BROMADENE®
BIOSIDUS S.A.U
Agente antineoplásico.
Composición.
Cada frasco ampolla contiene: Bortezomib 3.5 mg, Manitol 35.00 mg. Cada ampolla de solvente contiene: Solución fisiológica 3.5 ml.
Farmacología.
Bortezomib inyectable es un agente antineoplásico que se encuentra disponible sólo para uso como inyección intravenosa (IV). El Bortezomib es un ácido borónico dipeptídico modificado. El producto se presenta como un éster manitol borónico que, en forma reconstituida, consiste en el éster manitol en equilibrio con su producto de hidrólisis, el ácido borónico monomérico. La sustancia medicinal existe en su forma anhídrida cíclica como una boroxina trimétrica. El nombre químico de Bortezomib, el ácido borónico monomérico, es ácido [(1R)-3-metil-1[[(2S)-1-oxo-3-fenil-2-[(pirazinilcarbonil) amino] propil] amino] butil] borónico. El peso molecular es de 384,24. La fórmula molecular es C19H25BN4O4. La solubilidad de bortezomib, como el ácido borónico monomérico, en agua es de 3,3-3,8 mg/ml con un rango de pH de 2-6,5. Mecanismo de acción: El bortezomib es un inhibidor reversible de la actividad tipo quimotripsina, del proteasoma 26S en células de mamíferos. El proteasoma 26S es un gran complejo proteico que degrada las proteínas ubiquitinadas. La vía proteasoma-ubiquitina tiene un papel esencial en la regulación de la concentración intracelular de proteínas específicas, manteniendo, de esta manera, la homeostasis entre las células. La inhibición del proteasoma 26S evita esta proteólisis prevista que puede afectar a múltiples cascadas de señalización de la célula. Esta alteración de los mecanismos homeostáticos normales puede conducir a la muerte celular. Los experimentos han demostrado que el bortezomib es citotóxico para una variedad de tipos de células cancerígenas in vitro. El bortezomib causa un retraso en el desarrollo tumoral in vitro en modelos tumorales preclínicos, incluyendo el mieloma múltiple. Farmacocinética: Luego de la administración intravenosa de una dosis de 1,0 mg/m2 y 1,3 mg/m2, administrada a 24 pacientes con mieloma múltiple (n = 12, por cada nivel de dosis), las concentraciones plasmáticas máximas medias estimadas de bortezomib (Cmáx) fueron de 57 y 102 g/ml, respectivamente. En dosis posteriores, cuando se administró dos veces por semana, las concentraciones plasmáticas observadas máximas medias se extendieron de 67 a 106 ng/ml para la dosis de 1,0 mg/m2 y de 89 a 120 ng/ml para la dosis de 1,3 mg/m2. La vida media de eliminación promedio del bortezomib luego de múltiples dosificaciones osciló entre 40 y 193 horas luego de dosis de 1.0 mg/m2 y entre 76 y 108 horas luego de la dosis de 1,3 mg/m2. Los clearances corporales totales medios oscilaron entre 102 y 112 l/h luego de la primera dosis para la dosis de 1,0 mg/m2 y 1,3 mg/m2, respectivamente, y oscilaron de 15 a 32I L/h luego de las dosis posteriores para la dosis de 1,0 y 1,3 mg/m2, respectivamente. Distribución: El volumen de distribución del bortezomib medio se extendió de aproximadamente 498 a 1884 L/m2 luego de la administración de dosis únicas o repetidas de 1,0 mg/m2 o 1,3 mg/m2 a pacientes con mieloma múltiple. Esto sugiere que el bortezomib se distribuye ampliamente a los tejidos periféricos. La unión del bortezomib a las proteínas del plasma humano se promedió en un 83% por sobre el rango de concentración de 100-1000 ng/ml. Metabolismo: Los estudios in vitro con microsomas hepáticos humanos e isoenzimas del citocromo P450 expresadas en el ADNc humano indican que el bortezomib es metabolizado oxidativa y principalmente mediante las enzimas del citocromo P450, 3A4, 2C19 y 1A2. El metabolismo de Bortezomib por las enzimas CYP 2D6 y 2C9 es menor. La vía metabólica principal consiste en la desboronación para la formación de dos metabolitos desboronados que luego se someten a la hidroxilación en varios metabolitos. Los metabolitos desboronados por el bortezomib son inactivos como inhibidores del proteasoma 26S. Los datos agrupados del plasma de ocho pacientes a los 10 minutos y a los 30 minutos luego de la dosificación indican que los niveles plasmáticos de metabolitos son bajos en comparación con la droga madre. Eliminación: No se han caracterizado en humanos las vías de eliminación del bortezomib. Poblaciones especiales: Edad: Los análisis de los datos luego de la primera dosis del ciclo I (Día 1) en 39 pacientes con mieloma múltiple que habían recibido dosis intravenosas de 1,0 mg/m2 y 1,3 mg/m2 mostraron que ambas dosis normalizadas de AUC y Cmáx tienden a ser menores en pacientes más jóvenes. Los pacientes de < 65 años de edad (n= 26) tuvieron alrededor de un 25% menos de Cmáx y AUC normalizada por las dosis medias que los de > 65 años de edad (n =13). Sexo: Los valores de AUC y Cmáx normalizados de las dosis medias fueron comparables entre pacientes masculinos (n =22) y femeninos (n = 17) luego de la primera dosis del Ciclo 1 para las dosis de 1,0 y 1.3 mg/m2. Raza: El efecto de la raza a la exposición del bortezomib no se puede evaluar ya que la mayoría de los pacientes eran Caucásicos. Deterioro hepático: El efecto del deterioro hepático sobre la farmacocinética del bortezomib fue determinado en 51 enfermos de cáncer con diversos grados de deterioro hepático en un rango de dosis de 0,5 a 1,3 mg/m2. El deterioro hepático leve no alteró la farmacocinética de bortezomib. En pacientes con deterioro hepático moderado a severo fue observado un aumento del 50% en AUC con las dosis normalizadas de bortezomib con relación al grupo hepático de función normal. Deterioro renal: Se condujo un estudio farmacocinético en pacientes con diversos grados de deterioro renal que se clasificaron de acuerdo a sus valores de clearance de creatinina (CrCl) en los siguientes grupos: Normal (CrCl ≥60 mL/min/1,73 m2 N=12), Leve (CrCl=40-59 mL/min/1,73 m2 N=10), Moderado (CrCl=20-39 mL/min/1,73 m2 N=9), y Severo (CrCl < 20 mL/min/1,73 m2 N=3). Un grupo de pacientes en diálisis que fueron dosificados después de la diálisis también fueron incluidos en el estudio (N=8). Se administró a los pacientes dosis intravenosas de 0,7 a 1,3 mg/m2 de bortezomib dos veces a la semana. La exposición de bortezomib (AUC y Cmáx normalizadas por las dosis) fue comparable entre todos los grupos.
Indicaciones.
Bortezomib está indicado como monoterapia para el tratamiento del mieloma múltiple en progresión en pacientes que han recibido previamente al menos 1 tratamiento y que han sido sometidos o no son candidatos a trasplante de médula ósea.
Dosificación.
Monoterapia: Dosis recomendada: La dosis recomendada de BROMADENE® es 1,3 mg/m2/dosis administrada como una inyección en bolo durante 3 a 5 segundos dos veces a la semana durante dos semanas (los días 1, 4, 8 y 11), seguida por un período de descanso de 10 días (los días 12 a 21). Para una terapia prolongada de más de 8 ciclos, BROMADENE® puede ser administrado con el esquema estándar o con el esquema de mantenimiento de una vez por semana durante 4 semanas (los días 1, 8, 15 y 22) seguidos por un período de descanso de 13 días (los días 23 al 35). Por lo menos deben transcurrir 72 horas entre dosis consecutivas de BROMADENE®. Modificación de la Dosis y Reinicio de la Terapia: La terapia con BROMADENE® deberá suspenderse ante el inicio de cualquier toxicidad no hematológica de grado 3 o grado 4, excluyendo una neuropatía, tal como se debate a continuación (ver Precauciones). Una vez que se han resuelto los síntomas de la toxicidad se puede reiniciar el tratamiento con BROMADENE® con una reducción de la dosis del 25% (1,3 mg/m2/dosis reducida a 1.0 mg/m2/dosis; 1,0 mg/m2/dosis reducida a 0,7 mg/m2/dosis). La siguiente tabla contiene la modificación de la dosis recomendada para el tratamiento de pacientes que experimentan una neuropatía sensorial periférica y/o dolor neuropático relacionado con BROMADENE® solamente luego de una cuidadosa evaluación de los riesgos/beneficios.
Criterios de toxicidad comunes NCI1-: https://www.ucdmc.ucdavis.edu/clinicaltrials/StudyTools/Documents/NCI_Toxicity_Table.pdf. Precauciones para la administración: BROMADENE® es un antineoplásico. Se debe tener cuidado durante su manipulación y preparación. Se deben emplear técnicas de asepsia apropiadas. Se recomienda el uso de guantes y vestuario protector a fin de evitar el contacto con la piel. En ensayos clínicos se reportó irritación local de la piel en el 5% de los pacientes. Pero la extravasación de BROMADENE® no se asoció con daños del tejido. Reconstitución/ Preparación para su Administración Intravenosa: Previo al uso, los contenidos de cada vial deben reconstruirse con 3,5 ml de solución salina normal (0,9%), Cloruro de Sodio Inyectable, USP. El producto reconstruido deberá ser una solución límpida e incolora. Antes de su administración, y siempre que el envase y la solución lo permitan, deberá controlarse visualmente que los productos medicinales parenterales no contengan materia particulada ni haya decoloración. Si se observa alguna decoloración o materia particulada, no se deberá usar el producto reconstituido. Terapia Combinada: Dosis Recomendada: BROMADENE® (bortezomib) se administra como una inyección IV en bolo de 3 a 5 segundos en combinación con melfalán oral y prednisona oral durante 9 ciclos de tratamiento de 6 semanas como se muestra en la Tabla 2. En ciclos 1-4, BROMADENE® se administra dos veces por semana (días 1, 4, 8, 11, 22, 29 y 32). En los ciclos 5-9, BROMADENE® se administra una vez por semana (días 1, 8, 22, y 29).
Lineamiento para el Manejo de Dosis para la Terapia Combinada: Modificación de la dosis y re-inicio de la terapia cuando se administra BORTEZOMIB en combinación con malfalán y prednisona: Antes de iniciar un nuevo ciclo de terapia: El recuento de plaquetas debe ser ≥70 x 109/L y el ANC debe ser ≥ 1.0 x 109/L. Las toxicidades no hematológicas deberían haberse resuelto a Grado 1 o valor basal.
Para información adicional concerniente al melfalán y prednisona, ver la información de prescripción del fabricante.
Contraindicaciones.
BROMADENE® está contraindicado en pacientes con hipersensibilidad al bortezomib, boro o manitol.
Reacciones adversas.
Resumen de un Ensayo Clínico en Pacientes con Mieloma Múltiple No Tratado Previamente: La Tabla 5 describe los datos de seguridad de 340 pacientes con mieloma múltiple no tratado previamente que recibieron BROMADENE® (1,3 mg/m2) en combinación con melfalán (9mg/m2) y prednisona (60mg/m2) en un estudio randomizado prospectivo. El perfil de seguridad de BROMADENE® en combinación con melfalán/prednisona es consistente con los perfiles de seguridad conocidos tanto para BROMADENE® como melfalán/prednisona.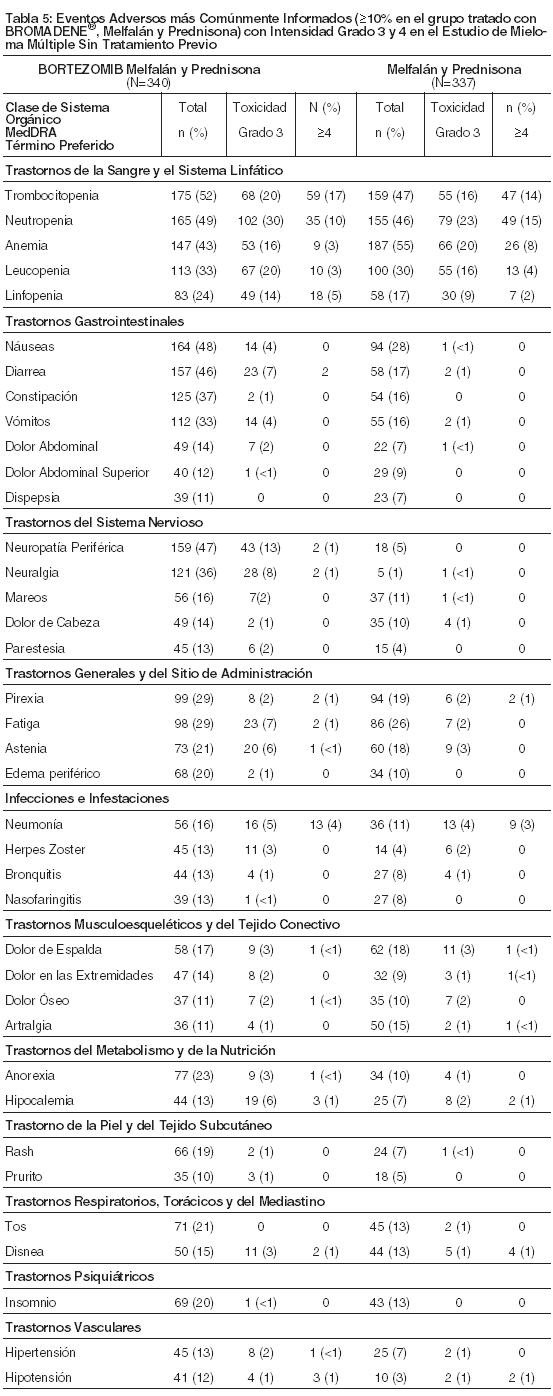
Estudio Randomizado de Mieloma múltiple Recidivante: Los datos de seguridad descriptos a continuación y en la Tabla 5 reflejan la exposición ya sea a BROMADENE® (n=331) o a dexametasona (n=332) en un estudio de pacientes con mieloma múltiple. Se administró BROMADENE® por vía intravenosa a dosis de 1,3 mg/m2 dos veces a la semana para 2 de 3 semanas (ciclo de 21 días). Después de ocho ciclos de 21 días los pacientes continuaron con la terapia por tres ciclos de 35 días en un régimen semanal. La duración del tratamiento fue hasta 11 ciclos (9 meses) con una duración media de 6 ciclos (4,1 meses); para la inclusión en el ensayo los pacientes deben haber tenido una enfermedad medible y 1 a 3 terapias previas. No hubo límite de edad superior para el ingreso. El clearance de creatinina puede tener valores tan bajos como 20 ml/min y los niveles de bilirrubina como 1,5 veces el límite superior normal. La frecuencia general de eventos adversos fue similar en hombres y en mujeres, y en pacientes de < 65 y ≥65 años de edad. La mayoría de los pacientes eran Caucásicos. Entre los 331 pacientes tratados con BROMADENE® los eventos más comúnmente informados por lo general fueron condiciones asténicas (61%), diarrea y náuseas (57% cada una), constipación (42%), neuropatía periférica NEC (36%), vómitos, pirexia, trombocitopenia y trastornos psiquiátricos (35% cada uno), anorexia y disminución del apetito (34%), parestesia y disestesia (27%), anemia y dolor de cabeza (26% cada uno) y los (21%). Los eventos adversos más comúnmente informados entre los 332 pacientes en el grupo tratado con dexametasona fueron trastornos psiquiátricos (49%), condiciones asténicas (45%), insomnio (27%), anemia (22%) y diarrea e infecciones pulmonares/respiratorias del tracto inferior (21% cada una). El catorce por ciento (14%) de los pacientes en el grupo tratados con BROMADENE® experimentó un efecto adverso Grado 4; las toxicidades más comunes fueron trombocitopenia (4%), neutropenia (2%) e hipercalcemia (2%). El dieciséis (16%) de los pacientes tratados con dexametasona experimentó un evento adverso Grado 4; la toxicidad más común fue hiperglucemia (2%). Eventos Adversos Serios (SAEs) y Eventos que Condujeron a la discontinuación del Tratamiento en el Estudio de Mieloma múltiple Recidivante: Los eventos adversos serios se definen como cualquier evento, independientemente de la causalidad, que ocasione la muerte, tenga riesgo de vida, requiera hospitalización o prolongue una hospitalización actual, cause discapacidad significativa, o se considere que constituye un evento médico importante. Un total de 144 pacientes (44%) del grupo de tratamiento con BROMADENE® experimentó un SAE durante el estudio, tal como sucedió con los 144 pacientes (43%) del grupo de los pacientes tratados con dexametasona. Los SAEs más comúnmente informados en el grupo del tratamiento con BROMADENE® fueron pirexia (6%), diarrea (5%), disnea y neumonía (4%) y vómitos (3%). En el grupo de tratamiento con dexametasona los SAEs más comúnmente informados fueron neumonía (7%), pirexia (4%) e hiperglucemia (3%). Un total de 145 pacientes, incluyendo 84 (25%) de 331 pacientes en el grupo de tratamiento con BROMADENE® y 61 (18%) de 332 pacientes en el grupo de tratamiento con dexametasona, discontinuó el tratamiento a causa de efectos adversos evaluados por el investigador como relacionados con la droga. Entre los 331 pacientes tratados con BROMADENE®, el evento relacionado con la droga más comúnmente informado que condujo a la discontinuación fue la neuropatía periférica (8%). Entre los 332 pacientes del grupo tratado con dexametasona, el evento relacionado con la droga más comúnmente informado que condujo a la discontinuación fue el trastorno psicótico e hiperglucemia (2% cada uno). Se consideró que 4 muertes estuvieron relacionadas con BROMADENE® en el estudio Fase 3: 1 caso de cada uno de los siguientes: shock cardiogénico, insuficiencia respiratoria, insuficiencia cardíaca congestiva y paro cardíaco. Se consideró que 4 muertes estuvieron relacionadas con la dexametasona: 2 casos de sepsis, 1 caso de meningitis bacteriana y 1 caso de muerte súbita en el hogar. Eventos Adversos más Comúnmente Informados en el Estudio de Mieloma múltiple Recidivante: En la Tabla 6 se observaron los efectos adversos más comunes del estudio de mieloma múltiple recidivante. Se incluyeron todos los eventos adversos con incidencia ≥10% en el grupo tratado con BROMADENE®.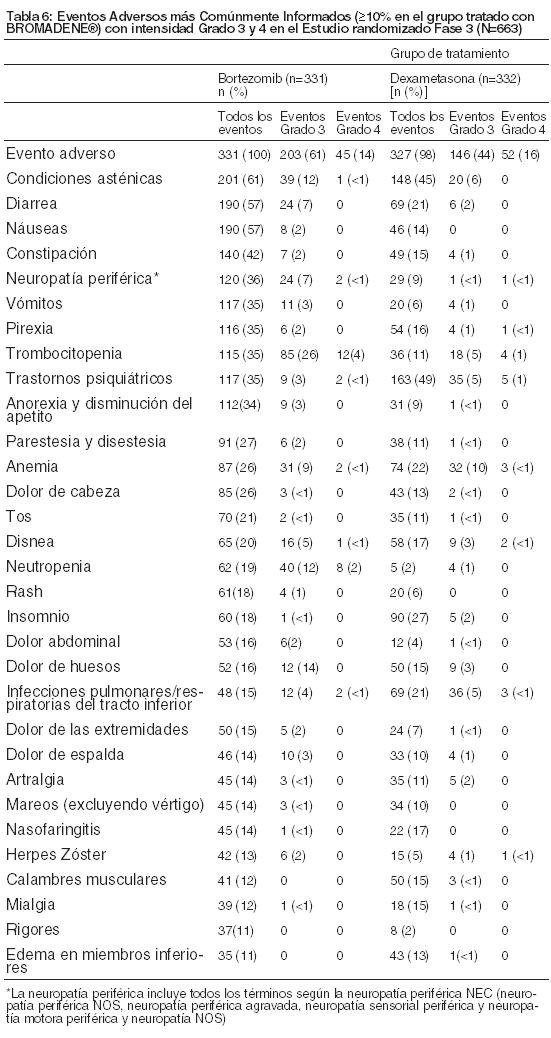
Estudios clínicos no randomizados Fase 2: Los dos estudios clínicos llevados a cabo evaluaron a 228 pacientes con mieloma múltiple que recibían una dosis de 1,3 mg/m2 de BROMADENE® dos veces por semana durante 2 semanas, seguido por un período de 10 días de descanso (duración de ciclo de tratamiento de 21 días) por un máximo de 8 ciclos de tratamiento. Los eventos adversos más comúnmente informados fueron condiciones asténicas (incluso fatiga, malestar y debilidad) (65%), náuseas (64%), diarrea (51%), disminución del apetito (incluso anorexia) (43%), constipación (43%), trombocitopenia (43%), neuropatía periférica (incluyendo neuropatía sensorial periférica y neuropatía periférica agravada) (37%), pirexia (36%), vómitos (36%) y anemia (32%). El catorce por ciento de los pacientes experimentó, por lo menos, un episodio de toxicidad grado 4, siendo la trombocitopenia (3%) y la neutropenia (3%) las toxicidades más comunes. Eventos Adversos Serios (SAEs): Los eventos adversos serios se definen como cualquier evento, independientemente de la causalidad, que: causa la muerte, amenaza la vida, requiere hospitalización o prolonga la hospitalización actual, causa un impedimento significativo o se considera un evento médico importante. Un total de 113 (50%) de los 228 pacientes experimentaron SAEs durante los estudios. Entre los SAEs más comúnmente informados se incluyen pirexia (7%), neumonía (7%), diarrea (6%), vómitos (5%), deshidratación (5%) y náuseas (4%). Los eventos adversos que el investigador consideraba que estaban relacionados con la droga y condujeron a la discontinuación, ocurrieron en un 18% de los pacientes. Las razones de la discontinuación incluyeron neuropatía periférica (5%), trombocitopenia (4%), diarrea (2%), y fatiga (2%). Se informaron dos muertes y el investigador consideró que estaban posiblemente relacionadas con la droga del estudio: un caso de paro cardiopulmonar y un caso de insuficiencia respiratoria. Los eventos adversos más comunes se observan en la Tabla 7. Se incluyeron todos los eventos adversos que ocurrieron en ≥10%. En los estudios de un solo grupo realizados, generalmente no es posible distinguir eventos adversos que son causados por la droga y aquellos que reflejan la enfermedad subyacente del paciente.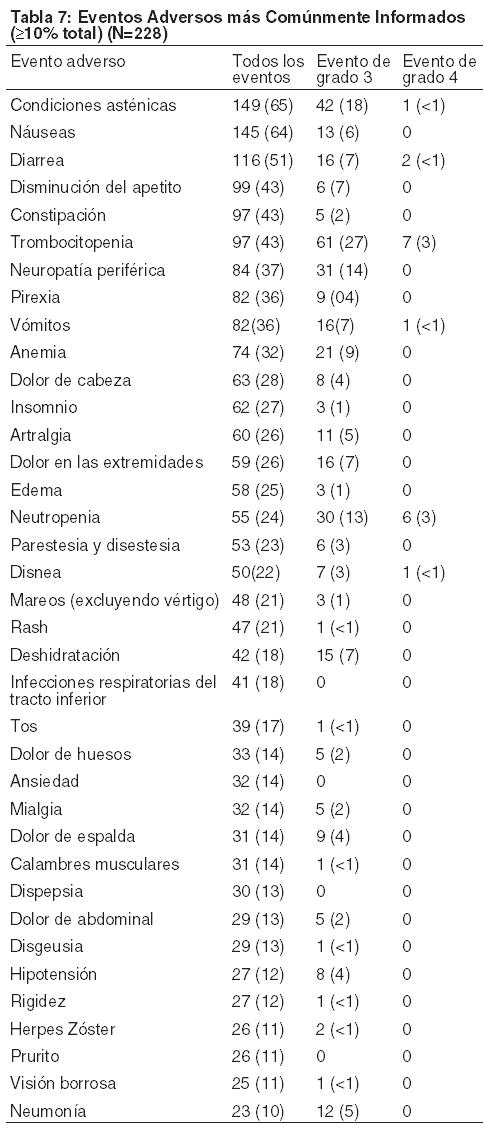
Estudio de Extensión Abierto Fase 2 en Mieloma múltiple Recidivante: En el estudio de extensión abierto Fase 2 de los 63 pacientes que se observan anteriormente no se observaron nuevas toxicidades acumulativas ni nuevas toxicidades a largo plazo con el tratamiento prolongado con BROMADENE®. Estos pacientes fueron tratados por un total de 5,3 a 23 meses, incluyendo el tiempo tratados con BROMADENE® en el estudio previo de BROMADENE®. Eventos Adversos Más Comúnmente Informados en el Resumen Integrado de Seguridad: Los eventos adversos más comunes se muestran en la Tabla 8. Se incluyen todos los eventos adversos que ocurrieron en ≥10%. En ausencia de un grupo comparador randomizado, generalmente no es posible distinguir entre los eventos adversos que son causados por la droga y los que reflejan la enfermedad subyacente del paciente. Por favor, vea análisis de las reacciones adversas específicas que se encuentra a continuación: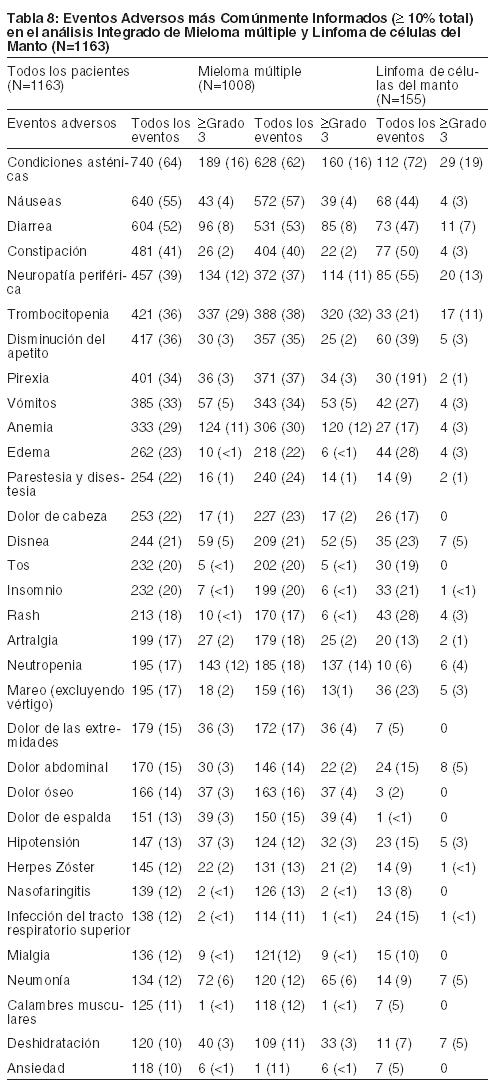
Descripción de los Eventos Adversos Seleccionados a partir de los Estudios Fase 2 y Fase 3 de mieloma múltiple y de Linfoma de células del Manto Fase 2. Eventos Gastrointestinales: Un total del 87% de los pacientes experimentó al menos un trastorno gastrointestinal. Los trastornos gastrointestinales más comunes incluyeron náuseas, diarrea, constipación, vómitos y disminución del apetito. Otros trastornos gastrointestinales incluyeron dispepsia y disgeusia. Los eventos gastrointestinales grado 3 ocurrieron en 18% de los pacientes; los eventos grado 4 fueron del 1%. Los eventos gastrointestinales se consideraron serios en el 11% de los pacientes. El cinco por ciento (5%) de los pacientes discontinuaron debido a un evento gastrointestinal. Se informaron náuseas más frecuentemente en los pacientes con mieloma múltiple (57%) en comparación con los pacientes con linfoma de células del manto (44%) [ver Advertencias y Precauciones]. Trombocitopenia: Entre los estudios, la trombocitopenia asociada con BROMADENE® se caracterizó por una disminución en el recuento de plaquetas, durante el período de dosificación (días 1 a 11) y un regreso a los valores basales durante el período de descanso de 10 días durante cada ciclo de tratamiento. En general, la trombocitopenia se informó en el 36% de los pacientes. La trombocitopenia fue Grado 3 en el 24%, ≥ Grado 4 en 5%, y seria en el 3% de los pacientes y el evento produjo la discontinuación de BROMADENE® en el 2% de los pacientes [ver Advertencias y Precauciones]. Se informó trombocitopenia más frecuentemente en los pacientes con mieloma múltiple (38%) en comparación con los pacientes con linfoma de células del manto (21%). La incidencia de trombocitopenia ≥Grado 3 también fue más alta en los pacientes con mieloma múltiple (32%) en comparación con los pacientes con linfoma de células del manto (11%). [ver Advertencias y Precauciones]. Neuropatía periférica: En general la neuropatía periférica NEC ocurrió en el 39% de los pacientes. La neuropatía periférica fue grado 3 para el 11% de los pacientes y grado 4 para < 1% de los pacientes. El ocho por ciento (8%) de los pacientes discontinuó BROMADENE® debido a neuropatía periférica. La incidencia de la neuropatía periférica fue más alta entre los pacientes con linfoma de células del manto (56%) en comparación con los pacientes con mieloma múltiple (37%). En el estudio de mieloma múltiple recidivante, entre los 87 pacientes que experimentaron neuropatía periférica Grado 2, el 51% había tenido una mejoría o se resolvió con un promedio de 3,5 meses desde la primera aparición. Entre los pacientes con neuropatía periférica en los estudios de mieloma múltiple Fase 2 que fue Grado 2 y llevó a la discontinuación o fue ≥ Grado 3, el 73% (24 de 33) informó mejoría o resolución luego del ajuste de la dosis de BROMADENE®, con un tiempo promedio de mejoría de un Grado o más desde la última dosis de BROMADENE® de 33 días [ver Advertencias y Precauciones]. Hipotensión: La incidencia de la hipotensión (hipotensión postural, hipotensión ortostática e hipotensión NOS) fue del 13% en los pacientes tratados con BROMADENE®. La hipotensión fue Grado 1 o 2 en la mayoría de los pacientes y Grado 3 en el 3% y ≥ Grado 4 en < 1%. El tres por ciento (3%) informó la hipotensión como un SAE y el 1% discontinuó debido a la hipotensión. La incidencia de la hipotensión fue similar en los pacientes con mieloma múltiple (12%) y en los que tenían linfoma de células del manto (15%). Además, el 2% de los pacientes experimentó hipotensión y tuvieron un evento sincopal. Puede ser necesario el ajuste de las dosis de las mediaciones antihipertensivas en los pacientes que reciben BROMADENE®. [ver Advertencias y Precauciones]. Neutropenia: El recuento de neutrófilos disminuyó durante el período de dosificación de BROMADENE® (días 1 a 11) y regresó a los valores basales durante el período de descanso de 10 días durante cada ciclo de tratamiento. En general, la neutropenia ocurrió en el 17% de los pacientes y fue de Grado 3 en el 9% de los pacientes y ≥Grado 4 en el 3%. Se informó la neutropenia como un evento serio en < 1% de los pacientes y < 1% de los pacientes discontinuaron debido a la neutropenia. La incidencia de neutropenia fue más alta en los pacientes con mieloma múltiple (18%) en comparación con los pacientes con linfoma de células del manto (6%). La incidencia de neutropenia ≥Grado 3 también fue más alta en los pacientes con mieloma múltiple (14%) en comparación con los pacientes con linfoma de células del manto (4%) [ver Advertencias y Precauciones]. Condiciones esténicas (fatiga, malestar, debilidad): Se informaron condiciones asténicas en el 64% de los pacientes. La astenia fue Grado 3 para el 16% y ≥ Grado 4 en < 1% de los pacientes. El cuatro por ciento (4%) de los pacientes discontinuó el tratamiento debido a la astenia. Se informaron condiciones asténicas en el 62% de los pacientes con mieloma múltiple y en el 72% de los pacientes con linfoma de células del manto. Pirexia: La pirexia ( > 38°C) se informó como un evento adverso para el 34% de los pacientes. El evento fue Grado 3 en el 3% y ≥ Grado 4 en < 1%. Se informó la pirexia como un evento adverso serio en el 6% de los pacientes y llevó a la discontinuación de BROMADENE® en < 1% de los pacientes. La incidencia de pirexia fue más alta en los pacientes con mieloma múltiple (37%) en comparación con los pacientes con linfoma de células del manto (19%). La incidencia de pirexia ≥Grado 3 fue del 3% en los pacientes con mieloma múltiple en comparación con el 1% de los pacientes con linfoma de células del manto. Infección del Virus del Herpes: Los médicos deben considerar el uso de profilaxis antiviral en sujetos tratados con BROMADENE®. En los estudios randomizados de mieloma múltiple no tratado previamente y recidivante, la reactivación de las infecciones del virus herpes zóster fue más frecuente en los sujetos tratados con BROMADENE® (13%) que en los grupos de control (4-5%). Se observó herpes simplex en 2-8% de los sujetos tratados con BROMADENE®, y en 1-5% en los grupos de control. En el estudio de mieloma múltiple no tratado previamente, la reactivación del virus herpes zóster en el grupo tratado con BROMADENE®, melfalán y prednisona fue menos frecuente en sujetos que recibían terapia antiviral profiláctica (3%) que en sujetos que no recibieron terapia antiviral profiláctica (17%). En la experiencia posterior a la comercialización, se informaron casos de meningoencefalitis por herpes y herpes oftálmico. Resumen Integrado de Seguridad (Mieloma múltiple y Linfoma de células del Manto): Los datos de seguridad de los estudios Fase 2 y 3 de 1,3 mg/m2 / dosis de BROMADENE® dos veces por semana durante 2 semanas seguido de un perodo de descanso de 10 días en 1163 pacientes con mieloma múltiple (N=1008) y linfoma de células del manto (N=155) se integraron y tabularon. En estos estudios, el perfil de seguridad de BROMADENE® fue similar en pacientes con mieloma múltiple y linfoma de células del manto. En el análisis integrado, los eventos adversos más comúnmente informados fueron las condiciones asténicas (incluyendo fatiga, malestar y debilidad) (64%), náuseas (55%), diarrea (52%), constipación (41%), neuropatía periférica NEC (incluyendo neuropatía periférica sensorial y neuropatía periférica agravada) (39%), trombocitopenia y disminución del apetito (incluyendo anorexia) (36% cada uno), pirexia (34%), vómitos (33%) y anemia (29%). El veinte por ciento (20%) de los pacientes experimentaron al menos 1 episodio de toxicidad ≥Grado 4, más comúnmente trombocitopenia (5%) y neutropenia (3%). Eventos Adversos Serios Adicionales a partir de Estudios Clínicos y con Posterioridad a la Comercialización: En los ensayos clínicos se han informado los siguientes SAEs clínicamente importantes que no se describen anteriormente en pacientes tratados con BROMADENE® administrado como monoterapia o bien en combinación con otros quimioterapéuticos. Estos estudios se realizaron en pacientes con malignidades hematológicas y en tumores sólidos. Trastornos del Sistema linfático y Sanguíneo: Coagulación intravascular diseminada, linfopenia, leucopenia. Trastornos cardíacos: angina de pecho, fibrilación auricular agravada, agitación atrial, bradicardia, paro sinusal, amiloidosis cardíaca, bloqueo atrioventricular completo, isquemia miocárdica, infarto de miocardio, pericarditis, efusión pericárdica, torsades de pointes, taquicardia ventricular. Trastornos auditivos y laberínticos: deterioro de la audición, vértigo. Trastornos visuales: Diplopía, y visión borrosa, infección conjuntival, irritación. Trastornos gastrointestinales: ascitis, disfagia, impacto fecal, gastroenteritis, gastritis hemorrágica, hematemesis, duodenitis hemorrágica, íleo paralítico, obstrucción del intestino grueso, obstrucción intestinal paralítica, peritonitis, obstrucción del intestino delgado, perforación del intestino grueso, estomatitis, melena, pancreatitis aguda, petequia de la mucosa oral, reflujo gastroesofágico. Trastornos generales y condiciones del sitio de administración: eritema en el sitio de la inyección, neuralgia, dolor en el sitio de la inyección, flebitis. Trastornos hepatobiliares: colestasis, hemorragia hepática, hiperbilirrubinemia, trombosis de la vena porta, hepatitis, insuficiencia hepática. Trastorno del sistema inmunológico: reacción anafiláctica, hipersensibilidad a la droga, hipersensibilidad mediada por el complejo inmunológico, angioedema, edema laríngeo. Infecciones e infestaciones: aspergilosis, bacteriemia, infección del tracto urinario, infección viral por herpes, listeriosis, shock séptico, toxoplasmosis, candidiasis oral, sinusitis, infección relacionada con el catéter. Complicaciones del procedimiento, envenenamiento y lesión: fractura de huesos, hematoma subdural, complicación relacionada con el catéter. Trastornos metabólicos y nutricionales: hipocalcemia, hiperuricemia, hipocaliemia, hipercaliemia, hiponatremia, hipernatremia. Trastornos del sistema nervioso: ataxia, coma, disartria, disautonomía, encefalopatía, parálisis craneal, convulsión grand mal (convulsión generalizada tónico-clónica), accidente cerebrovascular hemorrágico, disfunción motora, compresión del cordón espinal, paraplejía, ataque isquémico transitorio, parálisis, neuralgia post-herpética, síndrome reversible de leucoencefalopatía posterior. Trastornos psiquiátricos: agitación, confusión, cambio del estado mental, trastorno psicótico, idea de suicidio. Trastornos urinarios renales: cálculos renales, hidronefrosis bilateral, espasmo de vejiga, hematuria, cistitis hemorrágica, incontinencia urinaria, retención urinaria, insuficiencia renal (aguda y crónica), nefritis glomerular proliferativa. Trastornos respiratorios, torácicos y mediastínicos: síndrome de distrés respiratorio agudo, enfermedad pulmonar infiltrativa aguda difusa, neumonía por aspiración, atelectasia, enfermedad obstructiva crónica y exacerbada de las vías aéreas, disfagia, disnea, disnea por ejercicio, epistaxis, hemoptisis, hipoxia, infiltración pulmonar, efusión pleural, neumonitis, distrés respiratorio, hipertensión pulmonar. Trastornos de la piel y tejidos sub-cutáneos: urticaria, edema facial, rash (que puede ser prurítico), vasculitis leucocitoclástica. Trastornos vasculares: accidente cerebrovascular, hemorragia cerebral, trombosis de vena profunda, embolia periférica, embolia pulmonar, hipertensión pulmonar. Experiencia post-marketing: A continuación se listan las reacciones adversas a la droga clínicamente significativas que no han sido informadas anteriormente. Las frecuencias proporcionadas a continuación reflejan los porcentajes de reporte de reacciones adversas a la droga de la experiencia post-marketing mundial con BROMADENE®. Las frecuencias proporcionadas a continuación reflejan porcentajes de reporte y no se pueden hacer cálculos precisos de incidencia. Las reacciones adversas a la droga se clasifican por frecuencia, usando la siguiente convención: muy comunes ( < 1/10), comunes ( > 1/100 y < 1/10), poco comunes ( > 1/1000 y < 1/100), raros ( > 1/10000 y < 1/1000), muy raros ( < 1/10000, incluyendo reportes aislados).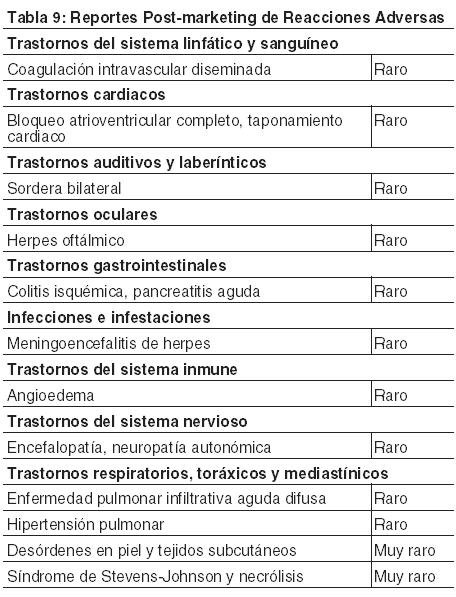
Precauciones.
En general, el perfil de seguridad de los pacientes tratados con BROMADENE® en monoterapia fue similar a la observada en pacientes tratados con BROMADENE® en combinación con melfalán y prednisona. Pacientes con deterioro hepático: Los pacientes con deterioro hepático leve no requieren un ajuste de dosis y deberán ser tratados con la dosis recomendada. Los pacientes con deterioro hepático moderado a severo deberían ser tratados con BROMADENE® en una dosis reducida de 0.9 mg/m2. Para pacientes con moderado a severo deterioro hepático, que requiere modificaciones de dosis subsecuentes, para manejar toxicidades, las reducciones de dosis deberían ser de 0.9 mg/m2 a 0.7 mg/m2, y de 0.7 mg/m2 a 0.5 mg/m2. Para una clasificación de daño hepático, ver la siguiente tabla.
Neuropatía Periférica: El tratamiento con BROMADENE® produce una neuropatía periférica que es, predominantemente, sensorial. Sin embargo, se han informado casos de neuropatía sensorial-motora periférica. Aquellos pacientes con signos o síntomas preexistentes (entumecimiento, dolor o una sensación de ardor en pies o manos) de neuropatía periférica pueden experimentar un empeoramiento durante el tratamiento con BROMADENE®. Deberá controlarse en los pacientes los síntomas de neuropatía, tales como una sensación de ardor, hiperestesia, hipoestesia, parestesia, malestar, dolor neuropático o debilidad. En los pacientes que experimentan un empeoramiento en la neuropatía periférica o nueva puede ser necesario un cambio en la dosis y en el régimen de BROMADENE® (ver Dosificación). Luego de los ajustes de la dosis, la mejoría o resolución de la neuropatía periférica se informó en el 51% de los pacientes con neuropatía periférica ≥ Grado 2 en el estudio de mieloma múltiple Fase 3. La mejoría o resolución de la neuropatía periférica se informó en el 73% de los pacientes que discontinuaron debido a neuropatía Grado 2 o que tuvieron neuropatía periférica ≥ Grado 3 en estudios de mieloma múltiple Fase 2 (ver Reacciones adversas). El resultado a largo plazo de la neuropatía periférica no se ha estudiado en el linfoma de células del manto. Hipotensión: En estudios de Fase 2 y 3 de mieloma múltiple, la incidencia de hipotensión ortostática/postural fue alrededor del 11 al 12% de pacientes. Estos casos se observan durante todo el tratamiento. Se deberá tener precaución al tratar pacientes con antecedentes de síncope, pacientes que reciben medicaciones que, se sabe, están asociadas con la hipotensión, y pacientes deshidratados. El tratamiento de la hipotensión ortostática/postural puede incluir un ajuste de medicamentos antihipertensivos, hidratación o la administración de mineralcorticoides. Trastornos Cardíacos: Se ha observado exacerbación o desarrollo agudo de la insuficiencia cardíaca congestiva y/o disminución de la fracción de eyección del ventrículo izquierdo ha sido reportado. Pacientes con factores de riesgo o enfermedades cardíacas pre-existentes se deben controlar minuciosamente. En los estudios Fase 3 de mieloma múltiple, la incidencia de cualquier trastorno cardíaco emergente del tratamiento fue del 15% y del 13% en los grupos tratados con BROMADENE® y dexametasona, respectivamente. La incidencia de eventos de insuficiencia cardíaca (edema pulmonar agudo, insuficiencia cardíaca, insuficiencia cardíaca congestiva, shock cardiogénico, edema pulmonar) fue similar en los grupos tratados con BROMADENE® y con dexametasona, 5% y 4%, respectivamente. Hubo casos aislados de prolongación del intervalo QT en estudios clínicos; no se ha establecido la causalidad. Desórdenes pulmonares: Se han reportado casos raros de enfermedad pulmonar infiltrativa aguda de etiología desconocida como neumonitis, neumonía intersticial, infiltración pulmonar y Síndrome de Distress Respiratorio Agudo (SDRA) en pacientes que reciben BROMADENE®. Algunos de estos eventos han sido fatales. Una proporción más alta de estos eventos han sido reportados en Japón. En el caso de síntomas nuevos o que se agraven los existentes, un diagnóstico precoz debería ser realizado y los pacientes deberían ser tratados apropiadamente. En un ensayo clínico, los primeros pacientes que recibieron altas dosis de citarabina (2g/m2 por día) a través de la infusión continua con daunorrubicina y BROMADENE® para el tratamiento a la leucemia mielogenosa aguda recidivante murieron de ARDS temprano en el curso de la terapia. Por eso este específico régimen de administración concomitante con altas dosis de citarabina (2g/m2 por día) a través de la infusión continua de 24 horas no es recomendado. Análisis de laboratorio: Se deben controlar con frecuencia los recuentos sanguíneos completos a lo largo del tratamiento con BROMADENE®. Eventos adversos gastrointestinales: El tratamiento con BROMADENE® puede ocasionar náuseas, diarrea, constipación y vómitos (ver Reacciones adversas) y a veces se requiere el uso de antieméticos y antidiarreicos. Para evitar la deshidratación se deberá administrar un reemplazo electrolítico y de fluidos. Trombocitopenia: BROMADENE® está asociado con la trombocitopenia (ver Reacciones adversas). Las plaquetas se encontraron más bajas el Día 11 de cada ciclo del tratamiento con BROMADENE® y por lo general se recuperaron hasta el valor basal para el próximo ciclo. El patrón cíclico de la disminución y recuperación del recuento de plaquetas se mantuvo consistente durante los 8 ciclos de la dosificación de dos veces por semana y no hubo evidencia de trombocitopenia acumulativa. El nadir del recuento de plaquetas medio medido fue aproximadamente 40% de la admisión. La severidad de la trombocitopenia relacionada con el recuento de plaquetas previo al tratamiento se muestra en la Tabla 4. En el estudio Fase 3 de mieloma múltiple, la incidencia de eventos significativos de hemorragias (≥Grado 3) fue similar tanto en el grupo tratado con BROMADENE® (%) como en el grupo tratado con dexametasona (5%). Se deben controlar los recuentos de plaquetas antes de cada dosis de BROMADENE®. La terapia con BROMADENE® se debe suspender cuando el recuento de plaquetas sea < 25.000/pL y se debe reiniciar con una dosis reducida (ver Dosificación y Reacciones adversas). Ha habido informes de hemorragia intracerebral y gastrointestinal asociada con BROMADENE®. Se pueden considerar las transfusiones.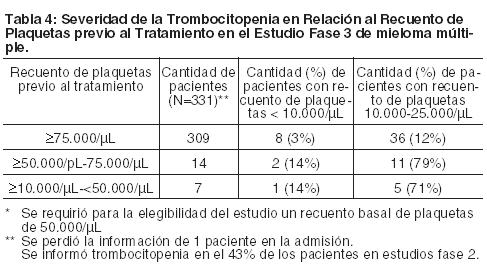
Síndrome de Lisis Tumoral: Debido a que BROMADENE® es un agente citotóxico y puede matar rápidamente las células malignas, puede ocurrir complicaciones del síndrome de lisis tumoral. Los pacientes con riesgo del síndrome de lisis tumoral son aquellos con una alta carga tumoral previa al tratamiento. Estos pacientes se deben controlar minuciosamente y se deben tomar las precauciones adecuadas. Eventos adversos hepáticos: Se han reportado raros casos de insuficiencia hepática aguda en pacientes que reciben múltiples medicaciones concomitantes y con serias condiciones médicas subyacentes. Otros eventos hepáticos reportados incluyeron el aumento