AZATEVA
TEVA ARGENTINA
Antineoplásico.
Composición.
Cada frasco ampolla de Azateva® contiene: Azacitidina 100 mg Excipientes: manitol 100 mg.
Indicaciones.
Azateva® está indicada para el tratamiento de pacientes adultos que no se consideran candidatos a trasplante de células madre hematopoyéticas (TCMH) y que padecen: Síndromes mielodisplásicos (SMD) intermedios 2 y de alto riesgo, según el sistema internacional de puntuación pronóstica (IPSS). Leucemia mielomonocítica crónica (LMMC) con el 10 al 29% de blastos medulares sin trastorno mieloproliferativo. Leucemia mieloide aguda (LMA) con 20 al 30% de blastos y displasia multilínea, según la clasificación de la Organización Mundial de la Salud (OMS). LMA con > 30% de blastos medulares según la clasificación de la OMS.
Dosificación.
El tratamiento con Azateva® debe iniciarse y monitorizarse bajo la supervisión de un médico con experiencia en el uso de fármacos quimioterapéuticos. Los pacientes deben ser tratados previamente con antieméticos para las náuseas y los vómitos. La dosis inicial recomendada para el primer ciclo de tratamiento, para todos los pacientes independientemente de los valores hematológicos iniciales, es de 75 mg/m2 de superficie corporal, por día, administrados por vía subcutánea o intravenosa, durante 7 días seguido de un periodo de reposo de 21 días (ciclo de tratamiento de 28 días). Se recomienda que los pacientes reciban tratamiento durante un mínimo de seis ciclos. El tratamiento debe continuarse mientras el paciente siga beneficiándose o hasta la progresión de la enfermedad. Se deben vigilar la respuesta/toxicidad hematológica y la toxicidad renal de los pacientes; puede ser necesario un retraso en el inicio del siguiente ciclo o una disminución de una dosis, como se explica más adelante. Análisis de laboratorio: Antes de iniciar el tratamiento y antes de cada ciclo de tratamiento, deben realizarse pruebas de función hepática y determinarse la creatinina sérica y el bicarbonato sérico. Deben efectuarse recuentos sanguíneos completos antes del inicio del tratamiento y cuando sea necesario para monitorizar la respuesta y la toxicidad, pero como mínimo, antes de cada ciclo de tratamiento. Ajuste de la dosis debido a toxicidad hematológica: La toxicidad hematológica se define como el recuento sanguíneo más bajo alcanzado (nadir) en un ciclo determinado, si el recuento de plaquetas es ≤50,0 × 109/l o el recuento absoluto de neutrófilos (RAN) es ≤1 × 109/l. La recuperación se define como un aumento de la/s línea/s celular/es en las que se observó una toxicidad hematológica, como mínimo, igual a la mitad de la diferencia absoluta entre el nadir y el recuento inicial, más el recuento nadir; es decir, recuento sanguíneo en la recuperación ≥ recuento nadir + (0,5 × [| recuento inicial - recuento nadir |]). Pacientes sin una disminución de los recuentos sanguíneos iniciales (es decir, leucocitos ≥3,0 x 109/l y RAN ≥1,5 x 109/l, y recuento plaquetario ≥75,0 x 109/l) antes del primer tratamiento: Si se observa toxicidad hematológica después del tratamiento con Azacitidina, el siguiente ciclo de tratamiento debe retrasarse hasta que el recuento plaquetario y el RAN se hayan recuperado. Si la recuperación se alcanza en un plazo de 14 días, no es necesario un ajuste de la dosis. Sin embargo, si la recuperación no se ha alcanzado en un plazo de 14 días, la dosis debe reducirse según la siguiente tabla. Después de las modificaciones de la dosis, la duración del ciclo debe volver a ser de 28 días.
Pacientes con recuentos sanguíneos iniciales reducidos (es decir, leucocitos < 3,0 x 109/l o RAN < 1,5 x 109/l o recuento plaquetario < 75,0 x 109/l) antes del primer tratamiento: Después del tratamiento con Azacitidina, si la disminución del recuento leucocitario del RAN o del recuento plaquetario con respecto al recuento antes del tratamiento es ≤50% o superior al 50%, pero con una mejoría en la diferenciación de cualquier línea celular, el siguiente ciclo no debe retrasarse y no debe efectuarse ningún ajuste de la dosis. Si la disminución del recuento leucocitario del RAN o del recuento plaquetario es superior al 50% con respecto al recuento antes del tratamiento, y no hay mejoría en la diferenciación de líneas celulares, el siguiente ciclo de tratamiento con Azacitidina debe retrasarse hasta que el recuento plaquetario y el RAN se hayan recuperado. Si la recuperación se alcanza en un plazo de 14 días, no es necesario un ajuste de la dosis. Sin embargo, si la recuperación no se ha alcanzado en un plazo de 14 días, debe determinarse la celularidad de la médula ósea. Si la celularidad de la médula ósea es > 50%, no debe efectuarse un ajuste de la dosis. Si la celularidad de la médula ósea es ≤50%, el tratamiento debe retrasarse y la dosis debe disminuirse, según la siguiente tabla:
Después de las modifi caciones de la dosis, la duración del ciclo siguiente debe volver a ser de 28 días. Poblaciones especiales: Pacientes de edad avanzada: No se recomienda ningún ajuste específico de la dosis en los pacientes de edad avanzada. Puesto que es más probable que los pacientes de edad avanzada presenten un deterioro de la función renal, puede ser conveniente vigilar la función renal. Insuficiencia renal: Se puede administrar Azacitidina a pacientes con insuficiencia renal sin la necesidad de ajustar la dosis inicial. Si se producen disminuciones inexplicadas de los niveles de bicarbonato sérico a menos de 20 mmol/l, la dosis deberá disminuirse en un 50% en el siguiente ciclo. Si se producen aumentos inexplicados de la creatinina sérica o del nitrógeno ureico en sangre (NUS) a ≥2 veces superiores a los valores iniciales y superiores al límite superior de la normalidad (LSN), el siguiente ciclo deberá retrasarse hasta que los valores vuelvan a la normalidad o a los valores iniciales, y la dosis deberá disminuirse en un 50% en el siguiente ciclo de tratamiento. Insuficiencia hepática: No se han realizado estudios formales en pacientes con insuficiencia hepática. Se deben vigilar atentamente las reacciones adversas en los pacientes con insuficiencia orgánica hepática grave. Antes del tratamiento inicial, no se recomienda ninguna modificación específica de la dosis inicial en los pacientes con insuficiencia hepática; las modificaciones posteriores de la dosis deben basarse en los valores hematológicos. Azateva® está contraindicado en los pacientes con tumores hepáticos malignos. Población pediátrica: No se ha establecido la seguridad y eficacia de Azacitidina en niños de 0 a 17 años. Recomendaciones para una manipulación segura: Azateva® es un medicamento citotóxico y, al igual que con otros compuestos potencialmente tóxicos, debe tenerse precaución al manipular y preparar suspensiones de Azacitidina. Deben aplicarse los procedimientos para la manipulación y eliminación correctas de medicamentos contra el cáncer. Si Azacitidina reconstituida entra en contacto con la piel, la zona deberá lavarse inmediatamente y a fondo con agua y jabón. Si entra en contacto con membranas mucosas, debe lavarse a fondo con agua. Procedimiento de reconstitución: Azateva® se debe reconstituir con agua para preparaciones inyectables. El periodo de validez del medicamento reconstituido puede prolongarse reconstituyéndolo con agua para preparaciones inyectables refrigerada (entre 2°C y 8°C). A continuación, se facilita información sobre la conservación del medicamento reconstituido. 1. Deben montarse los siguientes elementos: vial/es de Azateva®; vial/es de agua para preparaciones inyectables; guantes quirúrgicos no estériles; toallitas humedecidas en alcohol; jeringas para inyección de 5 ml con agujas. 2. Deben extraerse 4 ml de agua para preparaciones inyectables en la jeringa, asegurándose de purgar el aire atrapado dentro de la jeringa. 3. La aguja de la jeringa que contiene los 4 ml de agua para preparaciones inyectables debe introducirse a través del tapón de goma del vial de Azacitidina; a continuación, se inyecta en el vial el agua para preparaciones inyectables. 4. Después de extraer la jeringa y la aguja, el vial debe agitarse vigorosamente, hasta obtener una suspensión turbia uniforme. Después de la reconstitución, cada ml de suspensión contendrá 25 mg de Azacitidina (100 mg/4 ml). El producto reconstituido es una suspensión turbia y homogénea, sin aglomerados. La suspensión debe desecharse si contiene partículas grandes o aglomerados. No filtrar la suspensión después de la reconstitución ya que esto podría eliminar el principio activo. Se debe tener en cuenta que algunos adaptadores, agujas para perfusión y sistemas cerrados contienen filtros; por lo tanto, no se deben utilizar dichos sistemas para la administración del medicamento después de la reconstitución. 5. El tapón de goma debe limpiarse y se introduce una jeringa nueva con una aguja en el vial. A continuación, el vial debe invertirse, asegurándose que la punta de la aguja esté por debajo del nivel del líquido. Seguidamente, debe tirarse del émbolo hacia atrás para extraer la cantidad de medicamento necesaria para la dosis correcta, asegurándose de purgar el aire atrapado dentro de la jeringa. A continuación, debe extraerse del vial la jeringa con la aguja y la aguja debe desecharse. 6. Seguidamente, debe ajustarse firmemente a la jeringa una aguja subcutánea nueva (se recomienda el calibre 25) para inyectables. La aguja no debe purgarse antes de la inyección, a fin de reducir la incidencia de reacciones locales en el lugar de la inyección. 7. Cuando se necesite más de 1 vial, deben repetirse todos los pasos anteriores para la preparación de la suspensión. En el caso de dosis para las que se necesite más de 1 vial, la dosis se debe dividir en partes iguales, por ejemplo, dosis de 150 mg=6 ml; dos jeringas con 3 ml en cada jeringa. Debido a la retención en el vial y la aguja, es posible que no se pueda extraer toda la suspensión del vial. 8. El contenido de la jeringa de dosificación debe volver a resuspenderse inmediatamente antes de la administración. Debe permitirse que la jeringa cargada con la suspensión reconstituida alcance una temperatura de aproximadamente 20°C a 25°C durante un tiempo máximo de 30 minutos antes de la administración. Si el tiempo transcurrido es superior a 30 minutos, la suspensión debe desecharse correctamente y debe prepararse una dosis nueva. Para resuspender, haga rodar vigorosamente la jeringa entre las palmas de las manos, hasta obtener una suspensión uniforme y turbia. La suspensión debe desecharse si contiene partículas grandes o aglomerados. Forma de administración: Azateva® reconstituido debe inyectarse por vía subcutánea (introduzca la aguja con un ángulo de 45 a 90°), con una aguja de calibre 25, en el brazo, el muslo o el abdomen. Las dosis superiores a 4 ml deben inyectarse en dos lugares separados. Los lugares de inyección deben someterse a rotación. Las nuevas inyecciones deben administrarse como mínimo a 2,5 cm de distancia del lugar anterior y nunca en zonas sensibles, con equimosis, enrojecidas o endurecidas. Después de la reconstitución, no se debe filtrar la suspensión. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local. Cálculo de una dosis individual: La dosis total, según la superficie corporal (SC), puede calcularse de la siguiente manera: Dosis total (mg) = dosis (mg/m2) × SC (m2) La siguiente tabla se presenta sólo como un ejemplo para calcular dosis individuales de Azacitidina, basadas en un valor promedio de SC de 1,8 m2.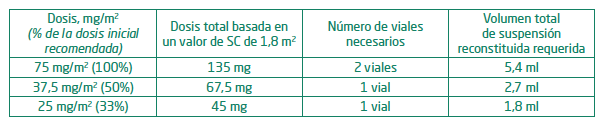
Estabilidad de la suspensión: Una vez reconstituida para administración subcutánea, Azateva® se puede conservar durante un máximo de 1 hora a 25°C. Preparación para administración intravenosa: Reconstituir el número de viales adecuados de Azateva® para alcanzar la dosis deseada. Reconstituir cada vial con 10 ml de agua para preparaciones inyectables. Agitar el vial hasta que todos los sólidos se disuelvan. La solución resultante tendrá una concentración de azacitadina de 10 mg/ml. La solución debe ser límpida. El fármaco parenteral debe ser inspeccionado visualmente previamente a la administración para detectar partículas y decoloración, siempre que la solución y el recipiente lo permitan. Extraer la cantidad requerida de solución de, Azateva® para administrar la dosis deseada e inyectarla en una bolsa de infusión de 50-100 ml de cloruro de sodio 0,9% para inyección o Ringer lactato para inyección. Incompatibilidad de la solución intravenosa: Azateva® es incompatible con soluciones de Dextrosa al 5%, Hespan, o soluciones que contengan bicarbonato. Estas soluciones tienen el potencial de incrementar la tasa de degradación de Azateva® y por lo tanto deben evitarse. Administración intravenosa: Azateva® en solución se administra por vía intravenosa. Administrar la dosis total en un período de 10 a 40 minutos. La administración debe completarse dentro de la hora posterior a la reconstitución del vial de Azateva®. Estabilidad de la solución: Azateva® reconstituida para administración intravenosa puede conservarse a 25°C, pero la administración debe completarse dentro de la hora posterior a la reconstitución del vial.
Precauciones.
Azacitidina está contraindicada en pacientes con hipersensibilidad conocida a la Azacitidina o a manitol. Está contraindicada en pacientes con tumores hepáticos malignos en estadio avanzado.
Presentación.
Azateva®: envases con 1 frasco ampolla.