BIKTARVY®
GADOR
Grupo farmacoterapéutico: Antivirales para uso sistémico; antivirales para el tratamiento de infecciones por VIH y combinaciones. Código ATC: J05AR20.
Composición.
Cada comprimido recubierto de BIKTARVY® contiene: Bictegravir (equivalente a 52.45 mg de Bictegravir sódico) 50 mg, Emtricitabina 200 mg, Tenofovir alafenamida (equivalente a 28.04 mg de Tenofovir alafenamida fumarato) 25 mg. Excipientes: Celulosa microcristalina 361.34 mg, Croscarmelosa sódica 49.54 mg, Estearato de magnesio 8.63 mg, Alcohol polivinílico1) 8.4 mg, Dióxido de titanio1) 4.6 mg, Polietilenglicol1 4.2 mg, Talco1) 3.1 mg, Óxido de hierro rojo (CIN°77491) 1) 0.5 mg, Óxido de Hierro negro (CIN°77499) 1) 0.1 mg. 1) Se refiere a los componentes del Opadry II marrón 85F165072. BIKTARVY® es un medicamento libre de gluten.
Farmacología.
Descripción: Los comprimidos de BIKTARVY® contienen bictegravir 50 mg, emtricitabina 200 mg, tenofovir alafenamida 25 mg y los siguientes excipientes: Celulosa microcristalina, croscarmelosa sódica, estearato de magnesio, alcohol polivinílico, dióxido de titanio, polietilenglicol, talco, óxido de hierro rojo, óxido de hierro negro. Los comprimidos recubiertos de BIKTARVY® son marcados en una de las caras del comprimido con "GSI" y en la otra cara del comprimido con "9883". Los comprimidos de BIKTARVY® se administran por vía oral. Farmacología clínica: Mecanismo de acción: Bictegravir es un inhibidor de la transferencia de las hebras de la integrasa (INI) que se une al sitio activo de la integrasa y bloquea la transferencia de hebras para la integración del ácido desoxirribonucleico (ADN) retroviral, que es esencial para el ciclo de replicación del VIH. Bictegravir muestra actividad frente al VIH-1 y al VIH-2. Emtricitabina es un inhibidor de la transcriptasa inversa análogo de nucleósidos (ITIAN) y un análogo nucleósido de 2'-desoxicitidina. Emtricitabina se fosforila por enzimas celulares para formar emtricitabina trifosfato. Emtricitabina trifosfato inhibe la replicación del VIH a través de su incorporación en el ADN viral mediante la transcriptasa inversa (TI) del VIH, lo que ocasiona la finalización de la cadena de ADN. Emtricitabina muestra actividad frente al VIH-1, VIH-2 y VHB. Tenofovir alafenamida es un inhibidor de la transcriptasa inversa análogo de nucleótido (ITIAN) y un profármaco fosfonamidato de tenofovir (análogo de 2'-desoxiadenosina monofosfato). Tenofovir alafenamida es permeable en las células y, debido a su mayor estabilidad plasmática y activación intracelular mediante hidrólisis por la catepsina A, tenofovir alafenamida es más eficaz que tenofovir disoproxilo fumarato en cuanto a la carga de tenofovir en las células mononucleares de sangre periférica (PBMC, peripheral blood mononuclear cells) (incluyendo linfocitos y otras células diana del VIH) y los macrófagos. Tenofovir intracelular se fosforila a continuación al metabolito farmacológicamente activo tenofovir difosfato. Tenofovir difosfato inhibe la replicación del VIH mediante su incorporación en el ADN viral por la TI del VIH, lo que produce la interrupción de la cadena de ADN. Tenofovir muestra actividad frente al VIH-1, VIH-2 y VHB. Propiedades farmacocinéticas: Absorción: Bictegravir se absorbe después de la administración oral y alcanza concentraciones plasmáticas máximas 2,0 a 4,0 horas después de la administración de B/F/TAF. En condiciones de ayuno, la administración de B/F/TAF con ingesta de poca grasa (~ 600 kcal, 27 % de grasa) o de mucha grasa (~ 800 kcal, 50 % de grasa) dio como resultado un aumento del AUC de bictegravir (24 %). Este modesto cambio no se considera clínicamente significativo y B/F/TAF se puede administrar con o sin alimentos. Después de la administración oral de B/F/TAF con o sin alimentos en adultos infectados con VIH-1, las medias de los parámetros farmacocinéticos de múltiples administraciones (CV%) de bictegravir fueron Cmáx = 6,15 mg/ml (22,9 %), AUCtau = 102 mg < 195 > h/ml (26,9 %) y Cvalle = 2,61 mg/ml (35,2 %). Emtricitabina se absorbe de forma rápida y extensa después de la administración oral con unas concentraciones plasmáticas máximas 1,5 a 2,0 horas después de la administración de B/F/TAF. La media de biodisponibilidad absoluta de emtricitabina en cápsulas duras de 200 mg fue del 93 %. La exposición sistémica de emtricitabina no se vio afectada cuando emtricitabina se administró con alimentos y B/F/TAF se puede administrar con o sin alimentos. Después de la administración oral de B/F/TAF con o sin alimentos en adultos infectados por el VIH-1, las medias de los parámetros farmacocinéticos de múltiples administraciones (CV%) de emtricitabina fueron Cmáx = 2,13 mg/ml (34,7 %), AUCtau = 12,3 mg < 195 > h/ml (29,2 %) y Cvalle = 0,096 mg/ml (37,4 %). Tenofovir alafenamida se absorbe de forma rápida después de la administración oral, alcanzándose las concentraciones plasmáticas máximas 0,5 a 2,0 horas después de la administración de B/F/TAF. En condiciones en ayunas, la administración de tenofovir alafenamida con ingesta de poca grasa (~ 600 kcal, 27 % de grasa) y de mucha grasa (~ 800 kcal, 50 % de grasa) dio como resultado un aumento del AUCúlt de un 48 % y un 63 %, respectivamente. Estos modestos cambios no se consideran clínicamente significativos y B/F/TAF se puede administrar con o sin alimentos. Después de la administración oral de B/F/TAF con o sin alimentos en adultos infectados por el VIH-1, los parámetros farmacocinéticos medios de administraciones múltiples (CV%) de tenofovir alafenamida fueron Cmáx = 0,121 mg/ml (15,4 %) y AUCtau = 0,142 mg < 195 > h/ml (17,3 %). Distribución: La unión in vitro de bictegravir a las proteínas plasmáticas humanas fue > 99 % (fracción libre ~ 0,25 %). El cociente entre la concentración de bictegravir in vitro en sangre humana y plasma fue 0,64. La unión in vitro de emtricitabina a proteínas plasmáticas humanas fue < 4 % y resultó independiente de la concentración en el rango de 0,02 a 200 mg/ml. A la concentración plasmática máxima, la relación de la concentración media del fármaco entre plasma y sangre fue ~ 1,0, y la relación de concentración media del fármaco entre semen y plasma fue ~ 4,0. La unión in vitro de tenofovir a proteínas plasmáticas humanas es inferior al 0,7 % y es independiente de la concentración en el rango de 0,01 25 mg/ml. La unión exvivo de tenofovir alafenamida a proteínas plasmáticas en las muestras recogidas durante los estudios clínicos fue de aproximadamente el 80 %. Biotransformación: El metabolismo es la principal vía de eliminación para bictegravir en seres humanos. Los estudios de fenotipado in vitro mostraron que bictegravir se metaboliza principalmente por CYP3A y UGT1A1. Después de la administración oral de una dosis única de [14C]-bictegravir, ~ 60 % de la dosis en las heces incluyó el precursor inalterado, el conjugado de desfluorohidroxi-BIC-cisteína y otros metabolitos oxidativos menores. En orina se recogió el 35 % de la dosis, y consistió principalmente en el glucurónido de bictegravir y en otros metabolitos oxidativos menores y sus conjugados de fase II. El aclaramiento renal del precursor inalterado fue mínimo. Tras la administración de [14C]-emtricitabina, se obtuvo una recuperación completa de la dosis de emtricitabina en la orina (~ 86 %) y las heces (~ 14 %). El 13 % de la dosis se recuperó en la orina en forma de tres aparentes metabolitos. La biotransformación de emtricitabina comprende la oxidación del radical tiólico, para dar los diastereómeros 3'-sulfóxido (~ 9 % de la dosis), y la conjugación con el ácido glucurónico, para formar el 2'-O-glucurónido (~ 4 % de la dosis). No hubo otros metabolitos identificables. El metabolismo es la ruta de eliminación principal de tenofovir alafenamida en los seres humanos, suponiendo > 80% de una dosis oral. Los estudios in vitro han mostrado que tenofovir alafenamida se metaboliza a tenofovir (metabolito principal) por medio de la catepsina A en las PBMC (incluyendo linfocitos y otras células diana del VIH) y los macrófagos, y por medio de la carboxilesterasa-1 en los hepatocitos. In vivo, tenofovir alafenamida se hidroliza en las células para formar tenofovir (metabolito principal), que se fosforila al metabolito activo, tenofovir difosfato. En los estudios clínicos humanos, una dosis oral de 25 mg de tenofovir alafenamida dio lugar a unas concentraciones de tenofovir difosfato más de 4 veces superiores en las PBMC y más del 90 % de las concentraciones menores de tenofovir en plasma, en comparación con una dosis oral de 300 mg de tenofovir disoproxil fumarato. Eliminación: Bictegravir se elimina principalmente por metabolismo hepático. La excreción renal de bictegravir intacto es una vía menor (~ 1 % de la dosis). La semivida de bictegravir en plasma fue de 17,3 horas. Emtricitabina se excreta principalmente a través de los riñones, por filtración glomerular y por secreción tubular activa. La semivida de emtricitabina en plasma fue de aproximadamente 10 horas. Tenofovir alafenamida se elimina después de la metabolización a tenofovir. Tenofovir alafenamida y tenofovir tienen una mediana de semivida plasmática de 0,51 y 32,37 horas, respectivamente. Tenofovir se elimina a través de los riñones por filtración glomerular y secreción tubular activa. La excreción renal de tenofovir alafenamida intacto es una vía menor, eliminándose en orina menos del 1 % de la dosis. Linealidad: La farmacocinética de múltiples administraciones de bictegravir es proporcional a la dosis en un intervalo de dosis de 25 a 100 mg. La farmacocinética de múltiples administraciones de emtricitabina es proporcional a la dosis en el intervalo de 25 a 200 mg. Las exposiciones a tenofovir alafenamida son proporcionales a la dosis en el intervalo de 8 mg a 125 mg. Otras poblaciones especiales: Insuficiencia renal: No se observaron diferencias clínicamente relevantes en la farmacocinética de bictegravir, tenofovir alafenamida, o tenofovir entre los sujetos sanos y los pacientes con insuficiencia renal grave (ClCr estimado < 30 ml/min). No existen datos farmacocinéticos de bictegravir o tenofovir alafenamida en pacientes con aclaramiento de creatinina inferior a 15 ml/min. La exposición sistémica media a emtricitabina fue mayor en pacientes con insuficiencia renal grave (CrCl < 30 ml/min) (33,7 mg < 195 > h/ ml) que en sujetos con función renal normal (11,8 mg < 195 > h/ml). Insuficiencia hepática No se observaron cambios clínicamente relevantes en la farmacocinética de bictegravir en sujetos con insuficiencia hepática moderada. La farmacocinética de emtricitabina no se ha estudiado en sujetos con insuficiencia hepática; sin embargo, emtricitabina no sufre un metabolismo significativo a través de las enzimas hepáticas, por lo que la repercusión de la insuficiencia hepática debería ser escasa. No se observaron cambios clínicamente relevantes en la farmacocinética de tenofovir alafenamida o en su metabolito tenofovir en los pacientes con insuficiencia hepática leve, moderada o grave. Edad, sexo y raza: La farmacocinética de bictegravir, emtricitabina y tenofovir no se ha evaluado por completo en los pacientes de edad avanzada (≥65 años de edad). Los análisis de población en los que se utilizaron datos farmacocinéticos combinados de ensayos en adultos no identificaron diferencias clínicamente relevantes debidas a la edad, sexo o raza en las exposiciones de bictegravir, emtricitabina o tenofovir alafenamida o tenofovir alafenamida. Microbiología: Actividad antiviral in vitro: La actividad antiviral de emtricitabina frente a aislados clínicos y de laboratorio del VIH-1 se evaluó en líneas celulares linfoblastoides, PBMC, células monocíticas/macrofágicas primarias y linfocitos T CD4+. Los valores de la concentración eficaz 50 % (CE50) de bictegravir oscilaron entre < 0,05 y 6,6 nM. La CE95 ajustada a proteínas de bictegravir fue 361 nM (0,162 microgramos/ml) para la cepa salvaje del virus VIH-1. Bictegravir mostró actividad antiviral en cultivos celulares frente al grupo del VIH-1 (M, N y O), incluidos los subtipos A, B, C, D, E, F y G (con valores de CE50 de < 0,05 a 1,71 nM), y frente al VIH-2 (CE50 = 1,1 nM). La actividad antiviral de emtricitabina frente a aislados clínicos y de laboratorio del VIH-1 se evaluó en líneas celulares linfoblastoides, en la línea celular MAGI CCR5 y en PBMC. Los valores de CE50 de emtricitabina oscilaron entre 0,0013 y 0,64 mM. Emtricitabina mostró actividad antiviral en cultivos celulares frente a los clados del VIH-1 A, B, C, D, E, F y G (valores de CE50 de 0,007 a 0,075 mM) y presentó actividad específica frente al VIH-2 (valores de CE50 de 0,007 a 1,5 mM). La actividad antiviral de tenofovir alafenamida frente a aislados clínicos y de laboratorio del subtipo B del VIH-1 se evaluó en líneas celulares linfoblastoides, PBMC, células monocíticas/macrofágicas primarias y linfocitos T CD4. Los valores de CE50 de tenofovir alafenamida oscilaron entre 2,0 y 14,7 nM. Tenofovir alafenamida mostró actividad antiviral en cultivos celulares frente a todos los grupos del VIH-1 (M, N y O), incluyendo los subtipos A, B, C, D, E, F y G (valores de CE50 de 0,10 a 12,0 nM) y presentó actividad específica frente al VIH-2 (valores de CE50 de 0,91 a 2,63 nM). Resistencia: In vitro: Se han seleccionado aislados de VIH-1 con sensibilidad reducida a bictegravir en cultivo celular. En una selección se observaron las sustituciones de los aminoácidos M50I y R263K, y la sensibilidad fenotípica a bictegravir se redujo en 1,3; 2,2 y 2,9 veces para M50I, R263K y M50I + R263K, respectivamente. En una segunda selección se observaron las sustituciones de los aminoácidos T66I y S153F, y la sensibilidad fenotípica a bictegravir se desplazó hasta 0,4; 1,9 y 0,5 veces para T66I, S153F y T66I + S153F, respectivamente. Los aislados del VIH-1 con sensibilidad reducida a emtricitabina han sido seleccionados en cultivo celular y tenían mutaciones M184V/I en la TI del VIH-1. Los aislados del VIH-1 con sensibilidad reducida a tenofovir alafenamida han sido seleccionados en cultivo celular y portaban la mutación K65R en la TI del VIH-1; además, se ha observado transitoriamente una mutación K70E en la TI del VIH-1. Los aislados del VIH-1 con la mutación K65R tienen bajo nivel de sensibilidad reducida a abacavir, emtricitabina, tenofovir y lamivudina. En estudios de selección de resistencia a medicamentos in vitro con tenofovir alafenamida no se ha observado desarrollo de alto nivel de resistencia después del cultivo prolongado. En pacientes que no habían recibido tratamiento previo (estudios GS-US-380-1489 y GS-US-380-1490) y virológicamente suprimidos (estudios GS-US-380-1844 y GS-US-380-1878), ningún paciente que recibió BIKTARVY® tuvo VIH-1 con resistencia genotípica o fenotípica emergente a bictegravir, emtricitabina o tenofovir alafenamida en la población de análisis de resistencia (n=13 con ARN del VIH-1 ≥ 200 copias/ml en el momento de confirmarse el fracaso virológico, la semana 48 o el abandono temprano del fármaco del estudio). En el momento del ingreso en el estudio, seis pacientes que no habían recibido tratamiento previo y un paciente virológicamente suprimido que recibió BIKTARVY® presentaron mutaciones preexistentes asociadas a resistencia a INI (6 sujetos con T97A y un sujeto que no había recibido tratamiento previo con Q148H + G140S); todos tuvieron ARN del VIH-1 < 50 copias/ml en la semana 48. Resistencia cruzada: La sensibilidad de bictegravir se analizó en 64 aislados clínicos resistentes a INI (20 con sustituciones simples y 44 con 2 o más sustituciones). De estos, todos los aislados mutantes simples y dobles carentes de Q148H/K/R y 10 de 24 aislados con Q148H/K/R con sustituciones asociadas a resistencia adicional a INI tenían una sensibilidad reducida ≤2,5 veces a bictegravir; se encontró una sensibilidad reducida > 2,5 veces a bictegravir en 14 de los 24 aislados que contenían sustituciones de G140A/C/S y Q148H/R/K en la integrasa. De ellos, 9 de los 14 cultivos aislados tenían mutaciones adicionales en L74M, T97A o E138A/K. Además, las líneas celulares mutantes con G118R y T97A + G118R tenían una sensibilidad reducida 3,4 y 2,8 veces a bictegravir, respectivamente. La relevancia de estos datos de resistencia cruzada in vitro aún está por establecerse en la práctica clínica. Bictegravir demostró una actividad antiviral equivalente frente a 5 clones mutantes del VIH-1 resistentes a ITINN, 3 resistentes a ITIAN y 4 resistentes a IP en comparación con la cepa salvaje. Los virus resistentes a emtricitabina con la sustitución M184V/I mostraron resistencia cruzada con lamivudina, pero conservaron la sensibilidad a didanosina, estavudina, tenofovir y zidovudina. Las mutaciones K65R y K70E tienen como resultado una sensibilidad reducida a abacavir, didanosina, lamivudina, emtricitabina y tenofovir, pero conservan la sensibilidad a zidovudina. El VIH-1 resistente a múltiples nucleósidos con una mutación de inserción doble T69S o con un complejo de mutación Q151M que incluye K65R, mostró una sensibilidad reducida a tenofovir alafenamida.
Indicaciones.
BIKTARVY® está indicado para el tratamiento de adultos infectados por el virus de la inmunodeficiencia humana de tipo 1 (VIH-1) sin resistencia viral actual o previa a los inhibidores de la integrasa, a emtricitabina o a tenofovir
Dosificación.
El tratamiento debe ser iniciado por un médico con experiencia en el tratamiento de la infección por el VIH. Posología: Un comprimido que se debe tomar una vez al día. Dosis omitidas: Si el paciente omite una dosis de BIKTARVY® en el plazo de 18 horas desde la hora normal de administración, debe tomar BIKTARVY® lo antes posible y continuar con la pauta habitual de administración. Si el paciente omite una dosis de BIKTARVY® durante más de 18 horas, no debe tomar la dosis omitida y simplemente debe continuar con la pauta habitual de administración. Si el paciente vomita en el plazo de 1 hora después de tomar BIKTARVY®, debe tomar otro comprimido. Si el paciente vomita transcurrida 1 hora de haber tomado BIKTARVY®, no necesita tomar otra dosis de BIKTARVY® hasta la próxima dosis habitual programada. Pacientes de edad avanzada: Existen datos limitados sobre el uso de BIKTARVY® en pacientes a partir de 65 años. No se requiere un ajuste de la dosis de BIKTARVY® en pacientes de edad avanzada. Insuficiencia renal: No se requiere un ajuste de la dosis de BIKTARVY® en pacientes con un aclaramiento de creatinina estimado (CrCl) ≥ 30 ml/min. No se recomienda iniciar el tratamiento con BIKTARVY® en pacientes con CrCl inferior a 30 ml/min, ya que no se dispone de datos suficientes sobre el uso de BIKTARVY® en esta población. Insuficiencia hepática: No se requiere un ajuste de la dosis de BIKTARVY® en pacientes con insuficiencia hepática leve (clase A de Child Pugh) o moderada (clase B de Child Pugh). No se ha estudiado BIKTARVY® en pacientes con insuficiencia hepática grave (clase C de Child Pugh); por tanto, no se recomienda el uso de BIKTARVY® en pacientes con insuficiencia hepática grave. Población pediátrica: No se ha establecido todavía la seguridad y eficacia de BIKTARVY® en niños menores de 18 años de edad. No se dispone de datos. Forma de administración: Vía oral. BIKTARVY® se puede tomar con o sin alimentos: El comprimido recubierto no se debe masticar, machacar ni partir. Formas farmacéuticas y concentraciones: Los comprimidos recubiertos de BIKTARVY® son marcados en una de las caras del comprimido con «GSI > > y en la otra cara del comprimido con «9883 > > . Los comprimidos de BIKTARVY® contienen bictegravir 50 mg, emtricitabina 200 mg y tenofovir alafenamida 25 mg.
Contraindicaciones.
Hipersensibilidad a los principios activos o a alguno de los excipientes. Administración concomitante con rifampicina y hierba de San Juan (Hypericum perforatum) (ver sección Interacciones).
Reacciones adversas.
Resumen del perfil de seguridad: La evaluación de las reacciones adversas se basa en datos de seguridad de todos los estudios de fase II y III realizados con BIKTARVY® y en la experiencia postcomercial. En estudios clínicos de pacientes que no habían recibido tratamiento previo tratados con BIKTARVY® durante 48 semanas, las reacciones adversas notificadas con más frecuencia fueron cefalea (5 %), diarrea (5 %) y náuseas (4 %). Tabla de reacciones adversas: Las reacciones adversas de la Tabla 4 se muestran según el sistema de clasificación de órganos y por orden de frecuencia. Las frecuencias se definen como sigue: frecuentes ≥ 1/100 a < 1/10) y poco frecuentes ≥ 1/1.000 a < 1/100).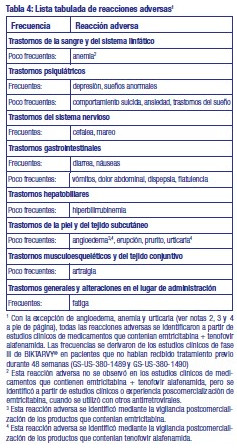
Descripción de las reacciones adversas seleccionadas: Parámetros metabólicos: El peso y los niveles de glucosa y lípidos en sangre pueden aumentar durante el tratamiento antirretroviral. Síndrome de reconstitución inmunitaria: Al inicio de la TARC, en los pacientes infectados por el VIH con deficiencia inmunitaria grave, puede aparecer una reacción inflamatoria frente a infecciones oportunistas latentes o asintomáticas. Se han notificado también trastornos autoinmunitarios (como la enfermedad de Graves y hepatitis autoinmune); no obstante, el tiempo hasta el inicio notificado es más variable y estos acontecimientos se pueden producir muchos meses después del inicio del tratamiento. Osteonecrosis: Se han notificado casos de osteonecrosis, especialmente en pacientes con factores de riesgo generalmente reconocidos, enfermedad avanzada por el VIH o exposición prolongada a la TARC. Se desconoce la frecuencia de esta reacción adversa. Cambios en la creatinina sérica: Se ha demostrado que bictegravir aumenta la creatinina sérica debido a la inhibición de la secreción tubular de creatinina, sin embargo, estos cambios no se consideran clínicamente relevantes ya que no reflejan un cambio en la tasa de filtración glomerular. Los aumentos en la creatinina sérica ocurrieron en la semana 4 de tratamiento y se mantuvieron estables hasta la semana 48. En los estudios GS-US-380-1489 y GS-US-380-1490, la mediana (Q1, Q3) de la creatinina sérica aumentó en 0,10 (0,03; 0,17) mg/dl; 0,11 (0,03; 0,18) mg/dl, y 0,11 (0,04; 0,19) mg/dl desde el inicio hasta la semana 48 en los grupos de BIKTARVY®, abacavir/dolutegravir/lamivudina, y dolutegravir + emtricitabina/tenofovir alafenamida, respectivamente. No hubo abandonos debido a acontecimientos adversos renales hasta la semana 48 en los estudios clínicos de BIKTARVY®. Cambios en la bilirrubina: En los estudios GS-US-380-1489 y GS-US-380-1490 se observaron aumentos de la bilirrubina total en el 12 % de los pacientes que no habían recibido tratamiento previo y que se trataron con BIKTARVY® durante 48 semanas. Los incrementos fueron principalmente de grado 1 (9 %) y de grado 2 (3 %) (1,0 a 2,5 × límite superior de la normalidad [LSN]), y no se asociaron a reacciones adversas hepáticas ni a otros resultados anómalos de laboratorio relacionados con el hígado. No hubo abandonos debido a acontecimientos adversos hepáticos hasta la semana 48 en los estudios clínicos de BIKTARVY®. Otras poblaciones especiales: Pacientes coinfectados por el virus de la hepatitis B: En 16 adultos coinfectados por VIH/VHB a quienes se administró BIKTARVY® (8 adultos infectados por VIH/VHB que no habían recibido tratamiento previo en el estudio GS-US-380-1490; 8 adultos suprimidos infectados por VIH/VHB en el estudio GS-US-380-1878), el perfil de seguridad de BIKTARVY® fue similar al de los pacientes monoinfectados por VIH-1. Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia al siguiente link: https://www.argentina.gob.ar/anmat/farmacovigilancia/ notificanos/eventosadversos y/o al Departamento de Farmacovigilancia de GADOR SA, enviando un correo electrónico a farmacovigilancia@ gador.com., o telefónicamente al 0800 220 2273.
Advertencias.
A pesar de que se ha probado que una supresión viral eficaz con un tratamiento antirretroviral reduce sustancialmente el riesgo de transmisión sexual, no se puede excluir un riesgo residual. Se deben tomar precauciones, conforme a las directrices nacionales, para prevenir la transmisión. Pacientes coinfectados por el VIH y el virus de las hepatitis B o C: Los pacientes con hepatitis B o C crónica tratados con terapia antirretroviral tienen un riesgo mayor de padecer reacciones adversas hepáticas graves y potencialmente mortales. Los datos de seguridad y eficacia de BIKTARVY® en pacientes coinfectados por el VIH-1 y el virus de la hepatitis C (VHC) son limitados. BIKTARVY® contiene tenofovir alafenamida, que es un componente activo contra el virus de la hepatitis B (VHB). El abandono del tratamiento con BIKTARVY® en pacientes coinfectados por VIH y VHB se puede asociar con exacerbaciones agudas graves de la hepatitis. Se debe efectuar un seguimiento estrecho, clínico y de laboratorio en pacientes coinfectados por VIH y VHB que abandonan el tratamiento con BIKTARVY®, durante al menos varios meses después de suspender el tratamiento. Enfermedad hepática: No se ha establecido la seguridad y eficacia de BIKTARVY® en pacientes con trastornos hepáticos significativos subyacentes. Los pacientes con insuficiencia hepática preexistente, incluida la hepatitis crónica activa, tienen una frecuencia aumentada de alteración de la función hepática durante la terapia antirretroviral combinada (TARC) y se deben controlar de acuerdo con las prácticas habituales. Si hay evidencia de empeoramiento de la enfermedad hepática en dichos pacientes, se tendrá que considerar la interrupción o abandono del tratamiento. Peso y parámetros metabólicos: Durante el tratamiento antirretroviral se puede producir un aumento en el peso y en los niveles de glucosa y lípidos en sangre. Tales cambios podrían estar relacionados en parte con el control de la enfermedad y en parte con el estilo de vida. Para los lípidos, hay en algunos casos evidencia de un efecto del tratamiento, mientras que para la ganancia de peso no hay una evidencia sólida que relacione esto con un tratamiento en particular. Para monitorizar los niveles de lípidos y de glucosa en sangre se hace referencia a las guías de tratamiento de la infección por el VIH. Los trastornos lipídicos se deben tratar como se considere clínicamente apropiado. Disfunción mitocondrial después de la exposición in útero: Los análogos de nucleós(t)idos pueden afectar a la función mitocondrial en un grado variable, siendo más marcado con estavudina, didanosina y zidovudina. Existen informes de disfunción mitocondrial en lactantes VIH negativo expuestos in utero y/o post-parto a análogos de nucleósidos; estos concernieron predominantemente al tratamiento con pautas que contenían zidovudina. Las principales reacciones adversas notificadas fueron trastornos hematológicos (anemia, neutropenia) y trastornos metabólicos (hiperlactatemia, hiperlipasemia). Estas reacciones fueron a menudo transitorias. Se han notificado de forma rara trastornos neurológicos de aparición tardía (hipertonía, convulsión, comportamiento anormal). Actualmente no se sabe si estos trastornos neurológicos son transitorios o permanentes. Estos hallazgos se deben considerar en cualquier niño expuesto in utero a análogos de nucleós(t)idos que presenten hallazgos clínicos graves de etiología desconocida, especialmente hallazgos neurológicos. Estos hallazgos no afectan a las recomendaciones nacionales actuales para utilizar tratamiento antirretroviral en mujeres embarazadas para prevenir la transmisión vertical del VIH. Síndrome de reconstitución inmunitaria: Cuando se instaura una TARC en pacientes infectados por el VIH con deficiencia inmunitaria grave puede aparecer una respuesta inflamatoria frente a patógenos oportunistas residuales o asintomáticos y provocar situaciones clínicas graves, o un empeoramiento de los síntomas. Normalmente estas reacciones se han observado en las primeras semanas o meses después del inicio de la TARC. Algunos ejemplos relevantes de estas reacciones incluyen: retinitis por citomegalovirus, infecciones micobacterianas generalizadas o localizadas y neumonía por Pneumocystis jirovecii. Se debe evaluar cualquier síntoma inflamatorio y establecer un tratamiento cuando sea necesario. Se han notificado también trastornos autoinmunitarios (como la enfermedad de Graves y hepatitis autoinmune) en caso de reconstitución inmunitaria; no obstante, el tiempo hasta el inicio notificado es más variable y estos acontecimientos se pueden producir muchos meses después del inicio del tratamiento. Infecciones oportunistas: Se debe informar a los pacientes que ni BIKTARVY® ni ningún otro tratamiento antirretroviral curan la infección por el VIH, y que pueden presentar infecciones oportunistas y otras complicaciones de la infección por el VIH. Por lo tanto, los pacientes deben permanecer bajo estrecha observación clínica por médicos con experiencia en el tratamiento de pacientes con enfermedades asociadas al VIH. Osteonecrosis: Se han notificado casos de osteonecrosis, especialmente en pacientes con infección avanzada por el VIH y/o con exposición prolongada a la TARC, aunque se considera que la etiología es multifactorial (incluyendo uso de corticosteroides, consumo de alcohol, inmunodepresión grave, índice de masa corporal elevado). Se debe aconsejar a los pacientes de que consulten al médico si experimentan molestias o dolor articular, rigidez articular o dificultad para moverse. Nefrotoxicidad: No se puede excluir un posible riesgo de nefrotoxicidad resultante de la exposición crónica a niveles bajos de tenofovir debido a la administración de tenofovir Alafenamida. Administración concomitante de otros medicamentos: BIKTARVY® no se debe administrar de forma concomitante y simultánea con antiácidos que contengan magnesio/aluminio, o suplementos de hierro en condiciones de ayuno. BIKTARVY® se debe administrar al menos 2 horas antes de la administración de antiácidos, o tomarse con alimentos 2 horas después de la administración de antiácidos que contengan magnesio y/o aluminio. BIKTARVY® se debe administrar al menos 2 horas antes de la administración de suplementos de hierro, o tomarse los dos juntos con alimentos. No se recomienda la administración concomitante de algunos medicamentos con BIKTARVY®: atazanavir, boceprevir, carbamazepina, ciclosporina (vía intravenosa u oral), oxcarbazepina, fenobarbital, fenitoína, rifabutina, rifapentina o sucralfato. BIKTARVY® no se debe administrar de forma concomitante con otros medicamentos antirretrovirales. Interacción con otros medicamentos y otras formas de interacción: Los estudios de interacciones se han realizado solo en adultos. BIKTARVY® no se debe administrar de forma concomitante con medicamentos que contengan tenofovir alafenamida, tenofovir disoproxilo, lamivudina o adefovir dipivoxil utilizados para el tratamiento de la infección por el VHB. Bictegravir: Bictegravir es un sustrato de CYP3A y UGT1A1. La administración concomitante de bictegravir y medicamentos que inducen fuertemente tanto CYP3A como UGT1A1, tales como rifampicina o la hierba de San Juan, pueden disminuir significativamente las concentraciones plasmáticas de bictegravir, lo cual puede dar como resultado una pérdida del efecto terapéutico de BIKTARVY® y el desarrollo de resistencias; por lo tanto, la administración concomitante está contraindicada. La administración concomitante de bictegravir con medicamentos que inhiben fuertemente tanto CYP3A como UGT1A1, tales como atazanavir, pueden aumentar mucho las concentraciones plasmáticas de bictegravir, por lo que no se recomienda la administración concomitante. Bictegravir es a la vez sustrato de P-gp y BCRP (breast cancer resistance protein). La relevancia clínica de esta característica no está establecida. Por lo tanto, se recomienda precaución cuando se combina bictegravir con medicamentos que se sabe que inhiben P-gp y/o BCRP (p.ej. macrólidos, ciclosporina, verapamilo, dronedarona, glecaprevir/pibrentasvir) (ver también la tabla a continuación). Bictegravir inhibe el transportador de cationes orgánicos 2 (OCT2, organic cation transporter 2) y el transportador de expulsión de múltiples fármacos y de toxinas 1 (MATE1, multidrug and toxin _xtrusión transporter 1) in vitro. La administración concomitante de BIKTARVY® con el sustrato de OCT2 y MATE1 metformina no produjo un aumento clínicamente significativo en la exposición a metformina. BIKTARVY® se puede administrar de forma conjunta con sustratos de OCT2 y MATE1. Bictegravir no es un inhibidor o inductor de CYP in vivo. Emtricitabina: Los estudios in vitro y clínicos de interacciones medicamentosas farmacocinéticas han mostrado que el potencial de interacciones mediadas por CYP entre emtricitabina y otros medicamentos es bajo. La administración concomitante de emtricitabina con medicamentos que se eliminan mediante secreción tubular activa puede aumentar las concentraciones de emtricitabina y/o del medicamento administrado de forma concomitante. Los medicamentos que reducen la función renal pueden aumentar las concentraciones de emtricitabina. Tenofovir alafenamida: Tenofovir alafenamida se transporta por la glicoproteína P (P gp) y la proteína de resistencia de cáncer de mama (BCRP). La administración concomitante de BIKTARVY® con medicamentos que afectan fuertemente a la actividad de la P gp y la BCRP puede conducir a cambios en la absorción de tenofovir alafenamida. Se prevé que los medicamentos que inducen la actividad de la P gp (p. ej., rifabutina, carbamazepina, fenobarbital) reduzcan la absorción de tenofovir alafenamida, dando lugar a una concentración plasmática reducida de tenofovir alafenamida, lo que puede producir una pérdida del efecto terapéutico de BIKTARVY® y la aparición de resistencias. La administración concomitante de BIKTARVY® con otros medicamentos que inhiben a la P gp y a la BCRP puede aumentar la absorción y la concentración plasmática de tenofovir alafenamida. Tenofovir alafenamida no es un inhibidor o inductor de CYP3A in vivo. Otras interacciones: Las interacciones entre BIKTARVY® o sus componentes individuales y medicamentos administrados de forma concomitante se enumeran en la Tabla 1 a continuación (el aumento se indica como "↑", la disminución como "↓" y la ausencia de cambios, como "↔"; todos los límites sin efecto son 70 %-143 %). (Ver Tabla 1).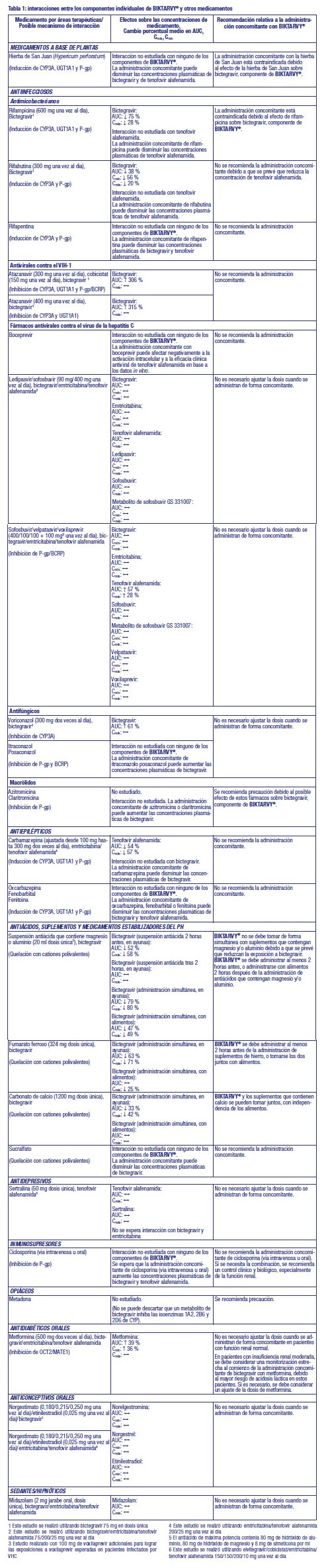
Estudios realizados con otros medicamentos: De acuerdo a los estudios de interacciones medicamentosas realizados con BIKTARVY® o con los componentes de BIKTARVY®, no se esperan interacciones clínicamente significativas con: amlodipina, atorvastatina, buprenorfina, drospirenona, famciclovir, famotidina, fluticasona, naloxona, norbuprenorfina, omeprazol o rosuvastatina. Toxicología preclínica: Bictegravir no fue mutagénico ni clastogénico en los estudios convencionales de genotoxicidad. Bictegravir no fue carcinogénico en un estudio de 6 meses en ratones transgénicos rasH2 (en dosis de hasta 100 mg/kg/día en machos y 300 mg/kg/día en hembras, lo que dio como resultado exposiciones aproximadamente 15 y 23 veces mayores en machos y hembras, respectivamente, que la exposición en seres humanos en la dosis recomendada para estos), ni en un estudio de 2 años en ratas (en dosis de hasta 300 mg/kg/día, lo que resultó en exposiciones de aproximadamente 31 veces la exposición en humanos). Los estudios de bictegravir en monos revelaron que el hígado es el principal órgano diana afectado por la toxicidad. La toxicidad hepatobiliar se describió en un estudio de 39 semanas con una dosis de 1.000 mg/kg/día, que dio lugar a exposiciones de aproximadamente 16 veces la exposición en humanos a la dosis recomendada en humanos, y fue parcialmente reversible después de un período de recuperación de 4 semanas. Los estudios en animales con bictegravir no han mostrado indicios de teratogenicidad ni un efecto sobre la función reproductiva. En descendientes de ratas y conejas tratadas con bictegravir durante el embarazo, no hubo efectos toxicológicamente significativos en los criterios de evaluación del desarrollo. Los datos de los estudios no clínicos de emtricitabina no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad, potencial carcinogénico, toxicidad para la reproducción y el desarrollo. Emtricitabina ha demostrado un potencial carcinogénico bajo en ratones y ratas. Los estudios no clínicos de tenofovir alafenamida en ratas y perros mostraron que los huesos y el riñón son los principales órganos afectadospor la toxicidad. La toxicidad ósea se observó en forma de reducción de la densidad mineral ósea en ratas y perros a unas exposiciones a tenofovir al menos 43 veces superiores a las esperadas después de la administración de B/F/TAF. Hubo una mínima infiltración de histiocitos presente en el ojo de perros con exposiciones a tenofovir alafenamida y tenofovir aproximadamente 14 y 43 veces superiores, respectivamente, a las esperadas después de la administración de B/F/TAF. Tenofovir alafenamida no fue mutagénico ni clastogénico en los ensayos convencionales de genotoxicidad. Dado que existe una menor exposición a tenofovir en ratas y ratones después de la administración de tenofovir alafenamida en comparación con tenofovir disoproxil fumarato, los estudios de carcinogenicidad y un estudio peri postnatal en ratas se realizaron solamente con tenofovir disoproxil fumarato. Los estudios convencionales de potencial carcinogénico y toxicidad para la reproducción y el desarrollo no mostraron riesgos especiales para los seres humanos. Los estudios de toxicidad para la reproducción en ratas y conejos no mostraron ningún efecto en los parámetros de apareamiento, fertilidad, embarazo y fetales. No obstante, tenofovir disoproxilo fumarato redujo el índice de viabilidad y peso de las crías en un estudio peri postnatal de toxicidad a dosis tóxicas para la madre. Uso en poblaciones específicas: Embarazo: No hay datos o estos son limitados (datos en menos de 300 embarazos) relativos al uso de bictegravir o de tenofovir alafenamida en mujeres embarazadas. Existe un elevado núme