Fenobarbital) depurarán al Valproato más rápidamente. Debido a estos cambios en el clearance del Valproato, se deberá intensificar el monitoreo de las concentraciones antiepilépticas cuando se agregan o retiran fármacos antiepilépticos concomitantes. Poblaciones Especiales: Neonatos: Dentro de los primeros dos meses de vida, los niños presentan una capacidad marcadamente disminuida para eliminar el Valproato en comparación con los niños mayores y los adultos. Esto se debe al menor clearance (quizás por el desarrollo demorado del sistema de la glucuroniltransferasa y otros sistemas enzimáticos comprometidos en la eliminación del Valproato) y al mayor volumen de distribución (en parte por la unión disminuida a las proteínas plasmáticas). Por ejemplo, en un estudio, la vida media en los niños de menos de 10 días de vida osciló entre 10 y 67 horas en comparación con un rango de 7 a 13 horas en niños mayores de 2 meses. Niños: Los pacientes pediátricos (por ejemplo entre 3 meses y 10 años) poseen un 50% más de clearance, expresado por peso (es decir, ml/min/kg), que los adultos. Los niños mayores de 10 años presentan parámetros farmacocinéticos similares a los de los adultos. Ancianos: La capacidad de los pacientes añosos (rango etario: 68 a 89 años) para eliminar al Valproato ha demostrado ser reducida en comparación con la de los adultos jóvenes (entre 22 y 26 años). El clearance intrínseco está reducido en un 39%; la fracción libre de Valproato está aumentada en un 44%. Por consiguiente, se deberá reducir la dosificación inicial en los ancianos (Dosificación). Sexo: No existen diferencias en el clearance de la fracción libre, ajustado según el área de superficie corporal, entre hombres y mujeres (4.8 ± 0,7 y 4,7 ± 0,07 l/h por 1.73 m2, respectivamente). Raza: Los efectos de la raza sobre la cinética del Valproato no han sido estudiados. Hepatopatía (Ver Contraindicaciones y Advertencias): La enfermedad hepática altera la capacidad para eliminar al Valproato. También está asociada con menores concentraciones de albúmina y mayores fracciones libres (aumento de 2 a 2.6 veces) de Valproato. Por consiguiente, el monitoreo de las concentraciones totales puede ser engañoso ya que las concentraciones libres pueden ser muy elevadas en pacientes con hepatopatía, mientras que las concentraciones totales pueden parecer normales. Nefropatías: Se ha informado de una ligera reducción (27%) en el clearance de Valproato libre en pacientes con insuficiencia renal (clearance de creatinina < 10 ml/min); sin embargo, la hemodiálisis generalmente reduce las concentraciones de Valproato en alrededor del 20%. Por lo tanto, no será necesario ajustar la dosis en pacientes con insuficiencia renal. En estos pacientes, la unión a las proteínas se ve considerablemente reducida, por lo que el monitoreo de las concentraciones totales puede llevar a conclusiones erróneas. Niveles Plasmáticos y Efecto Clínico: La relación entre concentración plasmática y respuesta clínica no está bien documentada. Un factor contribuyente es la unión no lineal y concentración-dependiente del Valproato a las proteínas, lo que afecta al clearance de la droga. Por lo tanto, el monitoreo de la concentración sérica total de Valproato no constituye un índice confiable de los tipos bioactivos de Valproato. Por ejemplo, debido a que la unión del Valproato a las proteínas plasmáticas depende de la concentración, la fracción libre aumenta desde aproximadamente 10% a 40 mcg/ml hasta 18.5% a 130 mcg/ml. En los ancianos, en los pacientes hiperlipidémicos y en aquellos con enfermedad hepática y renal, las fracciones libres son más elevadas de lo previsto. Epilepsia: Comúnmente, se considera que el rango terapéutico en la epilepsia es de 50 a 100 mcg/ml de Valproato total, a pesar de que algunos pacientes pueden ser controlados con concentraciones plasmáticas menores o mayores.
Indicaciones.
El Ácido Valproico está indicado como tratamiento único y combinado en pacientes con crisis parciales complejas que ocurran aisladas o asociadas con otro tipo de crisis. El Ácido Valproico está indicado como tratamiento único o combinado en el tratamiento de las crisis de ausencia simples y complejas, y como adyuvante en pacientes con crisis múltiples que incluyen crisis de ausencia. La ausencia simple se define como una muy breve obnubilación del sensorio o pérdida del conocimiento acompañada por ciertas descargas epilépticas generalizadas sin otros signos clínicos detectables. Se emplea el término de ausencia compleja cuando también se encuentran presentes otros signos. Ver Advertencias para consideraciones referentes a casos fatales de disfunción hepática.
Dosificación.
General: Las cápsulas de Depakene se administran por vía oral y deben ingerirse enteras, sin masticar para evitar la irritación de la boca y la garganta. El Ácido Valproico ha sido indicado como monoterapia y como terapia adyuvante en las convulsiones parciales complejas (CPC) en adultos y niños mayores de 10 años, y en crisis de ausencia simples y complejas en adultos y adolescentes. Como la dosificación del Ácido Valproico es titulada en forma creciente, las concentraciones de Fenobarbital, Carbamazepina y/o Fenitoína pueden verse afectadas (Ver Interacciones). Crisis parciales complejas (CPC): para adultos y niños de 10 o más años. Monoterapia (terapia inicial): El Ácido Valproico no ha sido estudiado sistemáticamente como terapia inicial. Los pacientes iniciarán el tratamiento en dosis de 10 a 15 mg/kg/día. La dosificación será incrementada de 5 a 10 mg/kg por semana hasta alcanzar la respuesta clínica óptima. Comúnmente esta respuesta es alcanzada a dosis diarias por debajo de 60 mg/kg/día. Si una respuesta clínica satisfactoria no fuese alcanzada, deberán medirse los niveles plasmáticos para determinar si éstos están dentro del rango terapéutico usualmente aceptado (50 a 100 mcg/ml). No se pueden realizar recomendaciones referentes a la seguridad del uso de Valproato a dosis por encima de 60 mg/kg/día. La probabilidad de trombocitopenia aumenta significativamente a concentraciones totales mínimas de Valproato por encima de 110 mcg/ml en mujeres y 135 mcg/ml en hombres. El beneficio de un mejor control de las crisis con mayores dosis deberá ser evaluado contra la posibilidad de una mayor incidencia de reacciones adversas. Cambio a monoterapia: Los pacientes deberán iniciar el tratamiento con dosis de 10-15 mg/kg/día. La dosis deberá ser aumentada de 5 a 10 mg/kg/semana para alcanzar la respuesta clínica óptima. Habitualmente esta respuesta se alcanza con dosis diarias por debajo de 60 mg/kg/día. Si la respuesta no fuese alcanzada deberán medirse los niveles plasmáticos para determinar si están dentro del rango terapéutico usualmente aceptado (50-100 mcg/ml). No se pueden realizar recomendaciones respecto a la seguridad del uso de Valproato a dosis por encima de los 60 mg/kg/día. La dosificación de las drogas antiepilépticas concomitantes pueden ser reducidas habitualmente en aproximadamente el 25% cada 2 semanas. Esta reducción puede iniciarse junto con el comienzo del tratamiento con Ácido Valproico, o postergarse por 1 ó 2 semanas si existiera algún temor a la aparición de convulsiones con esta reducción. La velocidad y duración de la suspensión de las drogas antiepilépticas concomitantes puede ser muy variable, y los pacientes deberán ser monitoreados durante este período debido a la frecuencia aumentada de convulsiones. Tratamiento adyuvante: El Divalproato de sodio puede ser agregado al régimen del paciente a una dosis de 10 a 15 mg/kg/día. La dosificación puede ser aumentada de 5 a 10 mg/kg/semana hasta alcanzar una respuesta clínica óptima. Habitualmente esta respuesta es alcanzada a dosis diarias por debajo de 60 mg/kg/día; si la respuesta no fuese alcanzada deberán medirse los niveles plasmáticos para determinar si están dentro del rango terapéutico aceptado (50-100 mcg/ml). No se pueden realizar recomendaciones con respecto a la seguridad de Divalproato con dosis por encima de los 60 mg/kg/día. Si la dosis diaria total excede los 250 mg deberá administrarse en dosis divididas. En un estudio de tratamiento adyuvante para CPC en que los pacientes recibían Carbamazepina o Fenitoína agregadas al Divalproato de sodio, no fue necesario realizar ajustes de las dosis de Carbamazepina o Fenitoína. Sin embargo, dado que el Valproato puede interactuar con éstas u otras drogas antiepilépticas en forma concomitante, así como con otras drogas (Ver Interacciones), se recomienda realizar determinaciones periódicas de las concentraciones plasmáticas de las drogas antiepilépticas concomitantes en el comienzo del tratamiento (Ver Interacciones). Crisis de ausencia simples y complejas: La dosis inicial recomendada es de 15 mg/kg/día aumentando a intervalos de una semana de 5 a 10 mg/kg/día hasta que las crisis sean controladas o los efectos colaterales descarten posteriores aumentos. La dosificación máxima recomendada es 60 mg/kg/día. Si la dosis diaria total excediera los 250 mg se deberá administrar en dosis divididas. No se ha establecido una buena correlación entre dosis diaria, concentración sérica y efecto terapéutico. Sin embargo, las concentraciones séricas terapéuticas de Valproato para la mayoría de los pacientes epilépticos oscilarán entre 50 y 100 mcg/ml. Algunos pacientes pueden ser controlados con concentraciones séricas menores o mayores que las mencionadas (Ver Farmacología). Debido a que la dosificación del Ácido Valproico se titula en forma creciente, las concentraciones sanguíneas de Fenobarbital y/o Fenitoína pueden verse afectadas (Ver Precauciones). Las medicaciones antiepilépticas no deberán suspenderse en forma abrupta en pacientes que reciben la droga para prevenir crisis mayores debido a la fuerte posibilidad de precipitar un estado de mal epiléptico con la consiguiente hipoxia y riesgo de muerte (Ver Advertencias).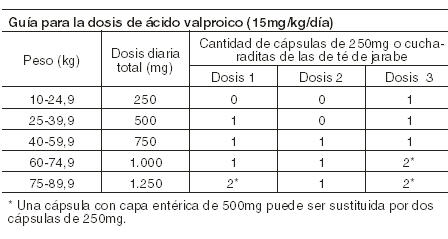
Recomendaciones Posológicas Generales: Posología en pacientes geriátricos: Debido a una disminución en el clearance de la fracción libre de Valproato y a una posible mayor susceptibilidad a la somnolencia en los ancianos, deberá reducirse la dosis inicial en estos pacientes. La dosificación deberá ir aumentándose más paulatinamente, con monitoreo periódico de la ingesta nutricional e hídrica, deshidratación, somnolencia y otros episodios adversos. Deberá considerarse la reducción de la dosis o la suspensión de Valproato en pacientes con ingesta deficiente de líquidos o alimentos y en pacientes con excesiva somnolencia. La dosis terapéutica definitiva deberá alcanzarse en base a la respuesta clínica y tolerancia del paciente (Ver Advertencias). Episodios adversos dosis-dependientes: La incidencia de episodios adversos (particularmente elevación de las enzimas hepáticas y trombocitopenia) puede estar relacionada con la dosis. La probabilidad de trombocitopenia aumenta significativamente con concentraciones totales de Valproato ≥110 mcg/ml en mujeres o ≥135 mcg/ml en hombres (Ver Precauciones). Se deberá evaluar el beneficio del mayor efecto terapéutico con dosis más altas frente a la posibilidad de una mayor incidencia de reacciones adversas. Irritación gastrointestinal: Los pacientes que sufren de irritación GI podrán beneficiarse con la administración de la medicación con las comidas o aumentando la dosis lentamente a partir de un nivel inicial bajo.
Contraindicaciones.
Depakene no será administrado a pacientes con enfermedad o disfunción hepática significativa. Depakene está contraindicado en pacientes con conocidos trastornos del ciclo de la urea (Ver Advertencias). El divalproato de sodio está contraindicado en pacientes que tienen enfermedades mitocondriales causadas por mutaciones en el ADN de la polimerasa gamma (POLG; por ejemplo, Síndrome de Alpers - Huttenlocher) y en niños menores de dos años de edad en los que se sospecha que tienen un trastorno relacionado con la POLG. (Ver Advertencias - Hepatotoxicidad). El Valproato de Sodio está contraindicado para su uso como profilaxis de la migraña en mujeres embarazadas (Ver Precauciones, y Advertencias - Uso en el embarazo). El Acido Valproico está contraindicado en pacientes con conocida hipersensibilidad a la droga. El Acido Valproico está contraindicado en pacientes con porfiria.
Reacciones adversas.
Epilepsia - Crisis parciales complejas (CPC): Los datos siguientes fueron obtenidos usando comprimidos de Divalproato de sodio. Basado en un estudio controlado con placebo de terapéutica coadyuvante para el tratamiento de crisis parciales complejas, fue generalmente bien tolerado y la mayoría de los efectos adversos fueron de leves a moderados en severidad. La intolerancia fue la razón principal de interrupción en los pacientes tratados con Divalproato de sodio (6%) comparado con el 1% de los pacientes tratados con placebo. La Tabla 1 enumera los efectos adversos emergentes del tratamiento informados por > 5% de los pacientes tratados con Divalproato de sodio, con una incidencia mayor que en el grupo de placebo, durante el estudio arriba mencionado. Dado que los pacientes también estaban siendo tratados con otras drogas antiepilépticas, no es posible determinar, en la mayoría de los casos, si los siguientes efectos adversos se debieron al Divalproato de sodio solamente o a la combinación con otras drogas antiepilépticas.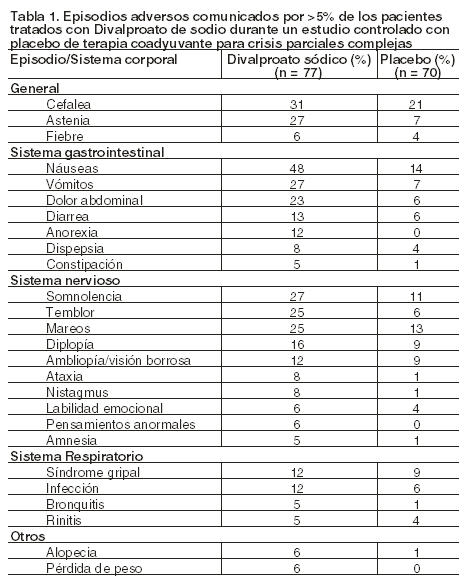
La Tabla 2 muestra los efectos adversos emergentes del tratamiento informados por > 5% de los pacientes en el grupo de Divalproato de sodio a altas dosis y, con una incidencia mayor que en el grupo de dosis bajas, en un estudio controlado de monoterapia con Divalproato de sodio para el tratamiento de crisis parciales complejas. Dado que a los pacientes se les estaba discontinuando otra droga antiepiléptica durante la primera fase del estudio, no es posible determinar si los siguientes efectos adversos son atribuibles sólo al Divalproato de sodio o a la combinación con otras drogas antiepilépticas.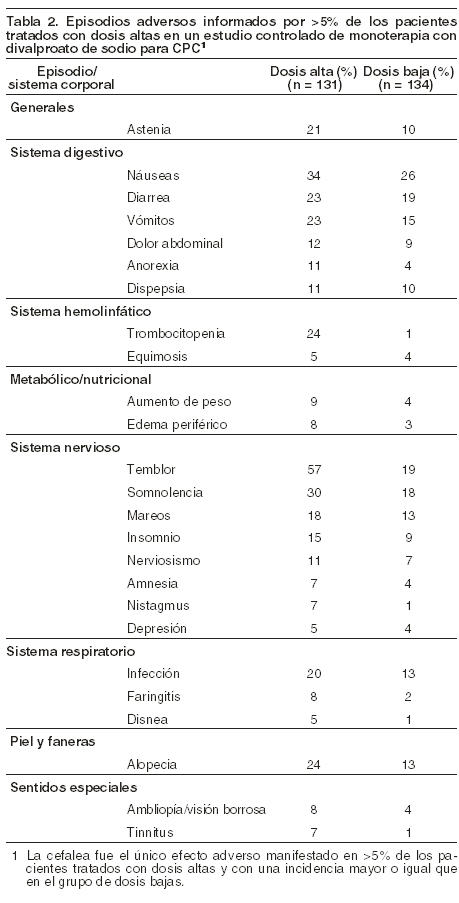
Los siguientes efectos adversos adicionales fueron informados por más del 1% pero menos del 5% de los 358 pacientes tratados con Divalproato de sodio en los estudios controlados de CPC. Generales: Dorsalgia, dolor de pecho, malestar. Sistema cardiovascular: Taquicardia, hipertensión, palpitaciones. Sistema digestivo: Aumento del apetito, flatulencia, hematemesis, eructos, pancreatitis, absceso periodontal. Sistema hemolinfático: Petequias. Trastornos metabólicos/nutricionales: Aumento de TGO y TGP. Sistema musculoesquelético: Mialgia, espasmos, artralgia, calambres en las piernas, miastenia. Sistema nervioso: Ansiedad, confusión, marcha anormal, parestesia, hipertonía, incoordinación, sueños anormales, trastornos de la personalidad. Sistema respiratorio: Sinusitis, tos incrementada, neumonía, epistaxis. Piel y faneras: Erupción, prurito, piel seca. Sentidos especiales: Perversión del gusto, visión anormal, sordera, otitis media. Sistema urogenital: Incontinencia urinaria, vaginitis, dismenorrea, amenorrea, poliaquiuria. Otras poblaciones de pacientes: Los efectos adversos informados con Valproato en ensayos en epilepsia, informes espontáneos y otras fuentes, se describen a continuación por sistema corporal. Sistema gastrointestinal: Los efectos adversos informados más frecuentemente en la iniciación del tratamiento son náuseas, vómitos e indigestión. Dichos efectos son transitorios y raramente requieren discontinuación de la terapéutica. Se ha informado diarrea, calambres abdominales, constipación y trastornos gingivales (principalmente hiperplasia gingival). También han sido informados anorexia con pérdida de peso y aumento del apetito con aumento de peso. La administración de Divalproato de sodio con capa entérica puede provocar una reducción de los efectos colaterales gastrointestinales en algunos pacientes. Sistema nervioso central: Se observaron efectos sedantes en pacientes que recibían Valproato solo, aunque con mayor frecuencia en pacientes que recibían terapéutica combinada. La sedación habitualmente desaparece con la reducción de la otra medicación antiepiléptica. Se ha observado temblor (posiblemente relacionado con la dosis), alucinaciones, ataxia, cefalea, nistagmo, diplopía, asterixis, "manchas delante de los ojos", disartria, mareos, confusión, hipoestesia, vértigo, incoordinación, deterioro de la memoria, trastornos cognitivos y parkinsonismo con el uso de Valproato. Se han registrado raros casos de coma en pacientes que recibían Valproato solo o en combinación con Fenobarbital. En raras ocasiones se manifestó encefalopatía con o sin fiebre o hiperamonemia inmediatamente después de instituir la monoterapia con Valproato, sin evidencia de disfunción hepática o niveles plasmáticos inadecuadamente altos. Aunque se ha informado de recuperación luego de la discontinuación de la droga, se registraron casos fatales en pacientes con encefalopatía hiperamoniémica, particularmente en pacientes con trastornos subyacentes del ciclo de la urea (Ver Advertencias - Trastornos del ciclo de la urea y Precauciones). Han habido reportes de postmarketing de atrofia cerebral y cerebelosa reversible e irreversible, asociada temporalmente con el uso de valproato. En algunos casos los pacientes se recuperaron con secuelas permanentes (Ver Advertencias y Precauciones). Atrofia cerebral observada en niños expuestos al valproato en el útero han conducido a varias formas de eventos neurológicos, incluyendo retrasos en el desarrollo y deterioro psicomotor (Ver Precauciones y Advertencias - Uso en el embarazo). Dermatológicos: Se ha observado un aumento transitorio en la pérdida del cabello, desórdenes del cabello (tales como textura anormal del pelo, cambios en el color, crecimiento anormal), erupción cutánea, fotosensibilidad, prurito generalizado, eritema multiforme y síndrome de Stevens-Johnson. Se han reportado casos aislados de necrólisis epidérmica tóxica, incluyendo un caso fatal en un lactante de 6 meses al que se le administraba Valproato y otras medicaciones concomitantes. También se ha informado de otro caso fatal de necrólisis epidérmica tóxica, en un paciente de 35 años de edad con SIDA que recibía muchas medicaciones concomitantes y quien tenía antecedentes de múltiples reacciones dermatológicas provocadas por fármacos. Se han informado reacciones cutáneas serias con la coadministración de Lamotrigina y Valproato (Ver Precauciones e Interacciones). Psiquiátricos: Se han comunicado casos de trastornos emocionales, depresión, psicosis, agresividad, hiperactividad psicomotora, hostilidad, agitación, alteración en la atención, comportamiento anormal, trastornos en el aprendizaje y deterioro del comportamiento. Musculoesquelético: Debilidad. Se han recibido informes de disminución de la masa ósea, potencialmente derivando en osteoporosis y osteopenia, durante el tratamiento prolongado con anticonvulsivantes, incluido Valproato. Algunos estudios han indicado que el aporte suplementario de calcio y vitamina D puede ser beneficioso para los pacientes que se encuentran en tratamiento crónico con Valproato. Hematológicos: Trombocitopenia e inhibición de la fase secundaria de la agregación plaquetaria reflejado en una alteración del tiempo de sangría, petequias, equimosis, formación de hematomas, epistaxis y hemorragia (Ver Precauciones - Generales e Interacciones). Se han observado casos de linfocitosis relativa, macrocitosis, hipofibrinogenemia, leucopenia, eosinofilia, anemia incluyendo la forma macrocítica con o sin deficiencia de folatos, supresión de la médula ósea, pancitopenia, anemia aplástica, agranulocitosis y porfiria intermitente aguda. Hepáticos: Es frecuente observar mínimas elevaciones de las transaminasas (por ejemplo TGO y TGP) y de la LDH, las que parecen ser dosis-dependientes. A veces los resultados de las pruebas de laboratorio incluyen elevaciones de la bilirrubina sérica y alteraciones en otras pruebas de la función hepática. Estos resultados pueden ser reflejo de hepatotoxicidad potencialmente severa (Ver Advertencias). Endócrinos: Menstruación irregular, amenorrea secundaria, hipertrofia mamaria, galactorrea y tumefacción de la glándula parótida, galactorrea e hiperandrogenismo (hirsutismo, virilismo, acné, alopecía con patrón masculino y/o andrógenos aumentados). Pruebas anormales de la función tiroidea incluyendo hipotiroidismo (Ver Precauciones). Se han informado casos aislados de enfermedad ovárica poliquística. No se ha establecido una relación causa/efecto. Pancreáticos: Pancreatitis aguda incluyendo casos fatales (Ver Advertencias). Metabólicos: Hiperamonemia (Ver Precauciones), hiponatremia y secreción inadecuada de la hormona antidiurética. Se han registrado casos raros de síndrome de Fanconi, principalmente en niños. Se ha informado de concentraciones disminuidas de carnitina, aunque no se determinó su importancia clínica. Se ha informado de hiperglicinemia (concentración plasmática elevada de glicina), la que fue asociada con la muerte de un paciente con hiperglicinemia no-cetósica preexistente. Genitourinarios: Enuresis, falla renal, nefritis túbuloinstersticial e infección del tracto urogenital. Sentidos Especiales: Pérdida de la audición, reversible e irreversible; sin embargo, no se pudo establecer una relación de causa/efecto. Otalgia. Neoplasias benignas, malignas e inespecíficas (incluyendo quistes y pólipos): Síndrome Mielodisplásico. Trastornos respiratorios, torácico y mediastínicos: Derrame Pleural. Otros: Se ha informado reacción alérgica, anafilaxis, edema de las extremidades, lupus eritematoso, rabdomiólisis, deficiencia de biotina/deficiencia de biotinidasa, dolor óseo, incremento de la tos, neumonía, otitis media, bradicardia, vasculitis cutánea, fiebre e hipotermia. Manía: Aunque la seguridad y eficacia del Ácido Valproico en el tratamiento de episodios maníacos asociados con el trastorno bipolar no ha sido evaluada, los siguientes efectos adversos, no mencionados más arriba, han sido informados por el 1% ó más de los pacientes tratados con Divalproato de sodio en dos estudios clínicos controlados con placebo: Generales: Escalofríos, dolor de cuello y rigidez de cuello. Sistema cardiovascular: Hipotensión, hipotensión postural, vasodilatación. Sistema digestivo: Incontinencia fecal, gastroenteritis, glositis. Sistema musculoesquelético: Artrosis. Sistema nervioso: Agitación, reacción catatónica, hipoquinesia, hiperreflexia, discinesia tardía, vértigo. Piel y faneras: Furunculosis, erupción maculopapulosa, seborrea. Sentidos especiales: Conjuntivitis, sequedad ocular, dolor ocular. Sistema genitourinario: Disuria. Migraña: Aunque la seguridad y eficacia del Ácido Valproico en el tratamiento de la profilaxis de la migraña no han sido evaluadas, los siguientes efectos adversos, no mencionados más arriba, han sido informados por el 1% ó más de los pacientes tratados con Divalproato de sodio en dos estudios clínicos controlados con placebo: Generales: Edema facial. Sistema digestivo: Boca seca, estomatitis. Sistema genitourinario: Cistitis, metrorragia y hemorragia vaginal. Información para los Pacientes: Los pacientes y/o sus tutores deberán ser advertidos de que el dolor abdominal, las náuseas, los vómitos y/o la anorexia pueden ser síntomas de pancreatitis que requieren evaluación clínica inmediata. Los pacientes y/o sus tutores deberán ser informados de los signos y síntomas asociados con la encefalopatía hiperamoniémica (Ver Precauciones - Hiperamoniemia) y deberán a su vez, informar al médico tratante en caso de que apareciera alguno de estos síntomas. Dado que el Valproato de sodio puede producir depresión del SNC, especialmente cuando se lo combina con otros depresores del mismo (por ejemplo: alcohol), se aconsejará a los pacientes evitar actividades riesgosas, tales como conducir automóviles u operar maquinarias peligrosas hasta asegurarse de que la droga no les provoque somnolencia. Debido a que el Ácido Valproico ha estado asociado a ciertos tipos de defectos congénitos, las pacientes en edad fértil serán advertidas de los riesgos asociados con el uso del Ácido Valproico durante el embarazo (Ver Advertencias).
Precauciones.
Disfunción hepática: Ver Contraindicaciones y Advertencias. Pancreatitis: Ver Advertencias. Generales: Dado que se han comunicado casos de trombocitopenia (Ver Advertencias), inhibición de la segunda fase de la agregación plaquetaria y anormalidades en los parámetros de coagulación (por ejemplo, fibrinógeno bajo), se recomienda realizar pruebas de coagulación y recuentos plaquetarios antes de iniciar el tratamiento y a intervalos regulares durante el mismo. En los pacientes tratados con Depakene, se recomienda controlar el recuento de plaquetas y los parámetros de coagulación antes de ser sometidos a procedimientos quirúrgicos. En un estudio clínico con Divalproato de sodio empleado como monoterapia en pacientes epilépticos, 34/126 pacientes (27%) que recibían un promedio de aproximadamente 50 mg/kg/día, presentaron por lo menos un valor plaquetario ≤75 x 109/litro. Se les retiró la medicación a aproximadamente la mitad de estos pacientes, con normalización posterior de los recuentos plaquetarios. En el resto de los pacientes, los recuentos plaquetarios se normalizaron con la continuación de la terapéutica. En este estudio, la probabilidad de trombocitopenia pareció aumentar significativamente con concentraciones totales de Valproato ≥110 mg/ml (mujeres) o ≥135 mg/ml (hombres). La presencia de hemorragias, hematomas o trastornos de la hemostasia / coagulación constituye una indicación para reducir la dosis o suspender el tratamiento. Dado que el Valproato puede interactuar con agentes que actúan como inductores enzimáticos, administrados en forma concomitante, se recomienda la determinación periódica de los niveles plasmáticos del Valproato y de dichas drogas concomitantes durante el curso inicial de la terapéutica, según indicación clínica (Ver Precauciones e Interacciones). El Valproato es eliminado parcialmente en la orina como un cetometabolito, lo que puede llevar a una falsa interpretación de cetonuria. Se ha informado de alteraciones en las pruebas de la función tiroidea asociadas con la administración de Valproato. Se desconoce su significado clínico. Existen estudios in vitro que sugieren que el Valproato estimula la replicación de los virus HIV y CMV bajo ciertas condiciones experimentales. Se desconocen sus consecuencias clínicas, si las hubiera. Además, estos hallazgos in vitro son de dudosa importancia para aquellos pacientes que reciben tratamiento antirretroviral de máxima supresión. Sin embargo, estos datos deberán tenerse en cuenta al interpretar los resultados del control rutinario de la carga viral en pacientes con HIV que reciben Valproato o durante el seguimiento clínico de pacientes con CMV. Pacientes con deficiencia subyacente de carnitina palmitotransferasa (CPT) Tipo II deben ser advertidos del mayor riesgo de rabdomiólisis cuando toman valproato. Hiperamoniemia: La hiperamoniemia se ha reportado en asociación con la terapia con Divalproato y puede presentarse aún con tests de función hepática normales. En pacientes que desarrollen letargia y vómitos inexplicados o cambios en el estado mental, deberá considerarse la encefalopatía hiperamoniémica por lo que deberán medirse los niveles de amonio. Asimismo, deberá considerarse la hiperamoniemia en pacientes con hipotermia (ver Precauciones - Hipotermia). Si el amonio estuviera aumentado se deberá discontinuar el tratamiento con Valproato. Deberán iniciarse intervenciones apropiadas para el tratamiento de la hiperamoniemia y tales pacientes deberán someterse a una investigación para trastornos subyacentes del ciclo de la urea (Ver Contraindicaciones y Advertencias - Trastornos del ciclo de la urea y Precauciones - Hiperamoniemia y Encefalopatía asociadas con el uso concomitante de Topiramato). Las elevaciones asintomáticas del amonio son más frecuentes, y cuando se presentan, requieren un estrecho monitoreo de los niveles plasmáticos de amonio. Si persiste la elevación, deberá considerarse la discontinuación del tratamiento con Valproato. Hiperamoniemia y Encefalopatía asociadas con el uso concomitante de Topiramato: La administración concomitante de Topiramato y Ácido Valproico se ha asociado a hiperamoniemia con o sin encefalopatía en pacientes quienes habían tolerado ambas drogas por separado. Los síntomas clínicos de encefalopatía hiperamoniémica a menudo incluyen alteraciones del nivel de conciencia y/o función cognitiva con letargia y vómitos. La hipotermia también puede ser una manifestación de la hiperamoniemia (ver Precauciones - Hipotermia). En la mayoría de los casos, los signos y síntomas desaparecen con la discontinuación de cualquiera de ambas drogas. Este evento adverso no es debido a una interacción farmacocinética. No se sabe si la monoterapia con Topiramato está asociada con hiperamoniemia. Los pacientes con trastornos congénitos del metabolismo, o una actividad reducida de las mitocondrias hepáticas pueden presentar riesgo aumentado de hiperamoniemia, con o sin encefalopatía. Aunque no ha sido estudiado, una interacción del Topiramato y del Ácido Valproico puede exacerbar defectos existentes o deficiencias enmascaradas en personas susceptibles (Ver Contraindicaciones y Advertencias - Trastornos del ciclo de la urea y Precauciones- Hiperamoniemia). Hipotermia: Se ha comunicado hipotermia, definida como el descenso no intencional de la temperatura corporal por debajo de 35°C, asociada con el tratamiento con Valproato junto con y en ausencia de hiperamoniemia. Esta reacción adversa también puede producirse en pacientes que reciben tratamiento concomitante de Topiramato con Valproato luego de iniciado el tratamiento con Topiramato o al aumentar la dosis diaria de Topiramato (ver Interacciones - Topiramato). Deberá considerarse la suspensión del tratamiento con Valproato en pacientes que desarrollen hipotermia, que puede manifestarse en una variedad de anormalidades clínicas tales como letargia, confusión, coma y alteraciones significativas en otros sistemas orgánicos importantes tales como el sistema cardiovascular y respiratorio. El tratamiento y la evaluación clínica deben incluir análisis de los niveles de amoníaco en sangre. Atrofia cerebral: Han habido reportes de postmarketing de atrofia cerebral y cerebelosa reversible e irreversible, asociada temporalmente con el uso de valproato. En algunos casos los pacientes se recuperaron con secuelas permanentes (ver Reacciones adversas). Las funciones motoras y cognitivas de pacientes con valproato debe ser monitoreadas rutinariamente y la droga debe ser discontinuada ante la presencia de sospecha o signos aparentes de atrofia cerebral. Reportes de atrofia cerebral con varias formas de problemas neurológicos incluyendo retrasos en el desarrollo y deterioro psicomotor han sido reportado en niños que fueron expuestos en el útero al valproato (ver Precauciones y Advertencias - Uso en el embarazo). Reacción de hipersensibilidad multiorgánica: Se han informado reacciones aisladas de hipersensibilidad multiorgánica con una estrecha relación temporal después de la iniciación de la terapéutica con Valproato en adultos y niños (mediana de tiempo hasta la detección 21 días; rango 1 a 40). Aunque escasos, muchos de estos casos necesitaron hospitalización y, al menos, un deceso ha sido reportado. Los signos y síntomas de este trastorno fueron diversos; sin embargo, generalmente, aunque no exclusivamente, los pacientes presentaron fiebre y erupción asociadas con compromiso de otros sistemas orgánicos. Otras manifestaciones asociadas pueden incluir linfadenopatía, hepatitis, pruebas anormales de la función hepática, anomalías hematológicas (por ejemplo, eosinofilia, trombocitopenia, neutropenia), prurito, nefritis, oliguria, síndrome hepatorrenal, artralgia y astenia. Debido a que el trastorno es variable en su expresión, pueden presentarse otros signos y síntomas de otros sistemas no mencionados anteriormente. Si se sospechara esta reacción, se deberá discontinuar el Valproato e iniciar un tratamiento alternativo. Aunque no es muy clara la existencia de una sensibilidad cruzada con otras drogas que pudiera producir este síndrome, la experiencia entre drogas asociadas con hipersensibilidad multiorgánica indicaría esta posibilidad. Carcinogénesis, mutagénesis, daño a la fertilidad: Carcinogénesis: Se desconoce el significado de los hallazgos en animales para los seres humanos. Mutagénesis: El Valproato no demostró ser mutagénico en un ensayo bacteriano in vitro (Test de Ames). Se informó de un aumento en la frecuencia de intercambio de cromátides hermanas en niños epilépticos tratados con Valproato, pero no se observó dicha asociación en otro estudio en adultos. Existe evidencia de que este aumento podría estar asociado con la epilepsia, pero se desconoce su significado biológico. Fertilidad: Se desconoce el efecto del Valproato sobre el desarrollo testicular, la producción espermática o sobre la fertilidad en seres humanos. Embarazo Categoría D: Ver Precauciones, y Advertencias - Uso en el embarazo. Lactancia: El Valproato se excreta en la leche materna. Se han informado concentraciones del 1 al 10% de las concentraciones séricas en la leche materna. Se desconoce su efecto sobre el lactante. Se deberá considerar la discontinuación de la lactancia cuando se administre Divalproato Sódico a mujeres durante este periodo. Pacientes Pediátricos: La experiencia ha indicado que los niños de menos de dos años están expue