Avastin®
ROCHE
Agente antineoplásico, anticuerpo monoclonal.
Composición.
Cada vial de 4 ml contiene 100 mg de bevacizumab (25 mg/ml), en un excipiente compuesto por trehalosa dihidrato 240 mg, fosfato disódico 4,8 mg, fosfato monosódico monohidratado 23,2 mg, polisorbato 20: 1,6 mg y agua para inyectables c.s.p. 4 ml. Cada vial de 16 ml contiene 400 mg de bevacizumab (25 mg/ml), en un excipiente compuesto por trehalosa dihidrato 960 mg, fosfato disódico 19,2 mg, fosfato monosódico monohidratado 92,8 mg, polisorbato 20: 6,4 mg y agua para inyectables c.s.p. 16 ml.
Farmacología.
Código ATC: L01XC07. Grupo farmacoterapéutico: Agente antineoplásico, anticuerpo monoclonal. Propiedades farmacodinámicas: Mecanismo de acción: Bevacizumab se une al factor de crecimiento del endotelio vascular (VEGF), factor clave de la vasculogénesis y la angiogénesis, inhibiendo así la unión de éste a sus receptores Flt-1 (VEGFR-1) y KDR (VEGFR-2), situados en la superficie de las células endoteliales. La neutralización de la actividad biológica del VEGF produce una regresión de la vascularización de los tumores, normaliza la vasculatura residual del tumor e inhibe la neovascularización tumoral, impidiendo así el crecimiento del tumor. Efectos farmacodinámicos: La administración de bevacizumab o del anticuerpo murino correspondiente en ratones inmunodeficientes (nude) xenotransplantados (modelos experimentales de cáncer) generó una amplia actividad antitumoral sobre varios tipos de cáncer humano, incluyendo colon, mama, páncreas y próstata. Se inhibió la progresión de la enfermedad metastásica y se redujo la permeabilidad microvascular. Eficacia clínica: Carcinoma metastásico de colon o recto (CCRm): La seguridad y la eficacia de la dosis recomendada (5 mg/kg de peso corporal cada dos semanas) en carcinoma metastásico de colon o recto fueron estudiadas en tres ensayos clínicos aleatorizados, controlados con comparador activo, en combinación con un esquema quimioterápico de primera línea basado en fluoropirimidinas. Avastin se asoció con dos regímenes quimioterápicos: AVF2107g: Un esquema semanal de irinotecán/5-fluorouracilo en bolo/ácido folínico (IFL) durante un total de 4 semanas conformando ciclos de 6 semanas (régimen de Saltz). AVF0780g: En combinación con 5-flourouracilo/ácido folínico (5-FU/FA) en bolo durante un total de 6 semanas conformando ciclos de 8 semanas (régimen de Roswell Park). AVF2192g: En combinación con 5-FU/FA en bolo durante un total de 6 semanas conformando un ciclo de 8 semanas (régimen de Roswell Park) en pacientes que no eran candidatos óptimos para un tratamiento de primera línea con irinotecán. Se llevaron a cabo tres ensayos adicionales con bevacizumab en pacientes con carcinoma metastásico de colon o recto: de primera línea (NO16966), de segunda línea sin tratamiento previo con bevacizumab (E3200), y de segunda línea con terapia previa con bevacizumab después de la progresión de la enfermedad en el tratamiento de primera línea (ML18147). En estos estudios, bevacizumab se administró en combinación con FOLFOX-4 (5-FU/LV/oxaliplatino), XELOX (capecitabina/oxaliplatino) y fluoropirimidina/irinotecán o fluoropirimidina/oxaliplatino, en los siguientes regímenes posológicos: NO16966: Avastin en una dosis de 7,5 mg/kg de peso corporal cada 3 semanas en combinación con capecitabina oral y oxaliplatino intravenoso (XELOX) o 5 mg/kg de Avastin cada 2 semanas asociado con leucovorina + 5-fluorouracilo en bolo, seguido de una perfusión de 5-fluorouracilo con oxaliplatino intravenoso (FOLFOX-4). E3200: Avastin en una dosis de 10 mg/kg de peso corporal cada 2 semanas en combinación con leucovorina y 5-fluorouracilo en bolo, seguido por una perfusión de 5-fluorouracilo con oxaliplatino intravenoso (FOLFOX-4) en pacientes no tratados previamente con bevacizumab. ML18147: Avastin en una dosis de 5 mg/kg de peso corporal cada 2 semanas o en una dosis de 7,5 mg/kg de peso corporal cada 3 semanas en combinación con fluoropirimidina/irinotecán o fluoropirimidina/oxaliplatino en pacientes con progresión de la enfermedad después del tratamiento con bevacizumab de primera línea. El régimen con irinotecán u oxaliplatino se cambió en función del uso indistinto de oxaliplatino o irinotecán como tratamiento de primera línea. AVF2107g: En este ensayo clínico Fase III, aleatorizado, doble-ciego y controlado con comparador activo se estudió Avastin en combinación con IFL como tratamiento de primera línea del carcinoma metastásico de colon o recto. Se aleatorizaron 813 pacientes para ser tratados con IFL + placebo (Brazo 1), o IFL + Avastin (5 mg/kg, cada 2 semanas, Brazo 2), (Tabla 4). Un tercer grupo de 110 pacientes recibieron 5-FU/FA en bolo + Avastin (Brazo 3). De acuerdo con lo especificado previamente, la incorporación de pacientes al estudio en el Brazo 3 se interrumpió una vez que se determinó y se consideró aceptable la seguridad de Avastin con el régimen de IFL. Todos los pacientes recibieron tratamiento hasta la progresión de la enfermedad. La media de edad fue de 59,4 años. En la escala de estado de desempeño ECOG (Eastern Cooperative Oncology Group), el 56,6% de los pacientes tenía un puntaje de 0, el 43% una ECOG1 y el 0,4% una ECOG2. Previamente, el 15,5% había recibido radioterapia y el 28,4% quimioterapia. La variable principal de eficacia del ensayo fue sobrevida global. La adición de Avastin a IFL dio lugar a un aumento estadísticamente significativo de la sobrevida global, la sobrevida libre de progresión y la tasa de respuesta global (Tabla 4). El beneficio clínico, medido como sobrevida global, se observó en todos los subgrupos preespecificados de pacientes, incluyendo aquellos definidos según la edad, sexo, estado de desempeño, localización del tumor primario, número de órganos afectados y duración de la enfermedad metastásica. Los resultados de eficacia de Avastin en combinación con quimioterapia IFL se muestran en la Tabla 4.
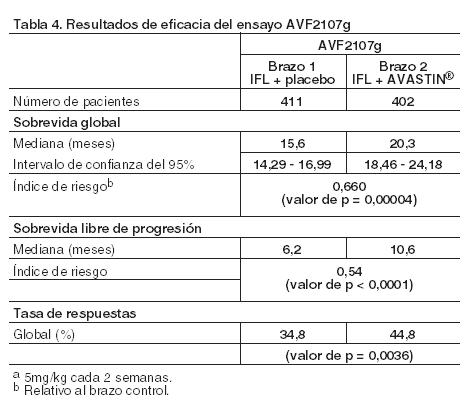
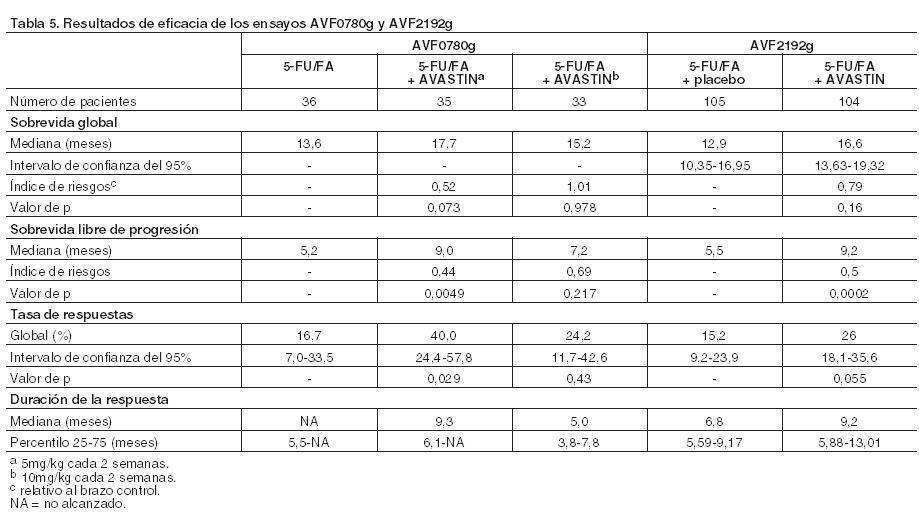
Entre los 110 pacientes aleatorizados al Brazo 3 (5-FU/FA + Avastin), antes de la interrupción de inclusión de pacientes en este brazo, la mediana de la sobrevida global fue de 18,3 meses y la de la sobrevida libre de progresión de 8,8 meses. AVF2192g: Ensayo clínico de Fase II, aleatorizado, doble-ciego, controlado con comparador activo, en el que se evaluaron la eficacia y la seguridad de Avastin en combinación con 5-FU/FA como tratamiento de primera línea del cáncer colorrectal metastásico en pacientes que no eran candidatos óptimos para terapia de primera línea con irinotecán. Se aleatorizaron 105 pacientes en el brazo de 5-FU/FA + placebo y 104 en el brazo de 5-FU/FA + Avastin (5 mg/kg cada 2 semanas). Todos los tratamientos se administraron hasta la progresión de la enfermedad. La adición de Avastin 5 mg/kg cada dos semanas a 5-FU/FA aumentó la tasa de respuestas objetivas, incrementó significativamente la duración de la sobrevida libre de progresión y mostró una tendencia a una sobrevida más prolongada si se compara con 5-FU/FA solo. AVF0780g: Ensayo clínico de Fase II, aleatorizado, abierto y controlado con comparador activo, para la evaluación de Avastin en combinación con 5-FU/FA para el tratamiento de primera línea del cáncer colorrectal metastásico. La mediana de edad fue de 64 años. El 19% de los pacientes había recibido previamente quimioterapia y el 14% radioterapia. Se aleatorizaron 71 pacientes para ser tratados con 5-FU/FA en bolo o 5-FU/FA + Avastin (5 mg/kg cada dos semanas). A un tercer grupo de 33 pacientes se administró 5-FU/FA en bolo + Avastin (10 mg/kg cada 2 semanas). Todos fueron tratados hasta la progresión de la enfermedad. Las variables principales del ensayo fueron la tasa de respuestas objetivas y la sobrevida libre de progresión. La adición de 5 mg/kg de Avastin cada dos semanas a 5-FU/FA produjo un aumento en la tasa de respuestas objetivas, la extensión de la sobrevida libre de progresión y una tendencia a sobrevida global más prolongada en comparación con 5-FU/FA solo (Tabla 5). Estos datos de eficacia concuerdan con los resultados obtenidos en el ensayo AVF2107g. Los datos de eficacia de los ensayos AVF0780 y AVF2192g en los que se investigó Avastin en combinación con 5-FU/FA se resumen en la Tabla 5.
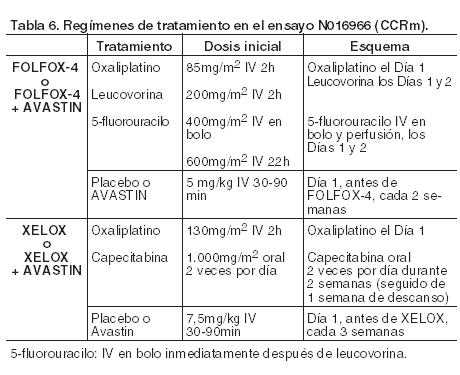
NO16966: Ensayo clínico de Fase III, aleatorizado, doble-ciego (para bevacizumab), en el que se investigó Avastin en una dosis de 7,5 mg/kg en combinación con capecitabina oral y oxaliplatino IV (XELOX), administrados en ciclos cada 3 semanas, o Avastin en una dosis de 5 mg/kg asociado con 5-FU/LV en bolo, seguido de una perfusión de 5-fluorouracilo con oxaliplatino IV (FOLFOX-4), administrados en ciclos cada 2 semanas. El ensayo tuvo dos fases: una inicial abierta de dos brazos (Parte I) en la que los pacientes fueron aleatorizados en dos grupos diferentes de tratamiento (XELOX y FOLFOX -4) y una posterior con un diseño factorial 2 x 2 de 4 brazos (Parte II) en la que los pacientes fueron distribuidos al azar en 4 grupos de tratamiento (XELOX + placebo, FOLFOX-4 + placebo, XELOX + Avastin, FOLFOX-4 + Avastin). En la Parte II la asignación del tratamiento fue doble-ciego con respecto a Avastin. Se aleatorizaron aproximadamente 350 pacientes en cada uno de los 4 brazos de la Parte II del ensayo clínico.
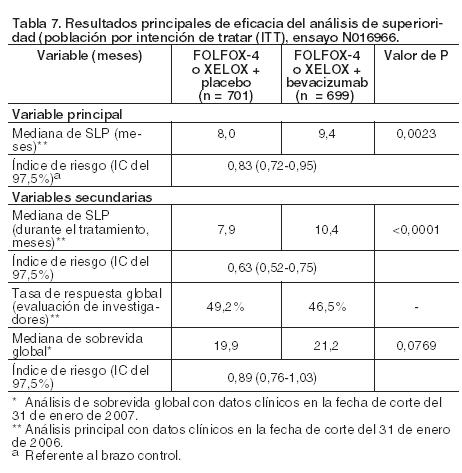
La variable principal de eficacia del ensayo fue la duración de la sobrevida libre de progresión. Los dos objetivos esenciales fueron: mostrar que XELOX era no inferior a FOLFOX-4 y que Avastin en combinación con FOLFOX-4 o XELOX era superior frente a la quimioterapia sola. Se cumplieron estos dos objetivos: En la comparación global se demostró la no inferioridad de los brazos de XELOX frente a los que contenían FOLFOX-4 en la población de pacientes incluidos por protocolo en términos de sobrevida libre de progresión y sobrevida global. En la comparación global se demostró la superioridad de los brazos que contenían Avastin frente a los brazos de quimioterapia sola en la población por intención de tratar en términos de sobrevida libre de progresión (Tabla 7). Los análisis secundarios de sobrevida libre de progresión, sobre la base de la evaluación de la respuesta durante el tratamiento, confirmaron el beneficio clínico significativamente superior para los pacientes tratados con Avastin (Tabla 7), que concordó con las ventajas estadísticamente significativas observadas en el análisis agrupado.
En el subgrupo de tratamiento con FOLFOX, la mediana de sobrevida libre de progresión fue de 8,6 meses en los pacientes tratados con placebo y de 9,4 meses en los que recibieron bevacizumab, índice de riesgo (hazard ratio HR) = 0,89, IC del 97,5% = [0,73; 1,08]; valor de p = 0,1871, siendo los resultados correspondientes en el subgrupo de tratamiento con XELOX de 7,4 frente a 9,3 meses, HR = 0,77, IC del 97,5% = [0,63; 0,94]; valor de p = 0,0026. En el subgrupo de tratamiento con FOLFOX, la mediana de sobrevida global fue de 20,3 meses en los pacientes tratados con placebo y de 21,2 meses en los que se administró bevacizumab HR = 0,94, IC del 97,5% = [0,75; 1,16], valor de p = 0,4937, siendo los resultados correspondientes en el subgrupo de tratamiento con XELOX de 19,2 frente a 21,4 meses, HR = 0,84, IC del 97,5% = [0,68; 1,04]; valor de p = 0,0698. ECOG E3200: En este ensayo clínico de Fase III, aleatorizado, abierto y controlado con comparador activo se investigó en pacientes con cáncer colorrectal avanzado tratados previamente (segunda línea) la administración de Avastin en una dosis de 10 mg/kg en combinación con 5-FU/LV en bolo y después 5-fluorouracilo en perfusión con oxaliplatino IV (FOLFOX-4), administrados en ciclos cada 2 semanas. En los brazos con quimioterapia se utilizó un régimen de FOLFOX-4 con el mismo esquema y dosis que se muestra en Tabla 6 para el ensayo NO16966. La variable principal de eficacia del ensayo fue la sobrevida global, que se definió como el tiempo que transcurre desde la aleatorización hasta la muerte por cualquier causa. Se distribuyeron al azar 829 pacientes (de los cuales 292 recibieron FOLFOX-4, 293 Avastin + FOLFOX-4 y 244 Avastin como monoterapia). La adición de Avastin a FOLFOX-4 dio como resultado una prolongación de la sobrevida global estadísticamente significativa. También se observaron mejoras estadísticamente significativas en la sobrevida libre de progresión y en la tasa de respuestas objetivas (Tabla 8).
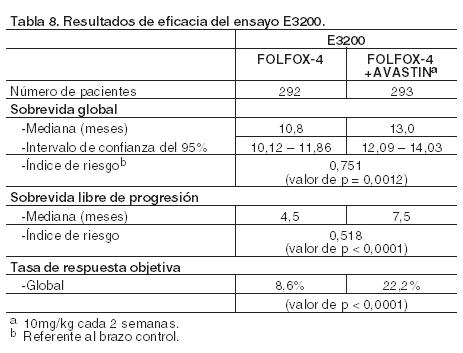
No se observaron diferencias significativas en la duración de la sobrevida global entre los pacientes que recibieron Avastin como monoterapia y los tratados con FOLFOX-4. La sobrevida libre de progresión y la tasa de respuestas objetivas fueron inferiores en el brazo de Avastin como monoterapia comparado con el brazo de FOLFOX-4. ML18147: En este ensayo de Fase III, abierto, aleatorizado y controlado, se evaluó Avastin en una dosis de 5 mg/kg cada 2 semanas o 7,5 mg/kg cada 3 semanas en combinación con quimioterapia basada en fluoropirimidinas en comparación con quimioterapia basada en fluoropirimidinas sola en pacientes con cáncer colorrectal metastásico en progresión después de un régimen de primera línea que contenía bevacizumab. Los pacientes con CCRm histológicamente confirmado y progresión de la enfermedad fueron aleatorizados 1:1 dentro de los 3 meses siguientes a la suspensión del tratamiento de primera línea con bevacizumab para recibir quimioterapia basada en fluoropirimidina/oxaliplatino o fluoropirimidina/irinotecán (cambio de la quimioterapia en función de la ya administrada en primera línea) con o sin bevacizumab. Los pacientes recibieron tratamiento hasta la progresión de la enfermedad o toxicidad inaceptable. La variable principal de valoración fue la sobrevida global (SG), definida como el tiempo transcurrido desde la aleatorización hasta la muerte por cualquier causa. Se aleatorizaron un total de 820 pacientes. La adición de bevacizumab a la quimioterapia basada en fluoropirimidinas resultó en una prolongación estadísticamente significativa de la sobrevida de los pacientes con CCRm cuya enfermedad había progresado después de un régimen de primera línea que contenía bevacizumab (ITT = 819) (véase Tabla 9).
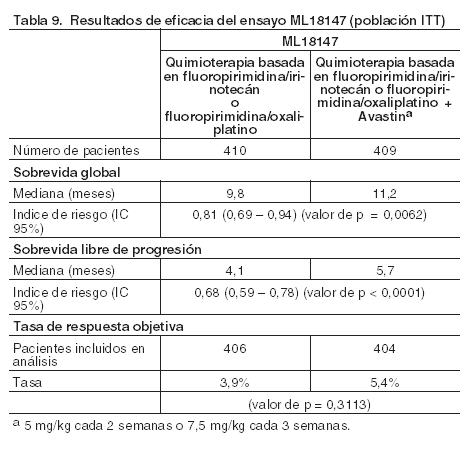
También se observó una mejoría estadísticamente significativa de la sobrevida libre de progresión. La tasa de respuesta objetiva fue baja en ambos grupos de tratamiento y no alcanzó significación estadística. En el estudio E3200 se utilizó una dosis equivalente a 5 mg/kg/semana de bevacizumab en pacientes sin tratamiento previo con bevacizumab, mientras que en el estudio ML18147 se empleó una dosis equivalente a 2,5 mg/kg/semana de bevacizumab en pacientes pretratados con bevacizumab. La comparación entre los ensayos clínicos de los datos de eficacia y seguridad está limitada por las diferencias entre los mismos, más notables en poblaciones de pacientes expuestas previamente a bevacizumab y a regímenes de quimioterapia. Ambas dosis equivalentes de 5 mg/kg/semana y 2,5 mg/kg/semana de bevacizumab proporcionaron un beneficio estadísticamente significativo en relación con la sobrevida global (Indice de riesgo 0,751 en el ensayo E3200 y de 0,81 en ML18147) y con la sobrevida libre de progresión (Indice de riesgo 0,518 en el ensayo E3200 y de 0,68 en ML18147). En términos de seguridad, hubo una mayor incidencia global de reacciones adversas de Grados 3 - 5 en el ensayo E3200 con respecto al ML18147. Cáncer de colon adyuvante (CCa): BO17920: Este ensayo de Fase III, aleatorizado, abierto, de 3 brazos, evaluó la seguridad y eficacia de Avastin administrado en una dosis equivalente a 2,5 mg/kg/semana, ya sea en un esquema de 2 semanas en combinación con FOLFOX4, o de 3 semanas asociado con XELOX en comparación con FOLFOX4 solo como quimioterapia adyuvante en 3.451 pacientes con estadio II de alto riesgo y estadio III de carcinoma de colon. Se observaron más recaídas y muertes debido a la progresión de la enfermedad en ambos grupos de Avastin en comparación con el grupo control. No se logró el objetivo principal de prolongar la sobrevida libre de enfermedad en pacientes con estadio III de cáncer de colon (n = 2.867) mediante la adición de Avastin a cualquier régimen de quimioterapia. El índice de riesgos para la sobrevida libre de enfermedad fue de 1,17 (IC 95%: 0,98 - 1,39) para el grupo FOLFOX4 + Avastin y de 1,07 (IC 95%: 0,90 - 1,28) para el grupo XELOX + Avastin. Cáncer de mama metastásico (CMm): Se diseñaron dos grandes ensayos Fase III con el fin de investigar el efecto del tratamiento de Avastin en combinación con dos agentes quimioterápicos en forma individual, en los que se midió como variable principal la sobrevida libre de progresión. En ambos ensayos se observó una mejoría clínica y estadísticamente significativa en este parámetro. A continuación se resumen los resultados de la sobrevida libre de progresión para los agentes quimioterápicos en forma individual incluidos en la indicación: Ensayo E2100 (paclitaxel): La mediana de sobrevida libre de progresión aumenta 5,6 meses, índice de riesgo 0,421 (valor de p < 0,0001, IC del 95%: 0,343; 0,516). Ensayo AVF3694g (capecitabina): La mediana de sobrevida libre de progresión aumenta 2,9 meses, índice de riesgo 0,69 (valor de p = 0,0002, IC del 95%: 0,56; 0,84). A continuación se proporcionan los detalles de cada ensayo y sus resultados. ECOG E2100: En este ensayo multicéntrico, abierto, aleatorizado y controlado con comparador activo, se evaluó Avastin en combinación con paclitaxel para el tratamiento del cáncer de mama metastásico o localmente recidivante en pacientes que no habían recibido previamente quimioterapia para la enfermedad metastásica y localmente recidivante. Los pacientes fueron aleatorizados para recibir paclitaxel solo (90 mg/m2 IV durante una hora una vez por semana, tres semanas consecutivas de cada cuatro) o en combinación con Avastin (10 mg/kg en infusión IV cada dos semanas). Se permitió ingresar a los pacientes que hubieran recibido tratamiento hormonal previo para la enfermedad metastásica. La terapia adyuvante con taxanos se autorizó sólo en aquellos casos en que hubiera sido completada por lo menos 12 meses antes de la incorporación al ensayo. De los 722 pacientes del ensayo, la mayoría tenía tumores HER2-negativos (90%), salvo un pequeño número con estado HER2-desconocido (8%) o HER2-positivo (2%), que había sido tratado previamente con trastuzumab o había sido considerado no apto para este tratamiento. Además, el 65% había recibido quimioterapia adyuvante, incluyendo un 19% y un 49% con tratamiento previo de taxanos y antraciclinas, respectivamente. Se excluyeron aquellos pacientes con metástasis en el sistema nervioso central, y se incorporaron los tratados previamente o con lesiones cerebrales resecadas. En el ensayo E2100, los pacientes se trataron hasta la progresión de la enfermedad. En aquellas situaciones en que se requirió la interrupción temprana de la quimioterapia, el tratamiento continuó con Avastin como monoterapia hasta la progresión de la enfermedad. Las características basales de los pacientes fueron similares entre los brazos del ensayo. La variable principal fue la sobrevida libre de progresión (SLP), evaluada por los investigadores del ensayo. También se realizó una revisión independiente de la variable principal. En la Tabla 10 se reúnen los resultados de este ensayo.
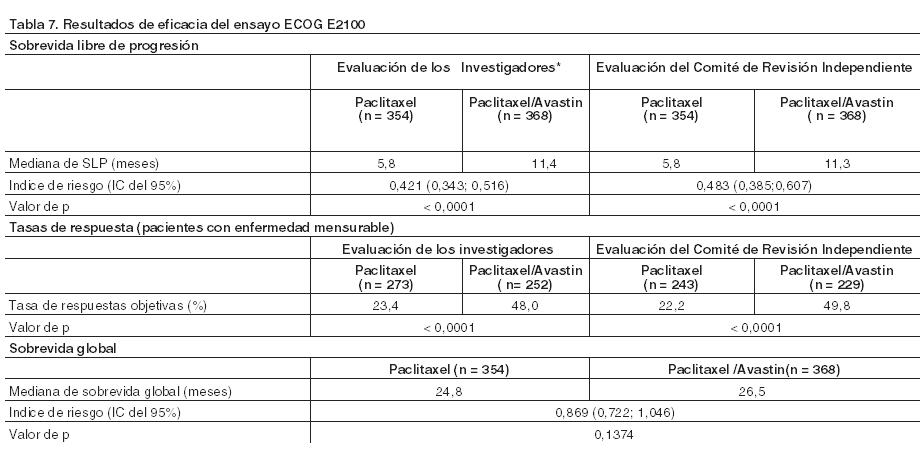
El beneficio clínico de Avastin, medido mediante la sobrevida libre de progresión, se observó en todos los subgrupos preespecificados en el ensayo (incluyendo el intervalo libre de enfermedad, el número de metástasis, la quimioterapia adyuvante previa y el estado de los receptores de estrógenos [RE]). AVF3694g: Este ensayo de Fase III, multicéntrico, aleatorizado, controlado con placebo fue diseñado para evaluar la eficacia y seguridad de Avastin en combinación con quimioterapia, comparado con quimioterapia más placebo, como tratamiento de primera línea para pacientes con cáncer de mama metastásico o localmente recurrente HER2-negativo. La quimioterapia fue elegida a criterio del investigador antes de la aleatorización en una proporción 2:1, para recibir Avastin y quimioterapia, o quimioterapia y placebo. Las quimioterapias elegidas que se administraron cada 3 semanas incluyeron capecitabina, taxanos (paclitaxel unido a proteínas, docetaxel), agentes basados en antraciclinas (doxorrubicina/ ciclofosfamida, epirrubicina/ciclofosfamida, 5-fluorouracilo/doxorrubicina/ciclofosfamida, 5-fluorouracilo/epirrubicina/ciclofosfamida). Avastin o placebo fueron administrados en una dosis de 15 mg/kg cada 3 semanas. Este ensayo incluyó una fase de tratamiento ciego, una fase opcional tras progresión abierta, y una fase de seguimiento de sobrevida. Durante la fase de tratamiento ciego, los pacientes recibieron quimioterapia y el medicamento de estudio (Avastin o placebo), cada 3 semanas hasta progresión de la enfermedad, toxicidad limitante del tratamiento, o fallecimiento. En caso de progresión de la enfermedad confirmada, los pacientes que entraron en la fase opcional abierta pudieron recibir Avastin, junto con una amplia gama de tratamientos de segunda línea. Se realizaron análisis estadísticos en forma independiente para: pacientes que recibieron capecitabina en combinación con Avastin o placebo; pacientes tratados con quimioterapia basada en taxanos o en antraciclinas en combinación con Avastin o placebo. La variable principal del ensayo fue la sobrevida libre de progresión evaluada por el investigador. Adicionalmente, esta variable fue también evaluada por un Comité de Revisión Independiente (CRI). En la Tabla 11 se presentan los resultados de los análisis definidos en el protocolo final para la sobrevida libre de progresión y la tasa de respuestas para la cohorte de capecitabina analizados independientemente en el ensayo AVF3694g. También, se dan a conocer los resultados de un análisis de sobrevida global exploratorio que incluye un seguimiento adicional de 7 meses (aproximadamente el 46% de los pacientes había fallecido). El porcentaje de los que recibieron Avastin en la fase abierta fue del 62,1% en el brazo de capecitabina + placebo y del 49,9% en el de capecitabina + Avastin.
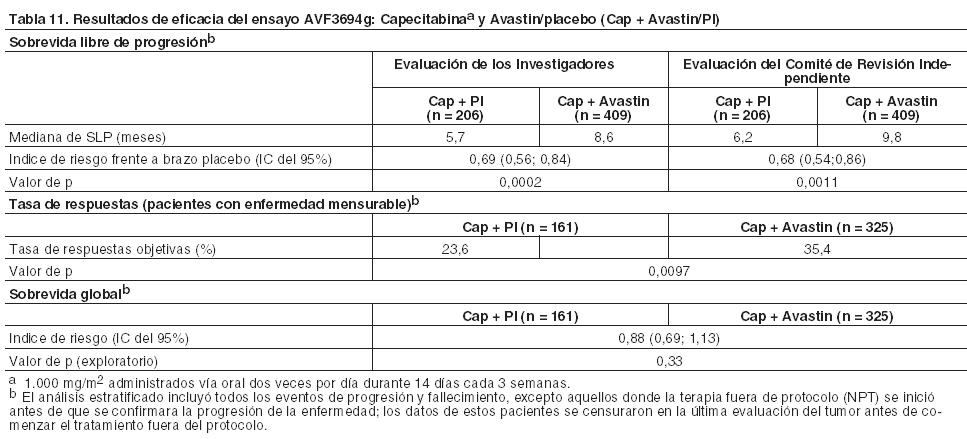
Se realizó un análisis no estratificado de la sobrevida libre de progresión (evaluado por el investigador) que no censuró para tratamiento, fuera de protocolo, antes de la progresión de la enfermedad. Los resultados fueron muy similares a los del objetivo principal de la sobrevida libre de progresión. Cáncer de pulmón no microcítico (CPNM): Primera línea de tratamiento para CPNM no escamoso, en combinación con quimioterapia basada en platino: En los ensayos E4599 y BO17704 se investigaron la seguridad y eficacia de Avastin asociado con quimioterapia basada en platino, en el tratamiento de primera línea de pacientes con cáncer de pulmón no microcítico (CPNM) con un tipo histológico sin predominio de células escamosas. En el ensayo E4599 se ha demostrado un beneficio en la sobrevida global con una dosis de bevacizumab de 15 mg/kg cada tres semanas. En el ensayo BO17704 se comprobó que tanto la dosis de 15 mg/kg cada 3 semanas como la de 7,5 mg/kg cada 3 semanas de bevacizumab aumentan la sobrevida libre de progresión y la tasa de respuestas. E4599: En este ensayo, multicéntrico, abierto, aleatorizado y controlado con comparador activo se evaluó la asociación de Avastin con quimioterapia como tratamiento de primera línea de pacientes con CPNM localmente avanzado (estadio IIIb con derrame pleural maligno), metastásico o recidivante con un tipo histológico sin predominio de células escamosas. Los pacientes fueron aleatorizados para recibir quimioterapia basada en platino (PC: paclitaxel 200 mg/m2 y carboplatino ABC = 6,0, ambos mediante perfusión IV) en el día 1 de cada ciclo de 3 semanas hasta 6 ciclos o PC en combinación con Avastin en una dosis de 15 mg/kg mediante infusión IV el día 1 de cada ciclo de 3 semanas. Después de la finalización de los seis ciclos de quimioterapia con carboplatino-paclitaxel o después de la interrupción prematura de la quimioterapia, los pacientes en el brazo de Avastin + carboplatino-paclitaxel continuaron recibiendo Avastin como monoterapia cada 3 semanas hasta la progresión de la enfermedad. Se aleatorizaron un total de 878 pacientes entre los dos brazos. Durante el ensayo, de los pacientes que recibieron el tratamiento en estudio, el 32,2% (136/422) recibió entre 7 - 12 administraciones de Avastin y el 21,1% (89/422) 13 ó más. El objetivo principal fue la duración de la sobrevida global. En la Tabla 12 se presentan los resultados.
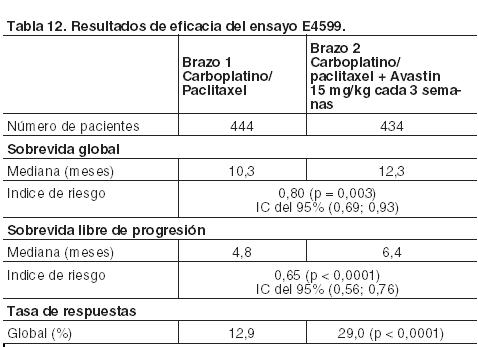
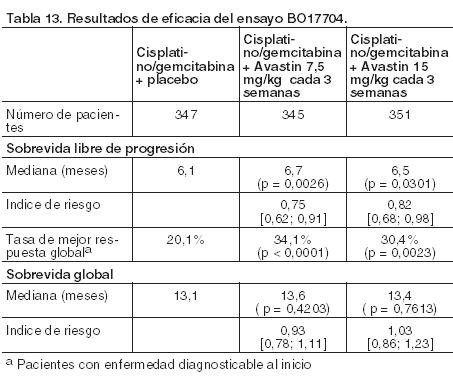
En un análisis exploratorio, el beneficio de Avastin en la sobrevida global fue más pronunciado en el subgrupo de pacientes que tenían histología de adenocarcinoma. BO17704: En este ensayo de Fase III, aleatorizado, doble-ciego de Avastin asociado con cisplatino y gemcitabina controlado frente a placebo, cisplatino y gemcitabina se incluyeron pacientes con CPNM localmente avanzado (estadio IIIb con metástasis de ganglios linfáticos supraclaviculares o con derrame pericárdico o pleural maligno), metastásico o recidivante con un tipo histológico sin predominio de células escamosas, que no habían recibido quimioterapia previa. La variable principal de eficacia fue la sobrevida libre de progresión (SLP), las variables secundarias del ensayo incluyeron la duración de la sobrevida global. Los pacientes fueron aleatorizados para la quimioterapia basada en platino, perfusión IV de 80 mg/m2 de cisplatino en el día 1 y perfusión IV de 1.250 mg/m2 de gemcitabina en los días 1 y 8 de cada ciclo de 3 semanas hasta 6 ciclos (CG) o CG en combinación con Avastin en una dosis de 7,5 o 15 mg/kg mediante infusión IV el día 1 de cada ciclo de 3 semanas. En los brazos que contenían Avastin, los pacientes podían recibir Avastin como monoterapia una vez cada 3 semanas hasta la progresión de la enfermedad o hasta que la toxicidad no fuera aceptable. Los resultados del ensayo muestran que el 94% (277/296) de los pacientes incluidos seguía recibiendo bevacizumab como monoterapia en el ciclo 7. Una alta proporción (aproximadamente el 62%) continuó con diferentes terapias anticancerosas no especificadas en el protocolo, lo cual podría haber impactado en el análisis de la sobrevida global. Los resultados de eficacia se presentan en la Tabla 13.
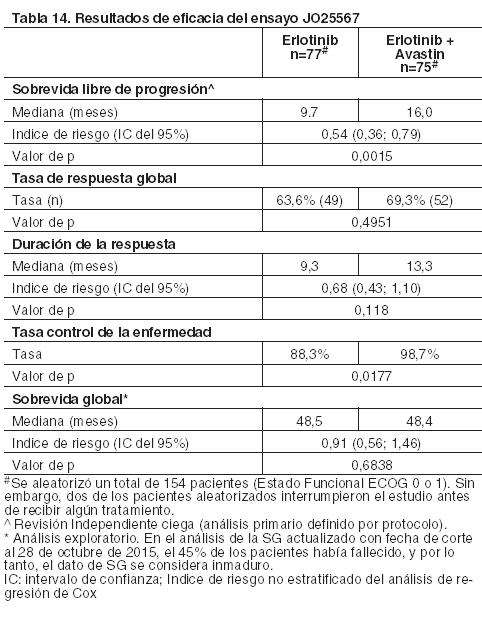
Primera línea de tratamiento para CPNM no escamoso, con mutaciones activadoras en EGFR en combinación con erlotinib: JO25567: En este ensayo de Fase II, aleatorizado, abierto, multicéntrico, llevado a cabo en Japón se evaluó la eficacia y seguridad de Avastin utilizado en combinación con erlotinib en pacientes con CPNM no escamoso con mutaciones activadoras en EGFR (deleción del exón 19 o mutación L858R del exón 21) que no habían recibido tratamiento sistémico anterior para estadio IIIB/IV o enfermedad recurrente. El criterio de valoración principal fue la sobrevida libre de progresión (SLP) basada en la evaluación de un Comité de Revisión Independiente. Los criterios de valoración secundarios incluyeron sobrevida global, tasa de respuestas, tasa de control de la enfermedad, duración de la respuesta, seguridad y la calidad de vida relacionada con la salud basada en el cuestionario FACT-L (Functional Assessment of Cancer Therapy for Patients with Lung Cancer). El estado de la mutación EGFR se determinó para cada paciente previamente a su selección y se aleatorizaron 154 pacientes que recibieron tratamiento con erlotinib + Avastin (150 mg diarios de erlotinib vía oral + Avastin [15 mg/kg IV cada 3 semanas]) o erlotinib en monoterapia (150 mg diarios vía oral) hasta la progresión de la enfermedad (PE) o toxicidad inaceptable. En ausencia de PE, la interrupción de uno de los componentes del tratamiento en el brazo erlotinib + Avastin no dio lugar a la suspensión del otro componente del mismo como se especifica en el protocolo del estudio. Los resultados de eficacia del estudio se muestran en la Tabla 14.
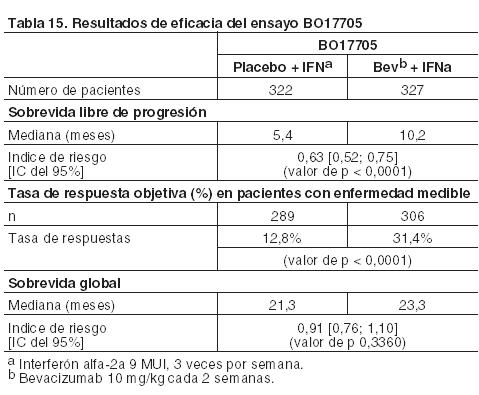
En el ensayo clínico abierto JO25567, la calidad de vida relacionada con la salud se evaluó mediante el puntaje total y los índices de resultado del cuestionario FACT-L, según la subescala de síntomas de cáncer de pulmón. Durante el tiempo libre de progresión, los puntajes FACT-L basales promedio se mantuvieron en ambos grupos de tratamiento. No hubo diferencias clínicamente significativas observadas entre los dos grupos de tratamiento en los resultados de FACT-L calidad de vida relacionada con la salud. Se destaca que los pacientes en el grupo de erlotinib más bevacizumab recibieron tratamiento más prolongado y la administración intravenosa de bevacizumab en comparación con la monoterapia con erlotinib oral en el grupo de control. Cáncer de células renales avanzado y/o metastásico (CRm): Avastin en combinación con interferón alfa-2a para el tratamiento de primera línea del cáncer de células renales avanzado y/o metastásico: BO17705: Se realizó un ensayo clínico de Fase III, aleatorizado, doble-ciego, en el que se evaluó la eficacia y seguridad de Avastin en combinación con interferón (IFN) alfa-2a frente a IFN alfa-2a como monoterapia en el tratamiento de primera línea del CRm. Los 649 pacientes aleatorizados (641 tratados) con carcinoma de células renales metastásico tenían un estado de desempeño de Karnofsky (KPS) ≥ 70%, carecían de metástasis en el sistema nervioso central y tenían una adecuada función orgánica. Los pacientes habían sido nefrectomizados por carcinoma primario de células renales. Se administró 10 mg/kg de Avastin cada 2 semanas hasta progresión de la enfermedad. Se administró IFN alfa-2a durante 52 semanas o hasta progresión de la enfermedad con una dosis inicial recomendada de 9 MUI tres veces por semana, permitiendo una reducción de la dosis hasta 3 MUI tres veces por semana en 2 etapas. Los pacientes fueron distribuidos según las características demográficas y el puntaje de Motzer y los brazos de tratamiento demostraron estar bien equilibrados según los factores pronóstico. El objetivo principal fue la sobrevida global, y dentro de los objetivos secundarios del ensayo se incluía la sobrevida libre de progresión. La adición de Avastin al IFN alfa-2a aumentó significativamente la sobrevida libre de progresión y la tasa de respuesta tumoral objetiva. Estos resultados se confirmaron a través de una revisión radiológica independiente. Sin embargo, el incremento de 2 meses en la sobrevida global (objetivo principal) no fue significativo (HR: 0,91). Una alta proporción de pacientes (aproximadamente 63% IFN/placebo y 55% Avastin/IFN) recibió después del ensayo, diferentes tratamientos anticancerosos no especificados, incluyendo agentes antineoplásicos, y que podrían haber impactado en el análisis de la sobrevida global. Los resultados de eficacia se presentan en Tabla 15.
Utilizando un modelo de regresión multivariado de Cox exploratorio retrospectivo se observó que los siguientes factores pronósticos basales estaban fuertemente asociados con la sobrevida independiente del tratamiento: sexo, recuento de glóbulos blancos y de plaquetas, pérdida de peso corporal en los 6 meses anteriores al ingreso en el ensayo, número de localizaciones metastásicas, suma del diámetro mayor de las lesiones diana, puntaje de Motzer. Cuando se realizó el ajuste de estos factores basales el resultado fue un tratamiento con un índice de riesgo de 0,78 (IC del 95% [0,63; 0,96], p = 0,0219), indicando una reducción del riesgo de muerte del 22% para los pacientes del brazo Avastin + IFN alfa-2a en comparación con los del brazo IFN alfa-2a. En 97 pacientes en el brazo de IFN alfa-2a y en 131 en el brazo de Avastin se redujo la dosis de IFN alfa-2a de 9 MUI a 6 o 3 MUI tres veces por semana, según lo especificado en el protocolo. Sobre la base de los resultados de la tasa de sobrevida libre de progresión (SLP libre de eventos) a lo largo del tiempo, la reducción de dosis de IFN alfa-2a no afectó a la eficacia de la combinación de Avastin e IFN alfa-2a, tal y como se demostró por un análisis de subgrupos. Los 131 pacientes en el brazo de bevacizumab + IFN alfa-2a que disminuyeron y mantuvieron la dosis de IFN alfa-2a a 6 o 3 MUI durante el ensayo, presentaron resultados de la tasa de sobrevida libre de progresión (SLP libre de eventos) a los 6, 12 y 18 meses del 73, 52 y 21%, respectivamente, en comparación con el 61, 43 y 17% de la población total de los pacientes que recibieron bevacizumab + IFN alfa-2a. AVF2938: Se realizó un ensayo clínico de Fase II, aleatorizado, doble-ciego, en el que se investigó Avastin 10 mg/kg en un esquema de 2 semanas frente a la misma dosis de Avastin en combinación con 150 mg de erlotinib diarios, en pacientes con carcinoma renal de células claras con metástasis. En este ensayo, un total de 104 pacientes fueron aleatorizados para recibir tratamiento, 53 con Avastin 10 mg/kg cada 2 semanas + placebo y 51 con Avastin 10 mg/kg cada 2 semanas asociado con erlotinib 150 mg diariamente. El análisis del criterio de valoración primario de eficacia no mostró diferencia entre el brazo de Avastin + placebo y el brazo de Avastin + erlotinib (mediana de sobrevida libre de progresión 8,5 frente a 9,9 meses). Siete pacientes en cada brazo mostraron una respuesta objetiva. La adición de erlotinib a bevacizumab no mostró una mejoría en la sobrevida global (SG), (índice de riesgo = 1,764; p = 0,1789), duración de la respuesta objetiva (6,7 frente a 9,1 meses) o el tiempo hasta la progresión de los síntomas (índice de riesgo = 1,172; p = 0,5076). AVF0890: Se realizó un ensayo clínico de Fase II aleatorizado para comparar la eficacia y seguridad de bevacizumab frente a placebo. Se aleatorizaron un total de 116 pacientes para recibir bevacizumab 3 mg/kg cada 2 semanas (n = 39), 10 mg/kg cada 2 semanas (n = 37) o placebo (n = 40). Un análisis provisional demostró que había un incremento significativo del tiempo hasta la progresión de la enfermedad en el grupo de 10 mg/kg en comparación con el grupo placebo (índice de riesgo = 2,55; p < 0,001). Existió una pequeña diferencia, al límite de la significación estadística, entre el tiempo a la progresión de la enfermedad en el grupo de 3 mg/kg y en el grupo placebo (índice de riesgo = 1,26; p = 0,053). Cuatro pacientes que habían recibido la dosis de 10 mg/kg de bevacizumab mostraron una respuesta objetiva parcial; la tasa de respuesta global (TRG) para la dosis de 10 mg/kg fue del 10 %. Glioblastoma (Grado IV según la OMS): AVF3708g: Estudio multicéntrico, abierto, randomizado y no comparativo (AVF3708g) que evaluó la eficacia y el perfil de seguridad de Avastin en el tratamiento de pacientes con glioblastoma. Los pacientes que habían experimentado una primera o segunda recaída después de radioterapia (que debía haber finalizado por lo menos 8 semanas antes de la administración de Avastin) y temozolomida fueron distribuidos al azar (1:1) para recibir Avastin en monoterapia (infusión i.v. de 10 mg/kg cada dos semanas) o una asociación de Avastin + irinotecán (125 mg/m2 por vía i.v. o para pacientes tratados simultáneamente con antiepilépticos inductores enzimáticos 340 mg/m2 por vía i.v. cada dos semanas) hasta la progresión de la enfermedad o toxicidad inaceptable. Los criterios de valoración primarios del estudio fueron la sobrevida libre de progresión a los 6 meses (SLP) y la tasa de respuestas objetivas (TRO) evaluada por un grupo de expertos independiente (IRF). Los otros criterios de valoración incluyeron la duración de la SLP y de la respuesta al tratamiento y la sobrevida global. La Tabla 16 presenta un resumen de los resultados de este estudio.
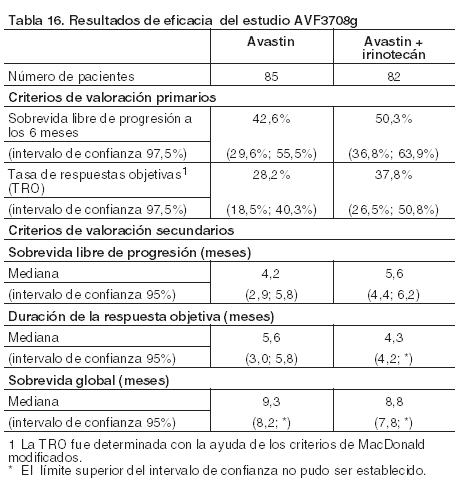
Las tasas de respuestas objetivas y la sobrevida libre de progresión (SLP) a los 6 meses en ambos brazos de tratamiento fueron significativamente mejores que las de los controles históricos. La sobrevida global mediana fue más prolongada en el grupo Avastin que en el de la asociación Avastin + irinotecán con 9,3 meses versus 8,8 meses, respectivamente. La mayoría de los pacientes que estaban recibiendo esteroides al inicio del tratamiento, incluyendo respondedores y no respondedores, fueron capaces de reducir la utilización de los mismos durante todo el transcurso del tratamiento con bevacizumab. La mayoría experimentó una respuesta objetiva o prolongada en la sobrevida libre de progresión (en la semana 24) y pudo mantener o mejorar sus funciones neurocognitivas mientras continuaba con el tratamiento del estudio comparado con su condición al inicio del mismo. La mayor parte de los que permanecían en el estudio, y a las 24 semanas estaban libres de progresión, demostraron estabilidad en el estado de desempeño de Karnofsky. Cáncer de Ovario Epitelial, Trompa de Falopio o Peritoneal Primario: Tratamiento de primera línea de cáncer de ovario: Se ha estudiado la seguridad y eficacia de Avastin en el tratamiento de primera línea en pacientes con cáncer de ovario epitelial, trompa de Falopio, o peritoneal primario en dos ensayos Fase III (GOG-0218 y BO17707) diseñados para evaluar el efecto de Avastin en combinación con carboplatino y paclitaxel en comparación con un régimen de quimioterapia sola. GOG-0218: Este ensayo de Fase III, multicéntrico, aleatorizado, doble-ciego, controlado con placebo y de tres brazos, evaluó el efecto de la adición de Avastin a un régimen de quimioterapia aprobado (carboplatino y paclitaxel) en pacientes con cáncer avanzado (estadios FIGO IIIB, IIIC y IV) de ovario epitelial, trompa de Falopio, o peritoneal primario. Se excluyeron del ensayo aquellas pacientes que habían recibido previamente bevacizumab o tratamiento sistémico para el cáncer de ovario (por ejemplo, quimioterapia, anticuerpos monoclonales, inhibidores de la tirosina quinasa, terapia hormonal) o radioterapia previa en el abdomen o pelvis. Se aleatorizaron en proporciones iguales un total de 1.873 pacientes en los siguientes tres brazos: Brazo CPP: Cinco ciclos de placebo (comenzando en el ciclo 2) en combinación con carboplatino (ABC 6) y paclitaxel (175 mg/m2) durante 6 ciclos seguido de placebo solo, hasta un total de 15 meses de tratamiento. Brazo CPB15: Cinco ciclos de Avastin (15 mg/ kg cada tres semanas, comenzando en el ciclo 2) en combinación con carboplatino (ABC 6) y paclitaxel (175 mg/m2) durante 6 ciclos seguido de placebo solo, hasta un total de 15 meses de tratamiento. Brazo CPB15+: Cinco ciclos de Avastin (15 mg/ kg cada tres semanas, comenzando en el ciclo 2) en combinación con carboplatino (ABC 6) y paclitaxel (175 mg/m2) durante 6 ciclos seguido del uso continuado de Avastin como monoterapia (15 mg/ kg cada tres semanas), hasta un total de 15 meses de tratamiento. La mayoría de las pacientes incluidas en el estudio fueron caucásicas (87% en los tres brazos); la mediana de la edad fue de 60 años en los brazos CPP y CPB15 y de 59 años en el brazo CPB15+; y el 29% en los brazos CPP o CPB15 y el 26% en el brazo CPB15+ tenían más de 65 años. En general, aproximadamente el 50% de las pacientes tenía al comienzo un puntaje de GOG PS de 0, el 43% de 1, y el 7% d
e 2. La mayoría tenía cáncer de ovario epitelial (82% en los brazos CPP y CPB15, 85% en el brazo CPB15+), seguido de cáncer peritoneal primario (16% en el brazo CPP, 15% en el brazo CPB15, 13% en el brazo CPB15+) y cáncer de trompa de Falopio (1% en el brazo CPP, 3% en el brazo CPB15, 2% en el brazo CPB15+). La mayoría tenía una histología de adenocarcinoma seroso (85% en los brazos CPP y CPB15, 86% en el brazo CPB15+). En general, aproximadamente el 34% de las pacientes tenía un estadio FIGO III con enfermedad residual macroscópica después de una citorreducción óptima con enfermedad residual macroscópica, el 40% un estadio III con una citorreducción subóptima, y el 26% un estadio IV. La variable principal fue la sobrevida libre de progresión evaluada por el investigador como progresión de la enfermedad y en base a las exploraciones radiológicas o a los niveles de CA-125, o al deterioro sintomático por protocolo. Además, se realizó un análisis preespecificado de los datos censurados para los eventos de progresión por CA-125, así como una revisión independiente de la sobrevida libre de progresión determinada por las exploraciones radiológicas. El ensayo alcanzó su variable principal de mejoría en la sobrevida libre de progresión. Las pacientes que recibieron bevacizumab con una dosis de 15 mg/kg cada tres semanas en combinación con quimioterapia y que continuaron recibiendo bevacizumab en monoterapia (CPB15+) tuvieron una mejoría clínica y estadísticamente significativa de la sobrevida libre de progresión en comparación con aquellas tratadas sólo con quimioterapia (carboplatino y paclitaxel) en el tratamiento de primera línea. En las pacientes que sólo recibieron bevacizumab en combinación con quimioterapia y que no continuaron recibiendo bevacizumab en monoterapia (CPB15), no se observó una mejoría clínicamente significativa. Los resultados de este ensayo se presentan en la Tabla 17.
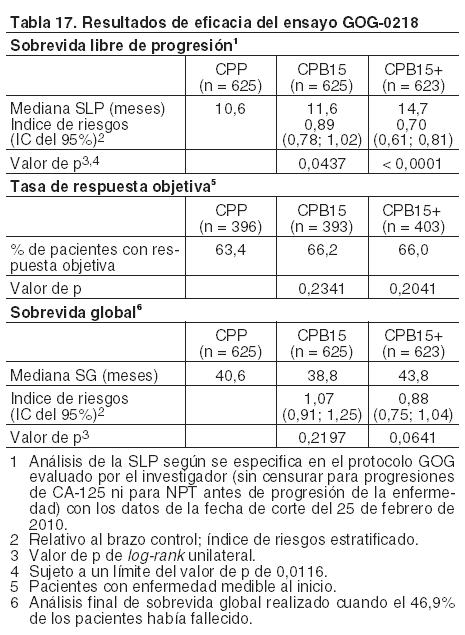
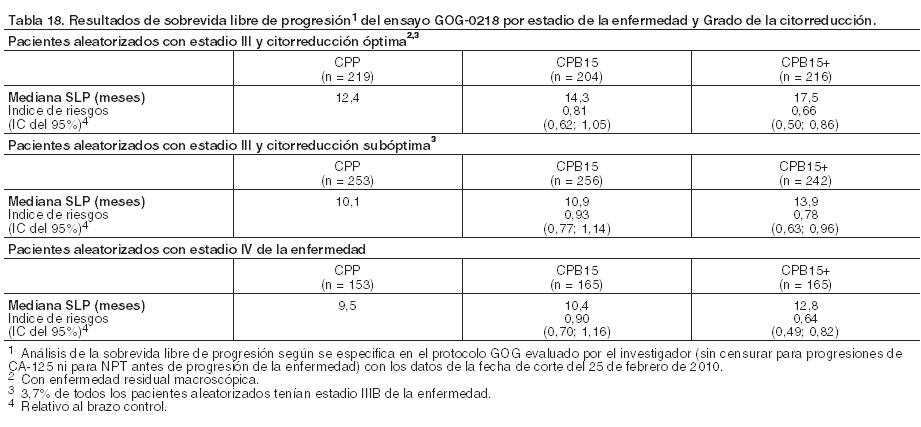
Se llevaron a cabo análisis preespecificados de sobrevida libre de progresión, todos ellos con datos clínicos de la fecha de corte del 29 de septiembre de 2009. Los resultados fueron los siguientes: El análisis de la sobrevida libre de progresión evaluada por el investigador según se específica en el protocolo (sin censurar la progresión por CA-125 o NPT) muestra un índice de riesgos estratificado de 0,71 (IC del 95%: 0,61-0,83, valor de p de log-rank unilateral < 0,0001) cuando se compara CPB15+ con CPP, con una mediana de sobrevida libre de progresión de 10,4 meses en el brazo CPP y de 14,1 meses en el brazo CPB15+. El análisis principal de la sobrevida libre de progresión evaluada por el investigador (censurando la progresión por CA-125 y NPT) muestra un índice de riesgos estratificado de 0,62 (IC del 95%: 0,52-0,75, valor de p de log-rank unilateral < 0,0001) cuando se compara CPB15+ con CPP, con una mediana de sobrevida libre de progresión de 12,0 meses en el brazo CPP y de 18,2 meses en el brazo CPB15+. El análisis de la sobrevida libre de progresión determinada por el Comité de revisión independiente (censurando para NPT) muestra un índice de riesgos estratificado de 0,62 (IC del 95%: 0,50-0,77, valor de p de log-rank unilateral < 0,0001) cuando se compara CPB15+ con CPP, con una mediana de sobrevida libre de progresión de 13,1 meses en el brazo CPP y de 19,1 meses en el brazo CPB15+. El examen por subgrupos de la sobrevida libre de progresión según el estadio de la enfermedad y el Grado de la citorreducción se presenta en la Tabla 18. Estos resultados demuestran la consistencia de los análisis de la sobrevida libre de progresión que se detallan en la Tabla 17.
BO17707 (ICON7): Este ensayo de Fase III, multicéntrico, aleatorizado, controlado, abierto y de dos brazos evaluó el efecto de la adición de Avastin a carboplatino más paclitaxel después de cirugía, en pacientes con estadio FIGO I o IIA (Grado 3 o sólo histología celular clara; n=142), o estadio FIGO IIB-IV (todos los Grados y todos los tipos de histología, n=1.386) de cáncer de ovario epitelial, trompa de Falopio, o peritoneal primario (NCI-CTCAE v.3). Se excluyeron del ensayo aquellas pacientes que habían recibido previamente bevacizumab o tratamiento sistémico para el cáncer de ovario (por ejemplo, quimioterapia, anticuerpos monoclonales, inhibidores de la tirosina quinasa, o terapia hormonal) o radioterapia previa en el abdomen o pelvis. Se aleatorizaron en proporciones iguales un total de 1.528 pacientes en los siguientes dos brazos: Brazo CP: Carboplatino (ABC 6) y paclitaxel (175 mg/m2) durante 6 ciclos de 3 semanas de duración. Brazo CPB 7,5+: Carboplatino (ABC 6) y paclitaxel (175 mg/m2) durante 6 ciclos de 3 semanas de duración más Avastin (7,5 mg/ kg cada tres semanas) hasta 12 meses (Avastin comenzó en el ciclo 2 de la quimioterapia, si el tratamiento se inició en las 4 semanas de la cirugía, o en el ciclo 1, si comenzó con más de cuatro semanas después de la cirugía). La mayoría de las pacientes incluidas en el estudio fueron caucásicas (96%), la mediana de la edad fue de 57 años en ambos brazos de tratamiento, el 25% tenía 65 años o más en cada brazo de tratamiento, y aproximadamente el 50% tenía un porcentaje de ECOG PS de 1; el 7% en cada brazo de tratamiento tenía un puntaje de ECOG PS de 2. La mayoría tenía cáncer de ovario epitelial (87,7%) seguido de cáncer peritoneal primario (6,9%) y cáncer de trompa de Falopio (3,7%) o una mezcla de los tres tipos de cáncer (1,7%). La mayoría tenía estadio FIGO III (68% en ambos brazos) seguido de estadio FIGO IV (13% y 14%), estadio FIGO II (10% y 11%) y estadio FIGO I (9% y 7%). Al inicio del estudio, la mayoría en cada brazo de tratamiento (74% y 71%) tenía tumores primarios poco diferenciados (Grado 3) (NCI-CTCAE v. 3). La incidencia de los subtipos histológicos de cáncer de ovario epitelial fue similar entre los brazos de tratamiento; el 69% de las pacientes en cada brazo tenía histología de adenocarcinoma seroso. La variable principal fue la sobrevida libre de progresión evaluada por el investigador usando criterios RECIST. El ensayo alcanzó su variable principal de mejoría en la sobrevida libre de progresión. Las pacientes que recibieron bevacizumab con una dosis de 7,5 mg/kg cada tres semanas en combinación con quimioterapia y que continuaron recibiendo bevacizumab en monoterapia hasta 18 ciclos, tuvieron una mejoría estadísticamente significativa de la sobrevida libre de progresión, en comparación con aquellas tratadas sólo con quimioterapia (carboplatino y paclitaxel) de primera línea. Los resultados de este ensayo se presentan en la Tabla 19.

El análisis principal de la sobrevida libre de progresión evaluada por el investigador con los datos de la fecha de corte del 28 de febrero de 2010, muestra un índice de riesgos no estratificado de 0,79 (IC del 95%: 0,68 - 0,91, valor de p de log-rank unilateral 0,0010), con una mediana de sobrevida libre de progresión de 16,0 meses en el brazo CP y de 18,3 meses en el brazo CPB7, 5+. El examen por subgrupos de la sobrevida libre de progresión según el estadio de la enfermedad y el Grado de la citorreducción se presenta en la Tabla 20. Estos resultados demuestran la consistencia del análisis principal de la sobrevida libre de progresión que se detalla en la Tabla 19.

Cáncer de ovario recurrente: La seguridad y eficacia de Avastin en el tratamiento de cáncer de ovario epitelial recurrente, carcinoma de trompa de Falopio, o carcinoma peritoneal primario se evaluó en dos ensayos Fase III (AVF4095g y MO22224) con diferentes poblaciones de pacientes y regímenes de quimioterapia. AVF4095g evaluó la eficacia y seguridad de bevacizumab en combinación con carboplatino y gemcitabina en pacientes con cáncer de ovario epitelial recurrente platino sensible, carcinoma de trompa de Falopio, o carcinoma peritoneal primario. MO22224 evaluó la eficacia y seguridad de bevacizumab asociado con paclitaxel, topotecán, o doxorrubicina liposomal pegilada en pacientes con cáncer de ovario epitelial recurrente platino resistente, carcinoma de trompa de Falopio, o carcinoma peritoneal primario. AVF4095g: En este ensayo de Fase III, randomizado, doble-ciego y controlado con placebo, se ha estudiado la seguridad y eficacia de Avastin en el tratamiento de pacientes con cáncer de ovario epitelial sensible al platino y recurrente, de trompa de Falopio, o peritoneal primario que no habían recibido previamente quimioterapia o tratamiento previo con bevacizumab. El estudio comparó el efecto de agregar Avastin a la quimioterapia de carboplatino y gemcitabina y continuar el tratamiento con Avastin en monoterapia hasta progresión, frente a la combinación de carboplatino y gemcitabina. En el estudio sólo se incluyeron pacientes con carcinoma de ovario, peritoneal primario o trompa de Falopio confirmado histológicamente que habían recaído después de > 6 meses del tratamiento con quimioterapia basada en platino y que no habían recibido quimioterapia durante la recaída ni tratamiento previo con bevacizumab, otros inhibidores VEGF o agentes dirigidos frente a receptores VEGF. Un total de 484 pacientes con enfermedad medible fueron aleatorizadas en proporciones 1:1 en los siguientes dos brazos: Carboplatino (ABC4, día 1) y gemcitabina (1.000 mg/m2 en los días 1 y 8) y placebo en forma concurrente cada 3 semanas durante 6 y hasta 10 ciclos, seguidos de placebo solo (cada 3 semanas) hasta progresión de la enfermedad o toxicidad inaceptable. Carboplatino (ABC4, día 1) y gemcitabina (1.000 mg/m2 en los días 1 y 8) y Avastin en forma concurrente (15 mg/kg día 1) cada 3 semanas durante 6 y hasta 10 ciclos seguidos de Avastin (15 mg/kg cada 3 semanas) en monoterapia hasta progresión de la enfermedad o toxicidad inaceptable. La variable principal fue la sobrevida libre de progresión basada en la evaluación del investigador usando los criterios modificados RECIST 1.0. Otras variables adicionales fueron respuesta objetiva, duración de la respuesta, sobrevida global y seguridad. También se llevó a cabo una revisión independiente de la variable principal. Los resultados de este ensayo se presentan en la Tabla 21.
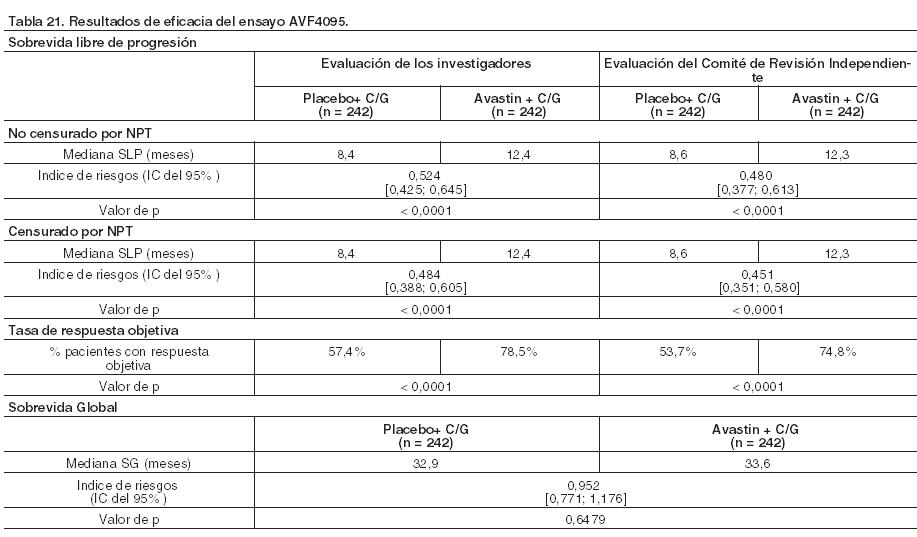
Los análisis de la sobrevida libre de progresión por subgrupos desde el último tratamiento con platino hasta recaída se presentan en la Tabla 22.
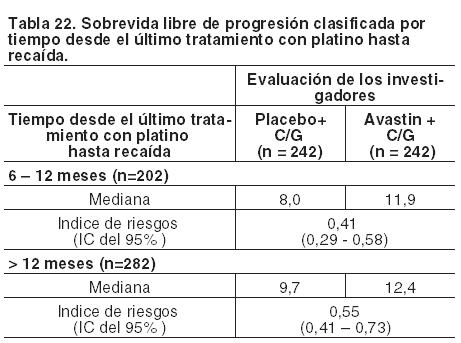
MO22224 (AURELIA): En este ensayo se evaluó la eficacia y seguridad de bevacizumab en combinación con quimioterapia en pacientes con cáncer de ovario epitelial recurrente platino resistente, carcinoma de trompa de Falopio, o carcinoma peritoneal primario. Este ensayo clínico fue diseñado como un Fase III, abierto, aleatorizado y de dos grupos para la evaluación de bevacizumab más quimioterapia (QT + BV) en comparación con quimioterapia sola (QT). Se incorporó un total de 361 pacientes a las cuales se les administró o bien quimioterapia (paclitaxel, topotecán o doxorrubicina liposomal pegilada [DLP]) sola o en combinación con bevacizumab: Grupo con quimioterapia sola (QT): Paclitaxel 80 mg/m2 en infusión intravenosa de 1 hora en los días 1, 8, 15 y 22 cada 4 semanas. Topotecán 4 mg/m2 en infusión intravenosa de 30 minutos en los días 1, 8 y 15 cada 4 semanas. Alternativamente, una dosis de 1,25 mg/m2 podría ser administrada durante 30 minutos en los días 1 - 5 cada 3 semanas. DLP 40 mg/m2 en infusión intravenosa de 1 mg/min en el día 1 cada 4 semanas. Después del ciclo 1, el fármaco se puede administrar como infusión durante 1 hora. Grupo con quimioterapia y bevacizumab (QT + BV): La quimioterapia elegida se combinó con bevacizumab 10 mg/kg en infusión intravenosa cada 2 semanas (o bevacizumab 15 mg/kg cada 3 semanas si se usa asociada con topotecán 1,25 mg/m2 en los días 1 - 5 en un ciclo de 3 semanas). Las pacientes seleccionadas tenían cáncer de ovario epitelial, carcinoma de trompa de Falopio o carcinoma peritoneal primario, que progresaron dentro de los 6 meses de terapia previa con platino consistente en un mínimo de 4 ciclos de tratamiento. Las pacientes debían tener una expectativa de vida mayor o igual a 12 semanas y ninguna radioterapia previa sobre la pelvis o el abdomen. La mayoría era FIGO estadio IIIC o IV y en ambos grupos tenía un puntaje de ECOG PS de 0 (QT: 56,4% en comparación con QT + BV: 61,2%). El porcentaje con un puntaje de ECOG PS de 1 o ≥ 2 fue del 38,7% y de un 5,0% en el grupo de QT, y de 29,8% y 9,0% en el de QT + BV. Se dispone de información sobre la raza del 29,3% de las pacientes, y casi todas eran blancas. La edad promedio era de 61,0 años (rango: 25 - 84). Un total de 16 pacientes (4,4%) era mayor de 75 años de edad. La tasa global de interrupción, debido a eventos adversos fue del 8,8% en el grupo QT y del 43,6% en el de QT + BV (principalmente debido a eventos adversos de Grados 2-3) y el tiempo promedio de interrupción en el grupo de QT + BV fue de 5,2 meses comparado con 2,4 meses en el de QT. La tasa de interrupción debida a eventos adversos en el subgrupo de pacientes mayores de 65 años de edad fue del 8,8% en el grupo QT y del 50,0% en el de QT + BV. El índice de riesgos (HR) para la sobrevida libre de progresión fue de 0,47 (IC del 95%: 0,35, 0,62) y de 0,45 (IC del 95%: 0,31, 0,67) para los subgrupos menores de 65 años y mayores de 65 años, respectivamente. El objetivo primario fue la sobrevida libre de progresión, con variables secundarias que incluyeron tasa de respuesta objetiva y sobrevida global. Los resultados se presentan en la Tabla 23.
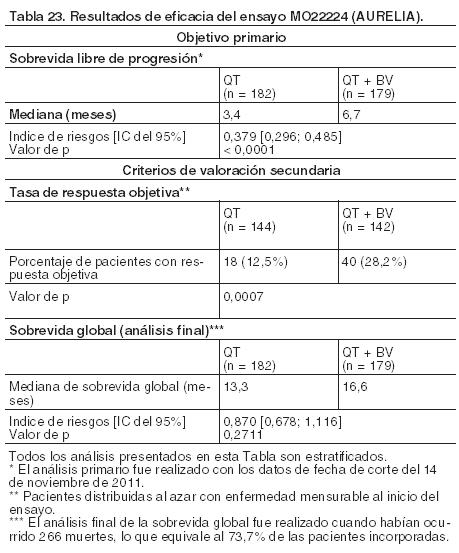
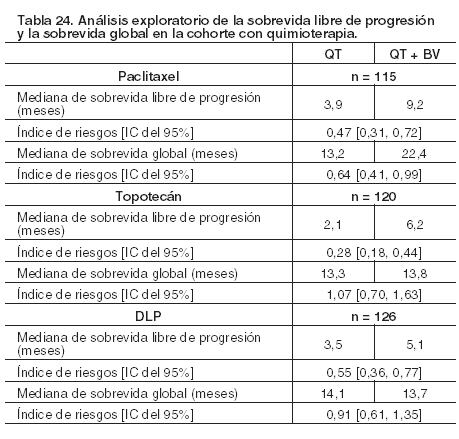
El ensayo alcanzó su objetivo primario de mejoría de la sobrevida libre de progresión. Las pacientes recurrentes platino resistentes tratadas únicamente con quimioterapia (paclitaxel, topotecán o DLP) en comparación con aquéllas que recibieron bevacizumab en una dosis de 10 mg/kg cada 2 semanas (o 15 mg/kg cada 3 semanas si se usaba asociado con 1,25 mg/m2 de topotecán en los días 1 - 5 cada 3 semanas) en combinación con quimioterapia y que continuaron con bevacizumab hasta progresión de la enfermedad o toxicidad inaceptable, tuvieron una mejoría estadísticamente significativa de la sobrevida libre de progresión. Los análisis exploratorios de sobrevida libre de progresión y sobrevida global de acuerdo con las distintas cohortes de quimioterapia (paclitaxel, topotecán y DLP) mostraron mejoría con la adición de bevacizumab. Los resultados se resumen en la Tabla 24.
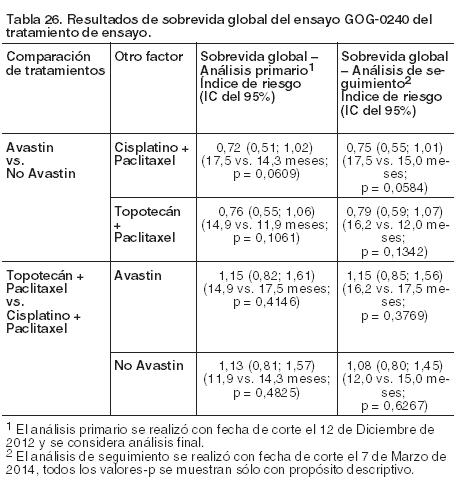
Cáncer de cuello uterino: GOG-0240: La seguridad y eficacia de Avastin en combinación con quimioterapia (paclitaxel y cisplatino o paclitaxel y topotecán) en el tratamiento de pacientes con carcinoma persistente, recurrente o metastásico del cuello uterino se evaluó en este ensayo de Fase III, aleatorizado, multicéntrico, abierto y de cuatro grupos. Un total de 452 pacientes fueron distribuidas al azar para recibir ya sea: Paclitaxel 135 mg/m2 intravenoso durante 24 horas en el día 1 y cisplatino 50 mg/m2 intravenoso en el día 2, cada 3 semanas; o paclitaxel 175 mg/m2 intravenoso durante 3 horas en el día 1 y cisplatino 50 mg/m2 intravenoso en el día 2, cada 3 semanas; o paclitaxel 175 mg/m2 intravenoso durante 3 horas en el día 1 y cisplatino 50 mg/m2 intravenoso en el día 1, cada 3 semanas. Paclitaxel 135 mg/m2 intravenoso durante 24 horas en el día 1 y cisplatino 50 mg/m2 intravenoso en el día 2 más bevacizumab 15 mg/kg intravenoso en el día 2, cada 3 semanas; o paclitaxel 175 mg/m2 intravenoso durante 3 horas en el día 1 y cisplatino 50 mg/m2 intravenoso en el día 2 más bevacizumab 15 mg/kg intravenoso en el día 2, cada 3 semanas; o paclitaxel 175 mg/m2 intravenoso durante 3 horas en el día 1 y cisplatino 50 mg/m2 intravenoso en el día 1 y bevacizumab 15 mg/kg intravenoso en el día 1, cada 3 semanas. Paclitaxel 175 mg/m2 intravenoso durante 3 horas en el día 1 y topotecán 0,75 mg/m2 intravenoso durante 30 minutos en los días 1 - 3, cada 3 semanas. Paclitaxel 175 mg/m2 intravenoso durante 3 horas en el día 1 y topotecán 0,75 mg/m2 intravenoso durante 30 minutos en los días 1 - 3 más bevacizumab 15 mg/kg intravenoso en el día 1, cada 3 semanas. Las pacientes elegibles tenían carcinoma de células escamosas persistente, recurrente o metastásico, carcinoma adenoescamoso, o adenocarcinoma de cuello uterino, que no era tratable mediante cirugía y/o radioterapia, y que no hubieran recibido tratamiento previo con bevacizumab u otros inhibidores VEFG o agentes dirigidos frente a receptores VEGF. La media de edad era de 46,0 años (rango: 20 - 83) en el grupo de quimioterapia sola y de 48 años (rango: 22 - 85) en el de quimioterapia más Avastin; y mayores de 65 años un 9,3% de las pacientes del grupo de quimioterapia sola y un 7,5% de aquéllas del de quimioterapia más Avastin. De las 452 pacientes aleatorizadas al inicio del estudio, la mayoría era de raza blanca (80,0% en el grupo de quimioterapia sola y 75,3% en el de quimioterapia más Avastin) y tenían carcinoma de células escamosas (67,1% en el grupo de quimioterapia sola y 69,6% en el de quimioterapia más Avastin), enfermedad persistente/recurrente (83,6% en el grupo de quimioterapia sola y 82,8% en el de quimioterapia más Avastin), 1 - 2 localizaciones metastásicas (72,0% en el grupo de quimioterapia sola y 76,2% en el de quimioterapia más Avastin), compromiso de los ganglios linfáticos (50,2% en el grupo de quimioterapia sola y 56,4% en el de quimioterapia más Avastin), y con un intervalo libre de platino ≥ 6 meses (72,5% en el grupo de quimioterapia sola y 64,4% en el de quimioterapia más Avastin). La variable principal de eficacia fue la sobrevida global. Las variables secundarias de eficacia incluían sobrevida libre de progresión y tasa de respuesta objetiva. Los resultados del análisis primario y de seguimiento se presentan para el tratamiento con Avastin y para el tratamiento de ensayo en las Tablas 25 y 26, respectivamente.

Población pediátrica: La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con bevacizumab en los diferentes grupos de la población pediátrica, en carcinoma de mama, adenocarcinoma del colon y del recto, carcinoma de pulmón (carcinoma de células pequeñas y de células no pequeñas), carcinoma de riñón y de la pelvis renal (excluyendo nefroblastoma, nefroblastomatosis, sarcoma de células claras, nefroma mesoblástico, carcinoma de la médula renal y tumor rabdoide del riñón), carcinoma ovárico (excluyendo rabdomiosarcoma y tumores de células germinales), carcinoma de trompa de Falopio (excluyendo rabdomiosarcoma y tumores de células germinales), carcinoma peritoneal (excluyendo blastomas y sarcomas) y cérvix y carcinoma de cuerpo uterino. En dos estudios con un total de 30 niños mayores de 3 años con glioma de alto Grado recidivante o progresivo no se observó actividad antitumoral cuando fueron tratados con bevacizumab e irinotecán. No se dispone de información suficiente para determinar la seguridad y la eficacia de bevacizumab en niños con glioma de alto Grado recién diagnosticado. En un estudio de un solo brazo (PBTC-022), 18 niños con glioma de alto Grado no pontino recurrente o progresivo (incluyendo 8 con glioblastoma [Grado 4 de la OMS], 9 con astrocitoma anaplásico [Grado 3] y 1 con oligodendroglioma anaplásico [Grado 3]) fueron tratados con bevacizumab (10 mg/kg) dos semanas y luego con bevacizumab en combinación con CPT-11 (125-350 mg/m2), una vez cada dos semanas hasta la progresión. No se lograron respuestas radiológicas parciales o totales objetivas (criterios de MacDonald). La toxicidad y las reacciones adversas fueron hipertensión arterial y fatiga, así como isquemia del sistema nervioso central con déficit neurológico agudo. En una serie retrospectiva realizada en una única Institución, 12 niños con glioma de alto Grado recidivante o progresivo (3 con Grado 4 de la OMS, 9 con Grado 3) fueron tratados consecutivamente (de 2005 a 2008) con bevacizumab (10 mg/kg) e irinotecán (125 mg/m2) cada 2 semanas. Se registraron 2 respuestas parciales y ninguna completa (criterios de MacDonald). Propiedades farmacocinéticas: Los datos farmacocinéticos de bevacizumab provienen de 10 ensayos clínicos realizados en pacientes con tumores sólidos en los que se administró bevacizumab en infusión IV. El ritmo de infusión se estableció en base a la tolerabilidad, con una duración de 90 minutos para la administración inicial. La farmacocinética de bevacizumab fue lineal en un intervalo de dosis de 1 a 10 mg/kg. Como se observó con otros anticuerpos, los datos farmacocinéticos de bevacizumab son descriptos por un modelo de dos compartimientos. En términos generales, en todos los ensayos clínicos, la disposición de bevacizumab estaba caracterizada por un clearance lento, un volumen de distribución limitado del compartimiento central (Vc), y una vida media de eliminación prolongada. Estos parámetros aseguran la presencia de niveles plasmáticos terapéuticos de bevacizumab estables, con un amplio rango de esquemas de administración (tales como, una vez cada 2 o cada 3 semanas). En un meta-análisis farmacocinético poblacional no se detectaron diferencias significativas en la farmacocinética de bevacizumab con respecto a la raza cuando se tiene en consideración el peso corporal o en relación con la edad (ninguna correlación entre el clearance de bevacizumab y la edad de los pacientes [la mediana de la edad fue de 59 años, y los percentilos 5 y 95 de 37 y 76 años, respectivamente]). Distribución: El valor medio del volumen central (Vc) fue de 2,73 litros para mujeres y de 3,28 litros para hombres, los cuales están en el rango descripto para las IgG y otros anticuerpos monoclonales. Cuando bevacizumab se administró junto con agentes antineoplásicos, el valor medio del volumen periférico (Vp) fue de 1,69 litros para mujeres y de 2,35 litros para hombres. Después de la corrección en función del peso corporal, los hombres tuvieron un mayor Vc (+ 20%) que las mujeres. Biotransformación: La evaluación del metabolismo de bevacizumab en conejos, después de la administración de una dosis única IV de 125I-bevacizumab indicó que su perfil metabólico era similar al esperado para una IgG nativa que no se uniera al VEGF. El metabolismo y la eliminación de bevacizumab son similares a los de la IgG endógena, es decir, el catabolismo se produce principalmente por vía proteolítica en todo el organismo, incluyendo las células endoteliales y no depende principalmente de la eliminación hepática y renal. La unión al receptor FcRn protege la IgG del metabolismo celular, resultando en una prolongada vida media de eliminación terminal. Eliminación: La farmacocinética de bevacizumab es lineal en dosis que oscilan entre los 1,5 y 10 mg/kg/semana. El valor del clearance es, por término medio, igual a 0,188 y 0,220 l/día para pacientes mujeres y hombres, respectivamente. Después de la corrección en función del peso corporal, los hombres tenían el clearance de bevacizumab más alto (+17%) que las mujeres. Según el modelo bicompartimental, la vida media de eliminación es de 18 días para una paciente femenina media y de 20 días para un paciente masculino medio. Bajos valores de albúmina y una alta carga tumoral son generalmente indicativos de la gravedad de la enfermedad. El clearance de bevacizumab fue aproximadamente un 30% más rápido en pacientes con niveles bajos de albúmina sérica y un 7% más rápido en aquellos con una alta carga tumoral cuando se comparó con un paciente con valores medios de albúmina y carga tumoral. Farmacocinética en poblaciones especiales: Se analizó la farmacocinética poblacional para determinar los efectos de las características demográficas. Los resultados mostraron que no existe una diferencia significativa en la farmacocinética de bevacizumab en relación con la edad. Pacientes pediátricos: La farmacocinética de bevacizumab se ha estudiado en un número limitado de pacientes pediátricos. Los datos farmacocinéticos resultantes sugieren que el volumen de distribución y el clearance de bevacizumab son comparables a los obtenidos en adultos con tumores sólidos. Pacientes con insuficiencia renal: No se han realizado ensayos para investigar la farmacocinética de bevacizumab en pacientes con insuficiencia renal, ya que el riñón no es un órgano principal para el metabolismo o excreción de bevacizumab. Pacientes con insuficiencia hepática: No se han efectuado ensayos para investigar la farmacocinética de bevacizumab en pacientes con insuficiencia hepática, ya que el hígado no es un órgano principal para el metabolismo o excreción de bevacizumab. Datos preclínicos sobre seguridad: En estudios de hasta 26 semanas de duración realizados con macacos (monos cinomolgus) se observó displasia ósea en animales jóvenes con cartílagos de crecimiento abiertos, en concentraciones séricas medias de bevacizumab inferiores a las esperadas con dosis recomendadas para los seres humanos. En conejos, se comprobó que bevacizumab inhibe la cicatrización en dosis inferiores a la dosis clínica recomendada. Se ha observado que los efectos sobre la cicatrización son completamente reversibles. No se han realizado estudios para evaluar el potencial mutagénico y carcinogénico de bevacizumab. No se han llevado a cabo estudios específicos en animales para evaluar el efecto sobre la fertilidad. Sin embargo, puede esperarse un efecto adverso sobre la fertilidad femenina, ya que en estudios de toxicidad con dosis repetidas realizados en animales, se registró una inhibición de la maduración de los folículos ováricos, una disminución/ausencia del cuerpo lúteo y un descenso asociado del peso de ovarios y útero, así como una reducción en el número de ciclos menstruales. Se ha observado que bevacizumab es embriotóxico y teratogénico en conejos. Entre los efectos descriptos se incluyen disminución del peso corporal materno y fetal, aumento del número de resorciones fetales y de la incidencia de malformaciones macroscópicas específicas y esqueléticas del feto. Las consecuencias negativas sobre el feto se verificaron con todas las dosis estudiadas. Con la dosis más baja empleada, las concentraciones séricas medias fueron aproximadamente 3 veces mayores que en seres humanos tratados con 5 mg/kg cada 2 semanas. Información sobre malformaciones fetales observadas en el entorno poscomercialización se proporciona en Precauciones; Fertilidad, embarazo y lactancia y en Reacciones adversas.
Indicaciones.
Carcinoma metastásico de colon o recto (CCRm): Bevacizumab está indicado en combinación con quimioterapia basada en fluoropirimidinas para el tratamiento de pacientes adultos con carcinoma metastásico de colon o recto. Cáncer de mama metastásico (CMm): Bevacizumab está indicado en combinación con paclitaxel para el tratamiento de primera línea de pacientes adultos con cáncer de mama metastásico. Para más información sobre el estado del receptor 2 del factor de crecimiento epidérmico humano (HER2), véase Farmacología; Propiedades farmacodinámicas. Bevacizumab está indicado, en combinación con capecitabina, para el tratamiento de primera línea de pacientes adultos con cáncer de mama metastásico, en los que no se considere apropiado el tratamiento con otras opciones de quimioterapia que incluyan taxanos o antraciclinas. Los pacientes que hayan recibido regímenes de tratamiento que contienen taxanos y antraciclinas en el entorno adyuvante en los últimos 12 meses deben ser excluidos del tratamiento con Avastin en combinación con capecitabina. Para más información sobre el estado del HER2, véase Farmacología; Propiedades farmacodinámicas. Cáncer de pulmón no microcítico (CPNM): Bevacizumab está indicado, asociado con quimioterapia basada en platino, para el tratamiento de primera línea de pacientes adultos con cáncer de pulmón no microcítico avanzado no resecable, metastásico o recidivante, a excepción de los que tengan un tipo histológico con predominio de células escamosas. Bevacizumab está indicado, en combinación con erlotinib, para el tratamiento de primera línea de pacientes adultos con cáncer de pulmón no microcítico no escamoso avanzado no resecable, metastásico o recidivante con mutaciones activadoras del receptor del factor de crecimiento epidérmico (EGFR) véase Farmacología; Propiedades farmacodinámicas. Cáncer de células renales avanzado y/o metastásico (CRm): Bevacizumab está indicado en combinación con interferón alfa-2a para el tratamiento de primera línea de pacientes adultos con cáncer de células renales avanzado y/o metastásico. Glioblastoma (Grado IV según la OMS): Bevacizumab está indicado como monoterapia en el tratamiento de pacientes con recidiva de glioblastoma (Grado IV según la OMS) después de una terapia anterior con temozolomida. Cáncer de ovario epitelial, trompa de Falopio o peritoneal primario: Bevacizumab está indicado en combinación con carboplatino y paclitaxel para el tratamiento de primera línea de pacientes adultas con cáncer avanzado (estadios de la Federación Internacional de Ginecología y Obstetricia [FIGO] IIIB, IIIC y IV) de ovario epitelial, trompa de Falopio, o peritoneal primario. Bevacizumab está indicado en combinación con carboplatino y gemcitabina para el tratamiento de pacientes adultas con cáncer de ovario epitelial, carcinoma de trompa de Falopio, o carcinoma peritoneal primario sensible al platino después de la primera recaída, que no hayan recibido tratamiento previo con bevacizumab, otros inhibidores del factor de crecimiento del endotelio vascular (VEGF) o agentes dirigidos frente a receptores VEGF. Bevacizumab está indicado en combinación con paclitaxel, topotecán, o doxorrubicina liposomal pegilada para el tratamiento de pacientes adultas con cáncer de ovario epitelial, carcinoma de trompa de Falopio, o carcinoma peritoneal primario recurrente platino resistente, que no hayan recibido más de dos regímenes previos de quimioterapia ni tratamiento previo con bevacizumab, otros inhibidores del factor de crecimiento del endotelio vascular (VEGF) o agentes dirigidos frente a receptores VEGF (véase Farmacología; Propiedades farmacodinámicas). Cáncer de cuello uterino: Bevacizumab está indicado en combinación con paclitaxel y cisplatino o carboplatino, o paclitaxel y topotecán para el tratamiento de pacientes adultas con carcinoma persistente, recurrente o metastásico del cuello uterino.
Dosificación.
General: El reemplazo por cualquier otro agente biológico requiere el consentimiento del médico prescriptor. Avastin debe administrarse bajo la supervisión de un médico con experiencia en el empleo de medicamentos antineoplásicos. Posología: Carcinoma metastásico de colon o de recto (CCRm): La dosis recomendada de Avastin es de 5 mg/kg o 10 mg/kg de peso corporal administrada como infusión intravenosa una vez cada 2 semanas o de 7,5 mg/kg o 15 mg/kg de peso corporal administrados una vez cada 3 semanas. Se recomienda continuar el tratamiento hasta la progresión de la enfermedad subyacente o hasta toxicidad inaceptable. Cáncer de mama metastásico (CMm): La dosis recomendada de Avastin es de 10 mg/kg de peso corporal una vez cada 2 semanas o de 15 mg/kg de peso corporal una vez cada 3 semanas administrada como infusión intravenosa. Se recomienda continuar el tratamiento hasta la progresión de la enfermedad subyacente o hasta toxicidad inaceptable. Cáncer de pulmón no microcítico (CPNM): Primera línea de tratamiento para CPNM no escamoso en combinación con quimioterapia basada en platino: Avastin se administra en combinación con quimioterapia basada en platino hasta 6 ciclos de tratamiento, seguido de Avastin en monoterapia hasta la progresión de la enfermedad. La dosis recomendada de Avastin es de 7,5 mg/kg o 15 mg/kg de peso corporal administrada como infusión intravenosa una vez cada 3 semanas. En los pacientes con CPNM se ha demostrado el beneficio clínico con las dosis tanto de 7,5 mg/kg como de 15 mg/kg (véase Farmacología; Propiedades farmacodinámicas). Se recomienda continuar el tratamiento hasta la progresión de la enfermedad subyacente o hasta toxicidad inaceptable. Primera línea de tratamiento para CPNM no escamoso con mutaciones activadoras en EGFR en combinación con erlotinib: Se debe llevar a cabo el test de la mutación de EGFR antes de iniciar el tratamiento con la combinación de Avastin y erlotinib. Cuando se evalúa el estado de mutación del EGFR de un paciente, es importante elegir una metodología adecuadamente validada y confiable para evitar la obtención de falsos negativos o falsos positivos. La dosis recomendada de Avastin cuando se utiliza en combinación con erlotinib es de 15 mg/kg de peso corporal administrada como infusión intravenosa una vez cada 3 semanas. Se recomienda continuar el tratamiento con Avastin en combinación con erlotinib hasta progresión de la enfermedad. Para la Dosificación de erlotinib, consultar el Prospecto Información para Profesionales del producto. Cáncer de células renales avanzado y/o metastásico (CRm): La dosis recomendada de Avastin es de 10 mg/kg de peso corporal administrada como infusión intravenosa una vez cada 2 semanas. Se recomienda continuar el tratamiento hasta la progresión de la enfermedad subyacente o hasta toxicidad inaceptable. Glioblastoma (Grado IV según la OMS): La dosis recomendada de Avastin es de 10 mg/kg de peso corporal cada 2 semanas como infusión intravenosa. Los datos existentes no avalan la dosis de 15 mg/kg cada 3 semanas. Se recomienda continuar el tratamiento hasta la progresión de la enfermedad subyacente o hasta toxicidad inaceptable. Cáncer de ovario epitelial, trompa de Falopio o peritoneal primario: Tratamiento de primera línea: Avastin se administra en combinación con carboplatino y paclitaxel durante 6 ciclos de tratamiento, seguido de un uso continuado de Avastin en monoterapia hasta la progresión de la enfermedad o hasta un máximo de 15 meses o toxicidad inaceptable, lo que ocurra primero. La dosis recomendada de Avastin es de 15 mg/kg de peso corporal una vez cada 3 semanas como infusión intravenosa. Tratamiento de la enfermedad recurrente platino sensible: Avastin se administra en combinación con carboplatino y gemcitabina durante 6 ciclos y hasta 10 ciclos de tratamiento seguido de un uso continuado de Avastin en monoterapia hasta la progresión de la enfermedad. La dosis recomendada de Avastin es de 15 mg/kg de peso corporal administrada como infusión intravenosa una vez cada 3 semanas. Tratamiento de la enfermedad recurrente platino resistente: Avastin se administra en combinación con uno de los siguientes fármacos: paclitaxel, topotecán (administrado semanalmente) o doxorrubicina liposomal pegilada. La dosis recomendada de Avastin es de 10 mg/kg de peso corporal administrada como infusión intravenosa una vez cada 2 semanas. Cuando Avastin se asocia con topotecán (administrado en los días 1 - 5, cada 3 semanas), la dosis recomendada de Avastin es de 15 mg/kg de peso corporal administrada como infusión intravenosa una vez cada 3 semanas. Se aconseja continuar el tratamiento hasta progresión de la enfermedad o toxicidad inaceptable (véase Farmacología; Propiedades farmacodinámicas, ensayo clínico MO22224). Cáncer de cuello uterino: Avastin se administra en combinación con uno de los siguientes regímenes quimioterápicos: paclitaxel y cisplatino o carboplatino, o paclitaxel y topotecán. La dosis recomendada de Avastin es de 15 mg/kg de peso corporal administrada como infusión intravenosa una vez cada 3 semanas. Se recomienda continuar con el tratamiento hasta progresión de la enfermedad o toxicidad inaceptable (véase Farmacología; Propiedades farmacodinámicas). Poblaciones especiales: Población pediátrica: No se ha establecido la seguridad y eficacia de bevacizumab en niños y adolescentes. Avastin no está aprobado para su uso en pacientes menores de 18 años. Bevacizumab no debe utilizarse en la población pediátrica para las indicaciones aprobadas. Los datos actualmente disponibles se incluyen en "Reacciones adversas; Farmacología; Propiedades farmacodinámicas, Propiedades farmacocinéticas y Datos preclínicos sobre seguridad", sin embargo no se puede hacer una recomendación posológica. Avastin no se debe utilizar en niños de 3 a 18 años con glioma de alto Grado recurrente o progresivo por motivos de eficacia (véase Farmacología - Propiedades farmacodinámicas, Población pediátrica). No se recomienda la reducción de la dosis en caso de aparición de reacciones adversas. Si es necesario, el tratamiento debe interrumpirse permanente o temporalmente como se indica en "Precauciones". Pacientes de edad avanzada: No es necesario un ajuste de la dosis en pacientes de edad avanzada. Pacientes con insuficiencia renal: No se han estudiado la seguridad y la eficacia en pacientes con insuficiencia renal (véase Farmacología; Propiedades farmacocinéticas). Pacientes con insuficiencia hepática: No se han estudiado la seguridad y la eficacia en pacientes con insuficiencia hepática (véase Farmacología; Propiedades farmacocinéticas). Forma de administración: La dosis inicial debe administrarse en infusión intravenosa durante 90 minutos. Si se tolera bien la primera infusión, la segunda puede administrarse durante 60 minutos. Si se tolera bien la infusión de 60 minutos, todas las infusiones siguientes se pueden administrar durante 30 minutos. No se debe administrar en infusión intravenosa rápida ni en bolo. Precauciones que deben tomarse antes de manipular o administrar este medicamento: Para consultar las instrucciones de dilución del medicamento antes de la administración, véase Precauciones especiales de eliminación y otras manipulaciones. Las infusiones de Avastin no deben administrarse o mezclarse con soluciones de glucosa, (véase Incompatibilidades). Este medicamento no debe mezclarse con otros fármacos, excepto los mencionados en el ítem Precauciones especiales de eliminación y otras manipulaciones.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de sus excipientes. Hipersensibilidad a los productos derivados de células de ovario de hámster chino (CHO), o a otros anticuerpos recombinantes humanos o humanizados. Embarazo (véase Precauciones - Fertilidad, embarazo y lactancia).
Reacciones adversas.
Resumen del perfil de seguridad: El perfil de seguridad global de Avastin está basado en los datos de aproximadamente 5.400 pacientes con varios tipos de cáncer, tratados en su mayoría con Avastin en combinación con quimioterapia en ensayos clínicos. Las reacciones adversas más graves fueron: Perforaciones gastrointestinales (véase Precauciones). Hemorragia, incluyendo hemorragia pulmonar/hemoptisis, más frecuente en pacientes con cáncer de pulmón no microcítico (véase Precauciones). Tromboembolismo arterial (véase Precauciones). En los ensayos clínicos las reacciones adversas observadas g
lobalmente con mayor frecuencia en pacientes tratados con Avastin fueron: hipertensión, fatiga o astenia, diarrea y dolor abdominal. Los análisis de los datos de seguridad clínica sugieren que la incidencia de hipertensión y proteinuria durante la terapia con Avastin probablemente sea dosis-dependiente. Tabla de reacciones adversas: Las reacciones adversas enumeradas en esta sección se clasifican por frecuencia en las siguientes categorías: Muy frecuentes (≥1/10); frecuentes (≥1/100 y < 1/10); poco frecuentes (≥1/1.000 y < 1/100); raras (≥1/10.000 y < 1/1.000); muy raras ( < 1/10.000); y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las Tablas 1 y 2 enumeran las reacciones adversas asociadas con el uso de Avastin en combinación con diferentes regímenes de quimioterapia en múltiples indicaciones. La Tabla 1 muestra reacciones adversas clasificadas por frecuencia. Se determinó que éstas tenían una relación causal con Avastin a través de: Incidencias relativas observadas entre los grupos de tratamiento de ensayo clínico (al menos con una diferencia del 10% de reacciones NCI-CTCAE de Grados 1-5 o al menos con una diferencia del 2% de reacciones NCI-CTCAE de Grados 3-5). Estudios de seguridad posautorización. La notificación espontánea. Los estudios epidemiológicos/no intervencionales o los estudios observacionales. La evaluación de notificaciones de casos individuales. La Tabla 2 muestra la frecuencia de reacciones adversas graves. Estas se definen como eventos adversos con al menos una diferencia del 2% en comparación con el grupo control en los ensayos clínicos para reacciones NCI-CTCAE de Grados 3-5. La Tabla 2 también incluye las reacciones adversas que son consideradas por Roche como clínicamente significativas o graves. En las Tablas 1 y 2, se incluyen las reacciones adversas poscomercialización, según corresponda. La información detallada sobre estas manifestaciones posteriores a la comercialización se muestra en la Tabla 3. Las reacciones adversas se incluyen en la categoría de frecuencia apropiada en las siguientes Tablas según la incidencia más alta observada en cualquier indicación. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Algunas se observan frecuentemente con la quimioterapia; sin embargo, Avastin puede exacerbar estas manifestaciones cuando se combina con agentes quimioterápicos. Los ejemplos incluyen el síndrome de eritrodisestesia palmo-plantar con doxorrubicina liposomal pegilada o capecitabina, neuropatía sensorial periférica con paclitaxel u oxaliplatino, alteraciones de las uñas o alopecia con paclitaxel y paroniquia con erlotinib.
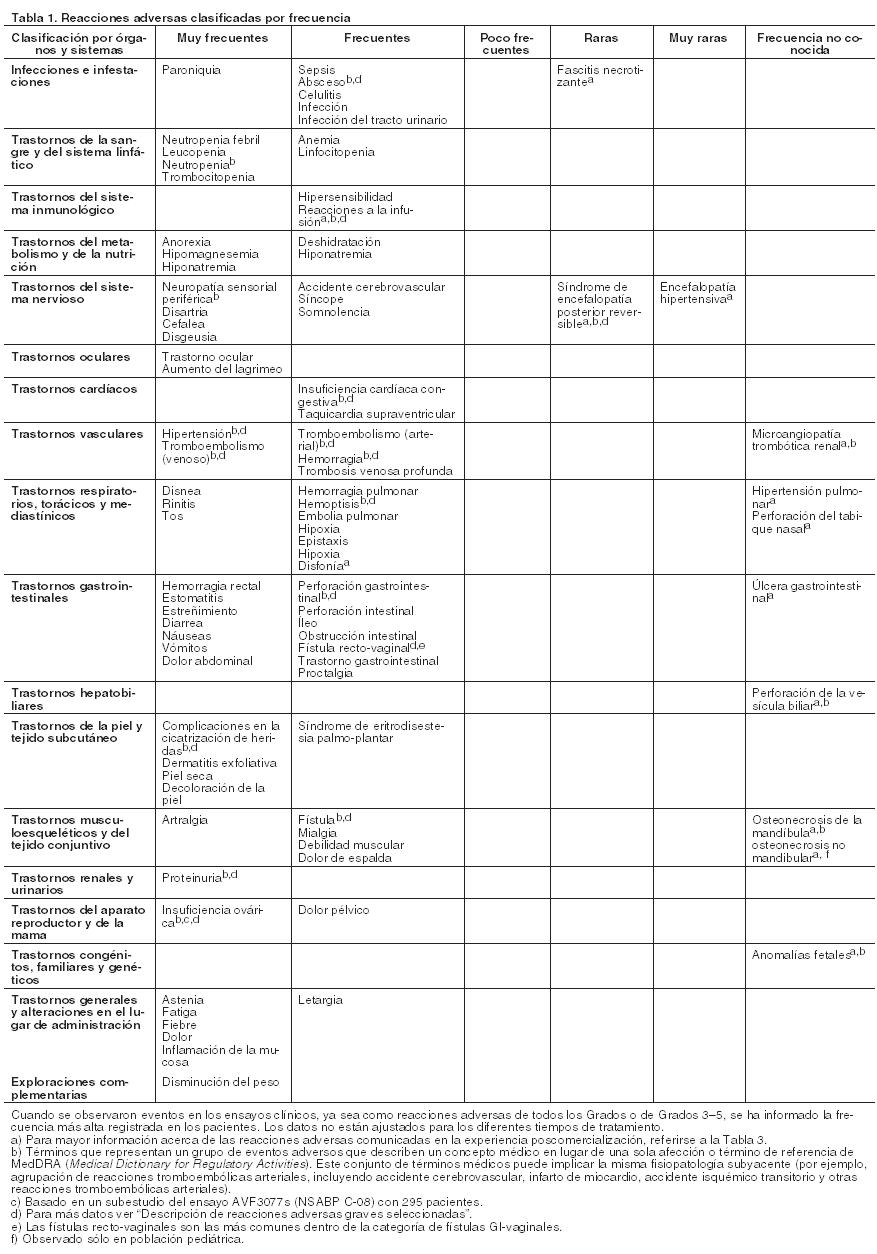
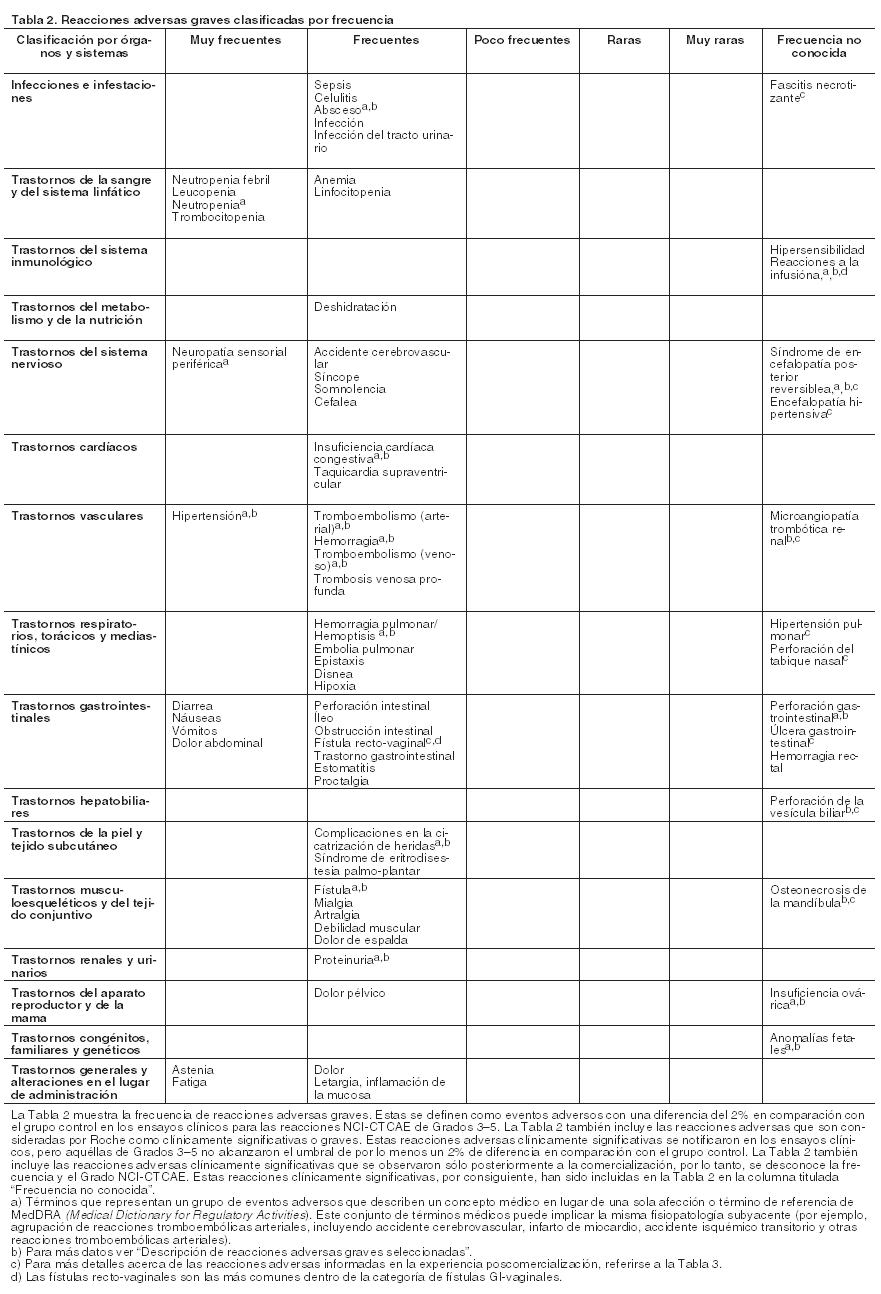
Descripción de reacciones adversas graves seleccionadas: Perforaciones gastrointestinales (GI) y Fístulas (véase Precauciones): Se ha asociado el uso de Avastin con casos graves de perforación gastrointestinal. En los ensayos clínicos se han notificado casos de perforaciones gastrointestinales con una incidencia menor del 1% en pacientes con cáncer de pulmón no microcítico, de hasta un 1,3% en pacientes con cáncer de mama metastásico, de hasta un 2% en aquellos con cáncer de células renales metastásico, con diagnóstico reciente de glioblastoma o con cáncer de ovario, y de hasta un 2,7% en pacientes con cáncer colorrectal metastásico (incluyendo fístula gastrointestinal y abscesos). Se han observado también casos de perforaciones gastrointestinales en pacientes con glioblastoma recaído. En un ensayo clínico en pacientes con cáncer cervical persistente, recurrente o metastásico (GOG-0240), se registraron perforaciones gastrointestinales (todos los Grados) en 3,2% de los pacientes, todos los cuales tenían una historia de radiación pélvica previa. Se registró diferencia en el tipo y gravedad de estas reacciones, comprendiendo desde la presencia de aire libre detectada en radiografía simple de abdomen, que se resolvió sin necesidad de tratamiento, hasta la perforación intestinal con absceso abdominal y desenlace mortal. Algunos casos ya presentaban inflamación intra-abdominal subyacente como consecuencia de úlcera gástrica, necrosis tumoral, diverticulitis o de colitis asociada con quimioterapia. Se ha comunicado desenlace mortal en aproximadamente un tercio de los casos graves de perforaciones gastrointestinales, lo que representa entre el 0,2% y el 1% de todos los pacientes tratados con Avastin. En los ensayos clínicos con Avastin, se informaron casos de fístula gastrointestinal (de todos los Grados) con una incidencia de hasta 2% en pacientes con cáncer colorrectal metastásico y cáncer de ovario, aunque también, pero con menos frecuencia, en aquéllos con otros tipos de cáncer. Fístulas GI-vaginales en el estudio GOG-240: En un estudio de pacientes con cáncer de cuello uterino persistente, recurrente o metastásico, la incidencia de fístulas GI-vaginales fue de 8,3% en los pacientes tratados con Avastin y 0,9% en los pacientes control, todos los cuales tenían antecedentes de radiación pélvica previa. La frecuencia de fístulas GI-vaginales en el grupo tratado con Avastin + quimioterapia fue mayor en pacientes con recurrencia de la enfermedad dentro del campo previamente radiado (16,7%) comparado con aquéllos con recurrencia de la enfermedad fuera del campo de la radiación previa (3,6%). Las frecuencias correspondientes en el grupo control que recibió únicamente quimioterapia fueron del 1,1% frente al 0,8%, respectivamente. Aquéllos, que desarrollan fístulas GI-vaginales, pueden tener obstrucciones intestinales y requerir una intervención quirúrgica, como así también ostomías. Fístulas no gastrointestinales (GI) (véase Precauciones): El uso de Avastin se ha asociado con casos graves de fístulas, incluyendo reacciones con desenlace mortal. En un estudio clínico con pacientes con cáncer cervical persistente, recurrente o metastásico (GOG-0240), 1,8% de las tratadas con Avastin y 1,4% de las pacientes control reportaron haber tenido fístulas no gastrointestinales vaginales, vesicales o fístulas en el tracto genital femenino. En varias indicaciones se observaron casos poco frecuentes (≥ 0,1% y < 1%) de fístulas que comprometen a otras partes del organismo diferentes del tracto gastrointestinal (por ejemplo, fístulas broncopleurales y biliares). También se han comunicado fístulas durante la experiencia poscomercialización. Las reacciones se notificaron en distintos momentos del tratamiento, desde la primera semana hasta pasado el primer año desde el inicio con Avastin, produciéndose la mayoría de los casos dentro de los primeros 6 meses de tratamiento. Cicatrización de heridas (véase Precauciones): Debido a que Avastin puede tener un impacto negativo en la cicatrización de heridas, se excluyeron de los ensayos clínicos de Fase III aquellos pacientes que se habían sometido a cirugía mayor en los últimos 28 días. En los ensayos clínicos de carcinoma metastásico de colon o recto, los pacientes que habían sido sometidos a cirugía mayor entre los 28 y los 60 días antes de iniciar la terapia con Avastin no presentaron un aumento del riesgo de hemorragia postoperatoria ni se registraron complicaciones en la cicatrización de heridas. Se observó que si los pacientes estaban siendo tratados con Avastin en el momento de la cirugía, presentaban mayor riesgo de hemorragia posoperatoria o problemas en la cicatrización de heridas en los 60 días siguientes a la cirugía mayor. La incidencia osciló entre el 10% (4/40) y el 20% (3/15). Se han notificado complicaciones graves en la cicatrización de heridas, incluyendo las relacionadas con una anastomosis, algunas de las cuales con resultado de muerte. En los ensayos de cáncer de mama localmente recidivante y metastásico se verificaron complicaciones en la cicatrización de heridas de Grados 3 - 5 en hasta 1,1% de las pacientes tratadas con Avastin comparado con hasta 0,9% en los brazos control (NCI-CTCAE v. 3). En el estudio AVF3708g de pacientes con glioblastoma recaído, la incidencia de complicaciones en la cicatrización de las heridas posoperatorias (incluidas dehiscencia de la herida en el lugar de la craneotomía y pérdida del líquido cerebroespinal) fue de 3,6% en los pacientes tratados con Avastin como único agente y de 1,3% en aquellos que recibieron Avastin más irinotecán. En ensayos clínicos de cáncer de ovario, se observaron complicaciones en la cicatrización de las heridas de Grados 3-5 hasta en un 1,2% de las pacientes del brazo de bevacizumab frente al 0,1% del brazo control (NCI-CTCAE v. 3). En el estudio BO21990 de pacientes con diagnóstico reciente de glioblastoma, la incidencia de complicaciones en la cicatrización de las heridas posoperatorias de Grados 3 - 5 (incluidas las que se manifestaron después de craneotomía) fue de 3,3% en los pacientes tratados con Avastin en combinación con quimioterapia y radioterapia y de 1,6% en aquellos que recibieron sólo quimioterapia y radioterapia. Hipertensión (véase Precauciones): En los ensayos clínicos, a excepción del estudio JO25567, la incidencia global de hipertensión (todos los Grados) fue de hasta un 42,1% en los pacientes tratados con Avastin comparado con hasta el 14% en aquellos que recibieron el comparador. La incidencia global NCI-CTC de la hipertensión de Grados 3 y 4 se produjo en 0,4% al 17,9% de los pacientes tratados con Avastin. La hipertensión de Grado 4 (crisis hipertensiva) se manifestó en hasta un 1,0% de los pacientes tratados con Avastin y quimioterapia comparada con hasta el 0,2% a los que se administró la misma quimioterapia sola. En el estudio JO25567, se observaron todos los Grados de hipertensión en el 77,3% de los pacientes que recibieron bevacizumab en combinación con erlotinib como primera línea de tratamiento para CPNM no escamoso con mutaciones activadoras en EGFR, en comparación con el 14,3% de los tratados con erlotinib solo. La hipertensión de Grado 3 se produjo en el 60,0% de los pacientes tratados con bevacizumab en combinación con erlotinib en comparación con el 11,7% a los que se administró erlotinib solo. No se produjeron acontecimientos de hipertensión de Grados 4 o 5. En general, la hipertensión se controló adecuadamente con antihipertensivos orales, tales como inhibidores de la enzima convertidora de angiotensina, diuréticos y bloqueadores de los canales de calcio. Rara vez fue necesaria la interrupción del tratamiento con Avastin o la hospitalización. Se han notificado casos muy raros de encefalopatía hipertensiva, algunos de los cuales fueron mortales. No existe una correlación entre el riesgo de hipertensión asociada con el tratamiento con Avastin y las características basales de los pacientes, la enfermedad subyacente o la terapia concomitante. Síndrome de Encefalopatía Posterior Reversible (véase Precauciones): Se han informado casos raros de pacientes tratados con Avastin que desarrollan signos y síntomas que concuerdan con el Síndrome de Encefalopatía Posterior Reversible (SEPR), un caso raro de trastorno neurológico. Su manifestación puede incluir convulsiones, dolor de cabeza, estado mental alterado, alteraciones visuales, o ceguera cortical, con o sin hipertensión asociada. Las características clínicas del SEPR son, a menudo, inespecíficas, y, por lo tanto, su diagnóstico requiere confirmación mediante técnicas de imagen cerebral, preferiblemente resonancia magnética (RM). En pacientes que desarrollan SEPR, se recomienda un reconocimiento temprano de los síntomas, con un tratamiento oportuno de los síntomas específicos, incluyendo control de la hipertensión (si está asociado con hipertensión grave no controlada), además de interrumpir bevacizumab. Los síntomas normalmente se resuelven o mejoran en los días posteriores a la interrupción del tratamiento, aunque algunos pacientes han experimentado algunas secuelas neurológicas. No se conoce la seguridad de reanudar la terapia con Avastin en pacientes que hayan experimentado previamente SEPR. A través de los ensayos clínicos, se han informado 8 casos de SEPR. Dos de los ocho casos no tuvieron confirmación radiológica por RM. Proteinuria (véase Precauciones): En los ensayos clínicos, se han comunicado casos de proteinuria en un intervalo desde el 0,7% hasta el 54,7% de los pacientes tratados con Avastin. La gravedad de la proteinuria varió desde clínicamente asintomática, transitoria, indicios de proteinuria hasta síndrome nefrótico, siendo la gran mayoría de los casos proteinuria de Grado 1 (NCI-CTCAE v. 3). Se registró proteinuria de Grado 3 en hasta el 8,1% de los pacientes tratados. La proteinuria de Grado 4 (síndrome nefrótico) se observó en hasta el 1,4% de los pacientes tratados. Los pacientes con antecedentes de hipertensión pueden tener un mayor riesgo de proteinuria durante el tratamiento con Avastin. Existen datos que sugieren que la proteinuria de Grado 1 (NCI-CTCAE v. 3) puede estar relacionada con la dosis de Avastin. Se recomienda realizar pruebas de proteinuria antes de comenzar con Avastin. En la mayoría de los ensayos clínicos donde los niveles de proteínas en la orina fueron ≥ 2 g/24 horas, el tratamiento con Avastin fue suspendido hasta la recuperación de niveles < 2 g/24 horas. Hemorragia (véase Precauciones): En los ensayos clínicos en todas las indicaciones, la incidencia global de reacciones hemorrágicas de Grados 3 - 5 según la escala NCI-CTCAE v. 3, osciló desde 0,4% hasta 6,9% en los pacientes tratados con Avastin, comparado con hasta un 4,5% de los del grupo de quimioterapia control. En el ensayo clínico (GOG-0240) en pacientes con carcinoma persistente, recurrente o metastásico del cuello uterino, se han notificado reacciones hemorrágicas de Grados 3 - 5 en hasta el 8,3% de los pacientes tratados con Avastin en combinación con paclitaxel y topotecán comparado con hasta el 4,6% de aquéllos que recibieron paclitaxel y topotecán. Las reacciones hemorrágicas observadas en los ensayos clínicos fueron en su mayoría hemorragias asociadas con el tumor y hemorragias mucocutáneas menores (por ejemplo, epistaxis). Hemorragias asociadas con el tumor (véase Precauciones): La hemorragia pulmonar/hemoptisis grave o masiva se ha observado principalmente en ensayos con pacientes con cáncer de pulmón no microcítico (CPNM). Los posibles factores de riesgo incluyen histología de células escamosas, tratamiento con fármacos antirreumáticos/antiinflamatorios o con anticoagulantes, radioterapia previa, tratamiento con Avastin, historial médico previo de aterosclerosis, localización del tumor central y cavitación de tumores antes o durante el tratamiento. Las únicas variables que mostraron una correlación estadísticamente significativa con la hemorragia fueron el tratamiento con Avastin y la histología de células escamosas. Los pacientes con CPNM con un tipo histológico diagnosticado de células escamosas o con histología de tipo celular mixto con predominio de células escamosas se excluyeron de los ensayos Fase III posteriores, mientras que se incluyeron aquéllos con histología tumoral desconocida. En pacientes con CPNM, excluyendo los que tenían una histología con predominio de células escamosas, se observaron reacciones de todos los Grados con una frecuencia de hasta el 9,3% en los tratados con Avastin más quimioterapia comparado con hasta el 5% en aquellos que recibieron quimioterapia sola. Las reacciones de Grados 3 - 5 se han observado en hasta el 2,3% de los pacientes tratados con Avastin más quimioterapia comparado con < 1% con quimioterapia sola (NCI-CTCAE v. 3). La hemorragia pulmonar/hemoptisis grave o masiva puede presentarse en forma repentina y hasta dos tercios de las hemorragias pulmonares graves tuvieron un desenlace mortal. En pacientes con cáncer colorrectal se han notificado hemorragias gastrointestinales, incluyendo hemorragia rectal y melena, y se evaluaron como asociadas con el tumor. También se observaron casos raros de hemorragias vinculadas con el tumor en otros tipos y localizaciones tumorales, incluyendo casos en el sistema nervioso central (SNC) en pacientes con metástasis en el SNC (véase Precauciones) y en pacientes con glioblastoma. No se evaluó de manera prospectiva en los ensayos clínicos aleatorizados la incidencia de hemorragia en el SNC en pacientes con metástasis no tratadas localizadas en el SNC que recibieron bevacizumab. En un análisis exploratorio retrospectivo de los datos de 13 ensayos aleatorizados finalizados en pacientes con distintos tipos de tumores, 3 de 91 (3,3%) con metástasis cerebral experimentaron hemorragia del SNC (todas de Grado 4) cuando fueron tratados con bevacizumab, en comparación con 1 caso (Grado 5) de 96 (1%) que no recibió bevacizumab. En dos ensayos posteriores en pacientes con metástasis cerebral tratadas (que incluyeron alrededor de 800 pacientes), cuando se realizó el análisis de seguridad provisional se notificó un caso de Grado 2 de hemorragia en el SNC (1,2%) en los 83 pacientes a los que se administró bevacizumab (NCI-CTCAE v. 3). La hemorragia intracraneana puede sobrevenir en pacientes con glioblastoma recaído. En el estudio AVF3708g, la hemorragia en el SNC fue informada en 2,4% (2/84) de los pacientes del grupo Avastin solo (Grado 1), y en 3,8% (3/79) de aquellos tratados con Avastin más irinotecán (Grados 1, 2 y 4). Durante todos los ensayos clínicos, se observó hemorragia mucocutánea hasta en un 50% de los pacientes tratados con Avastin. Los más frecuentes fueron casos de epistaxis de Grado 1 según la escala (NCI-CTCAE v. 3) que duraron menos de 5 minutos, se resolvieron sin necesidad de tratamiento médico y no requirieron ningún cambio en el régimen establecido con Avastin. Los datos clínicos de seguridad sugieren que la incidencia de hemorragias mucocutáneas menores (por ejemplo, epistaxis) puede ser dependiente de la dosis. Asimismo, con menor frecuencia se produjeron casos de hemorragias mucocutáneas menores en otras localizaciones, tales como hemorragia gingival o vaginal. Tromboembolismo (véase Precauciones): Tromboembolismo arterial: En los pacientes tratados con Avastin en todas las indicaciones, se observó un aumento en la incidencia de reacciones tromboembólicas arteriales, incluyendo accidentes cerebrovasculares, infartos de miocardio, ataques isquémicos transitorios y otras reacciones tromboembólicas arteriales. En los ensayos clínicos, la incidencia global de las reacciones tromboembólicas arteriales fue de hasta un 3,8% en los brazos que incluyeron Avastin comparado con hasta el 2,1% en los brazos de quimioterapia control. Se notificó desenlace fatal en el 0,8% de los pacientes tratados con Avastin comparado con el 0,5% de los que recibieron quimioterapia sola. Se comunicaron accidentes cerebrovasculares (incluyendo ataques isquémicos transitorios) en hasta el 2,7% de los pacientes tratados con Avastin en combinación con quimioterapia comparado con hasta el 0,5% de aquellos a los que se administró quimioterapia sola. Se notificó infarto de miocardio en hasta el 1,4% de los pacientes tratados con Avastin en combinación con quimioterapia comparado con hasta el 0,7% de aquellos que recibieron quimioterapia sola. En un ensayo clínico para evaluar Avastin en combinación con 5-fluorouracilo/ácido folínico, AVF2192g, se incluyeron pacientes con cáncer colorrectal metastásico que no eran candidatos para el tratamiento con irinotecán. En este ensayo se observaron reacciones tromboembólicas arteriales en el 11% (11/100) de los pacientes comparado con el 5,8% (6/104) en el grupo de quimioterapia control. En el ensayo clínico AVF3708g no controlado, en pacientes con glioblastoma en recaída, se registraron eventos tromboembólicos arteriales en hasta el 6,3% (5/79) de los pacientes que recibieron Avastin en combinación con irinotecán, en comparación con hasta el 4,8% (4/84) a los que se les administró Avastin solo. Tromboembolismo venoso: La incidencia de reacciones tromboembólicas venosas en los ensayos clínicos fue similar en pacientes tratados con Avastin en combinación con quimioterapia comparada con aquellos que recibieron sólo quimioterapia control. Las reacciones tromboembólicas venosas incluyen trombosis venosa profunda, embolismo pulmonar y tromboflebitis. En los ensayos clínicos en todas las indicaciones, la incidencia global de reacciones tromboembólicas venosas osciló desde 2,8% hasta 17,3% de los pacientes tratados con Avastin en comparación con el 3,2% hasta 15,6% en los grupos control. Se han notificado reacciones tromboembólicas venosas de Grados 3 - 5 (NCI-CTCAE v. 3) en hasta un 7,8% de los pacientes tratados con quimioterapia más bevacizumab en comparación con hasta un 4,9% en aquellos que recibieron quimioterapia sola (en todas las indicaciones, excluyendo cáncer persistente, recurrente o metastásico del cuello uterino). En el ensayo clínico (GOG-0240) en pacientes con carcinoma persistente, recurrente o metastásico del cuello uterino, se han informado eventos de tromboembolismo venoso de Grados 3 - 5 en hasta el 15,6% de los pacientes tratados con Avastin en combinación con paclitaxel y cisplatino comparado con hasta el 7,0% de aquéllos que recibieron paclitaxel y cisplatino. Los pacientes que han sufrido una reacción tromboembólica venosa pueden tener un riesgo mayor de recurrencia con Avastin en combinación con quimioterapia que con quimioterapia sola. En el ensayo clínico BO21990, se observaron eventos tromboembólicos venosos de Grados 3 - 5 en el 7,6% de los pacientes con diagnóstico reciente de glioblastoma, tratados con Avastin en combinación con quimioterapia y radioterapia, en comparación con el 8,0% de los que recibieron sólo quimioterapia y radioterapia. Insuficiencia Cardíaca Congestiva (ICC): En los ensayos clínicos con Avastin, se observó insuficiencia cardíaca congestiva (ICC) en todas las indicaciones de cáncer estudiadas hasta la fecha, aunque tuvo lugar predominantemente en pacientes con cáncer de mama metastásico. En cuatro ensayos Fase III en pacientes con cáncer de mama metastásico (AVF2119g, E2100, BO17708 y AVF3694g) se notificó hasta en un 3,5% de las pacientes tratadas con Avastin en combinación con quimioterapia ICC de Grado 3 o superior (NCI-CTCAE v. 3) en comparación con hasta un 0,9% en los brazos control. En los pacientes del ensayo AVF3694g que recibieron antraciclinas en forma concomitante con bevacizumab, las incidencias de ICC de Grado 3 o superior en los brazos control y con bevacizumab fueron similares a las de otros ensayos en cáncer de mama metastásico: 2,9% en el brazo de antraciclina más bevacizumab y 0% en el brazo de antraciclina más placebo. Además, en el ensayo AVF3694g las incidencias de ICC de cualquier Grado fueron similares entre el brazo de antraciclina más Avastin (6,2%) y el de antraciclina más placebo (6,0%). Después de la terapia clínica apropiada, se registró una mejoría de los síntomas y/o de la función ventricular izquierda en la mayoría de los pacientes que desarrollaron ICC durante los ensayos en cáncer de mama metastásico. En la mayoría de los ensayos clínicos con Avastin, se excluyeron los pacientes con ICC preexistente de Grados II - IV de la NYHA (New York Heart Association); por lo tanto, no se dispone de información relacionada con el riesgo de agravamiento de la ICC en esta población. La exposición previa a antraciclinas y/o radiación previa sobre la pared torácica puede ser un posible factor de riesgo para el desarrollo de ICC. En un ensayo clínico de pacientes con linfoma difuso de células B grandes, se observó un incremento de la incidencia de ICC cuando recibieron bevacizumab con una dosis acumulada de doxorrubicina superior a 300 mg/m2. Este ensayo clínico de Fase III comparó rituximab/ciclofosfamida/doxorrubicina/vincristina/prednisona (R-CHOP) más bevacizumab con R-CHOP sin bevacizumab. Mientras que la incidencia de ICC fue, en ambos grupos, superior a la observada previamente para la terapia de doxorrubicina, la tasa fue mayor en el grupo R-CHOP más bevacizumab. Estos resultados sugieren que se debería considerar una observación clínica estrecha con evaluaciones cardiológicas apropiadas en aquellos pacientes expuestos a dosis de doxorrubicina acumuladas mayores de 300 mg/m2 cuando se combine con bevacizumab. Reacciones de hipersensibilidad/reacciones a la infusión (véanse Precauciones y Reacciones adversas, Experiencia poscomercialización): En algunos ensayos clínicos, se notificaron reacciones anafilácticas y de tipo anafilactoide con mayor frecuencia en los pacientes que habían recibido Avastin en combinación con quimioterapia que en los tratados con quimioterapia sola. La incidencia de estas reacciones en algunos ensayos clínicos con Avastin es frecuente (hasta un 5% en los pacientes a los que se administró bevacizumab). Infecciones: En el ensayo clínico (GOG-0240) en pacientes con carcinoma persistente, recurrente o metastásico del cuello uterino, se han notificado infecciones de Grados 3 - 5 en hasta el 24% de los pacientes tratados con Avastin en combinación con paclitaxel y topotecán comparados con hasta el 13% de aquéllos que recibieron paclitaxel y topotecán. En el ensayo clínico Fase III BO21990, aleatorizado, doble-ciego, controlado con placebo, multicéntrico, de Avastin en combinación con quimioterapia más radioterapia para el tratamiento de pacientes con diagnóstico reciente de glioblastoma, la incidencia de infecciones de todos los Grados fue de 54,4% y de Grados 3-5 del 12,8% en el brazo de bevacizumab más quimioterapia y radioterapia, en comparación con 39,1% y 7,8% en el de quimioterapia más radioterapia sola, respectivamente. Insuficiencia ovárica/Fertilidad (véase Precauciones): En el ensayo NSABP C-08, Fase III de Avastin en el tratamiento adyuvante de pacientes con cáncer de colon, se evaluó en 295 mujeres premenopáusicas la incidencia de nuevos casos de insuficiencia ovárica, definida como amenorrea de 3 o más meses, nivel de FSH ≥ 30 MUI/ml y un valor negativo de b-HCG para test de embarazo. Nuevos casos de insuficiencia ovárica se notificaron en un 2,6% de las pacientes del grupo mFOLFOX-6 en comparación con un 39% del grupo mFOLFOX-6 más bevacizumab. En un 86,2% de estas mujeres evaluadas se recuperó la función ovárica después de discontinuar bevacizumab. Se desconoce el efecto a largo plazo del tratamiento con bevacizumab en la fertilidad. Anomalías de laboratorio: La disminución del recuento de neutrófilos y de glóbulos blancos y la presencia de proteínas en la orina pueden estar asociadas con el tratamiento con Avastin. A través de los ensayos clínicos, en pacientes tratados con Avastin, aparecieron las siguientes anomalías de laboratorio de Grados 3 y 4 (NCI-CTCAE v. 3) con al menos un 2% de diferencia en comparación con los grupos control correspondientes: hiperglucemia, disminución de hemoglobina, hipopotasemia, hiponatremia, disminución del recuento de glóbulos blancos, aumento del Índice Normalizado Internacional (INR). Los ensayos clínicos han demostrado que los incrementos transitorios de la creatinina sérica (que oscila entre 1,5 a 1,9 veces el nivel basal), con y sin proteinuria, están asociados con el uso de Avastin. El aumento observado en la creatinina sérica no se vinculó con una mayor incidencia de manifestaciones clínicas de insuficiencia renal en los pacientes tratados con Avastin. Otras poblaciones especiales: Pacientes de edad avanzada: En los ensayos clínicos aleatorizados, la edad mayor de 65 años estaba asociada con un aumento del riesgo de reacciones tromboembólicas arteriales, incluyendo accidentes cerebrovasculares, ataques isquémicos transitorios e infartos de miocardio. Otras reacciones durante el tratamiento con Avastin que se observaron con una mayor frecuencia en pacientes mayores de 65 años fueron leucopenia y trombocitopenia de Grados 3 - 4 (NCI-CTCAE v. 3); y neutropenia, diarrea, náuseas, cefalea y fatiga de todos los Grados en comparación con los menores de 65 años (véanse Reacciones adversas y Precauciones, Tromboembolismo). En un ensayo clínico, la incidencia de hipertensión de Grado ≥ 3 fue dos veces más alta en pacientes mayores de 65 años que en el grupo más joven (menores de 65 años). En un ensayo en pacientes con cáncer de ovario recurrente platino resistente, también se informaron casos de alopecia, inflamación de las mucosas, neuropatía sensorial periférica, proteinuria e hipertensión, los cuales se produjeron con una tasa de por lo menos 5% superior en el grupo QT + BV en las pacientes de 65 años de edad o mayores tratadas con bevacizumab en comparación con las menores de 65 años de edad igualmente tratadas. No se observó un aumento en la incidencia de otras reacciones, incluyendo perforación gastrointestinal, complicaciones en la cicatrización, insuficiencia cardíaca congestiva y hemorragia, en los pacientes de edad avanzada (mayores de 65 años) tratados con Avastin en comparación con los menores de 65 años igualmente tratados. Población pediátrica: No se ha establecido la seguridad de Avastin en niños y adolescentes menores de 18 años. Avastin no está aprobado para su uso en pacientes menores de 18 años. Existen publicaciones, en las que se han observado casos de osteonecrosis no mandibular en pacientes menores de 18 años tratados con Avastin. Experiencia poscomercialización:
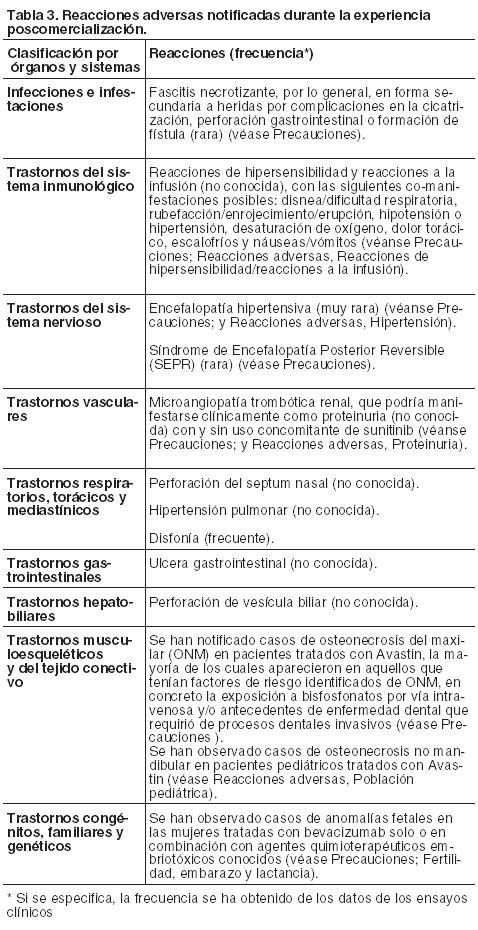
Comunicación de reportes de reacciones adversas: Es importante comunicar las presuntas reacciones adversas después de la autorización del medicamento. Esto permite la monitorización continua de la relación riesgo/beneficio. Se solicita a los profesionales de la salud informar de cualquier sospecha de eventos adversos asociados con el uso de Avastin® al Área de Farmacovigilancia de Roche al siguiente teléfono 0800-77-ROCHE (76243). En forma alternativa, esta información puede ser reportada ante ANMAT. Ante cualquier inconveniente con el producto, el paciente puede llenar la ficha que está en la Página Web de la ANMAT: http://www.anmat.gov.ar/farmacovigilancia/Notificar.asp o llamar a ANMAT responde al 0800-333-1234.
Precauciones.
A fin de mejorar la trazabilidad de los medicamentos biológicos, el nombre comercial y el número de lote del producto deben estar claramente registrados (o mencionados) en la historia clínica del paciente. Perforaciones gastrointestinales y Fístulas (véase Reacciones adversas): Los pacientes pueden tener un riesgo aumentado de perforación gastrointestinal y perforación de la vesícula biliar durante el tratamiento con Avastin. En aquéllos con carcinoma metastásico de colon o de recto, el proceso inflamatorio intraabdominal puede ser un factor de riesgo para perforaciones gastrointestinales, por lo que se debe tener precaución cuando se trate a estos pacientes. La radiación previa es un factor de riesgo para la perforación gastrointestinal en pacientes con carcinoma persistente, recurrente o metastásico del cuello uterino tratadas con Avastin y todas las pacientes con perforación gastrointestinal tenían antecedentes de radiación previa. Se debe interrumpir en forma permanente el tratamiento en quienes desarrollen una perforación gastrointestinal. Fístulas gastrointestinales-vaginales en el estudio GOG-0240: Las pacientes con carcinoma persistente, recurrente y metastásico del cuello uterino tratadas con Avastin pueden tener un mayor riesgo de fístulas entre la vagina y cualquier parte del tracto gastrointestinal (fístulas GI-vaginales) (véase Reacciones adversas). La radiación previa es un factor de riesgo importante para el desarrollo de fístula GI-vaginal y todas las pacientes con fístula GI-vaginal han tenido antecedentes de radiación previa. La recurrencia del cáncer en la zona previamente irradiada es un importante factor de riesgo adicional para el desarrollo de fístulas GI-vaginales. Fístulas no gastrointestinales (véase Reacciones adversas): Los pacientes pueden tener un riesgo aumentado de desarrollar fístulas durante el tratamiento con Avastin. En pacientes con fístula traqueoesofágica (TE) o con cualquier fístula de Grado 4 [(US National Cancer Institute- Common Toxicity Criteria (NCI-CTCAE), versión 3.0)] se debe interrumpir permanentemente el tratamiento con Avastin. Se dispone de información limitada acerca del uso continuado de Avastin en pacientes con otro tipo de fístulas. En aquellos casos de fístula interna que no se presente en el tracto gastrointestinal, se debe considerar la interrupción del tratamiento con Avastin. Complicaciones en la cicatrización (véase Reacciones adversas): Avastin puede influir negativamente en el proceso de cicatrización. Se han reportado complicaciones graves en la cicatrización de heridas, incluyendo las relacionadas con el cierre de una anastomosis, con desenlace fatal. No debe iniciarse la terapia por lo menos durante los 28 días siguientes a una intervención de cirugía mayor o hasta que la herida quirúrgica haya cicatrizado completamente. Se interrumpirá la administración de Avastin en aquellos pacientes que presenten complicaciones de la cicatrización durante el tratamiento, hasta que la herida haya cicatrizado en forma total. Debe aplazarse la terapia cuando se vayan a realizar intervenciones quirúrgicas programadas. En pacientes tratados con Avastin rara vez se ha reportado fascitis necrotizante, incluyendo casos fatales; por lo general, en forma secundaria a heridas por complicaciones en la cicatrización, perforación gastrointestinal o formación de fístula. Debe interrumpirse el tratamiento con Avastin en pacientes que desarrollen fascitis necrotizante e iniciarse inmediatamente la terapia adecuada (véase Reacciones adversas). Hipertensión (véase Reacciones adversas): Se ha observado una mayor incidencia de hipertensión en pacientes tratados con Avastin. Los datos de seguridad clínica sugieren que es probable que esta incidencia sea dependiente de la dosis. Se debe controlar adecuadamente la hipertensión preexistente antes de comenzar el tratamiento con Avastin. No existe información del efecto de Avastin en pacientes con hipertensión no controlada al inicio de la terapia. Generalmente se recomienda monitorizar la tensión arterial durante el tratamiento. En la mayoría de los casos, la hipertensión se estabilizó satisfactoriamente utilizando el tratamiento antihipertensivo estándar adecuado para la situación individual del paciente afectado. En aquéllos que reciban quimioterapia basada en cisplatino no se aconseja la utilización de diuréticos para tratar la hipertensión. El tratamiento con Avastin debe interrumpirse en forma permanente si la hipertensión clínicamente significativa no se puede controlar adecuadamente con la medicación antihipertensiva, o si el paciente desarrolla crisis hipertensivas o encefalopatía hipertensiva. Síndrome de Encefalopatía Posterior Reversible (SEPR) (véase Reacciones adversas): Se han notificado casos raros de pacientes tratados con Avastin que han desarrollado signos y síntomas que concuerdan con el Síndrome de Encefalopatía Posterior Reversible (SEPR), un trastorno neurológico raro que se puede presentar con los siguientes signos y síntomas, entre otros: convulsiones, cefalea, estado mental alterado, alteraciones visuales, o ceguera cortical, con o sin hipertensión asociada. Un diagnóstico del SEPR requiere confirmación mediante técnicas de imagen cerebral, preferiblemente por resonancia magnética. En los pacientes que desarrollen SEPR, está recomendado el tratamiento de los síntomas específicos, incluyendo el control de la hipertensión, junto con la interrupción de Avastin. No se conoce la seguridad de reanudar la terapia con Avastin en pacientes que hayan experimentado previamente el SEPR. Proteinuria (véase Reacciones adversas): Los pacientes con antecedentes de hipertensión pueden tener un mayor riesgo de proteinuria durante el tratamiento con Avastin. Existen datos que sugieren que la proteinuria de todos los Grados (NCI-CTCAE v. 3) puede estar relacionada con la dosis. Se recomienda monitorizar la proteinuria mediante análisis de orina empleando tiras reactivas antes y durante la terapia. La proteinuria de Grado 4 (síndrome nefrótico) se observó en hasta el 1,4% de los pacientes tratados. Se debe interrumpir en forma permanente el tratamiento en pacientes que desarrollen síndrome nefrótico (NCI-CTCAE v. 3). Tromboembolismo arterial (véase Reacciones adversas): En ensayos clínicos, la incidencia de reacciones de tromboembolismo arterial, incluyendo accidentes cerebrovasculares (ACVs), ataques isquémicos transitorios (AITs) e infartos de miocardio (IMs), fue mayor en los pacientes que recibieron Avastin en combinación con quimioterapia en comparación con aquellos tratados sólo con quimioterapia. Los pacientes tratados con Avastin junto con quimioterapia que tengan antecedentes de tromboembolismo arterial, diabetes o sean mayores de 65 años tienen un riesgo aumentado de sufrir reacciones tromboembólicas arteriales durante el tratamiento. Se debe tener precaución cuando se traten estos pacientes con Avastin. Se debe interrumpir permanentemente el tratamiento de los pacientes que sufran reacciones tromboembólicas arteriales. Tromboembolismo venoso (véase Reacciones adversas): Los pacientes tratados con Avastin pueden tener un riesgo de sufrir reacciones tromboembólicas venosas, incluyendo embolismo pulmonar. Las pacientes con carcinoma persistente, recurrente o metastásico del cuello uterino tratadas con Avastin en combinación con paclitaxel y cisplatino pueden tener un mayor riesgo de eventos de tromboembolismo venoso. El tratamiento con Avastin se debe interrumpir en pacientes con reacciones tromboembólicas venosas, que amenacen la vida (Grado 4) incluyendo el embolismo pulmonar (NCI-CTCAE v.3). Los pacientes con reacciones tromboembólicas ≤ Grado 3 requieren una monitorización rigurosa (NCI-CTCAE v. 3). Hemorragia: Los pacientes tratados con Avastin tienen un mayor riesgo de hemorragia, especialmente asociada con el tumor. Se debe interrumpir en forma permanente el tratamiento con Avastin en pacientes que desarrollen hemorragia de Grados 3 ó 4 durante la terapia con Avastin (NCI-CTCAE v.3) (véase Reacciones adversas). Sobre la base de las técnicas de imagen o de los signos y síntomas, los pacientes con metástasis no tratadas localizadas en el SNC fueron excluidos de los ensayos clínicos con Avastin. Por lo tanto, el riesgo de hemorragia en el SNC en tales pacientes no se ha evaluado de manera prospectiva en los ensayos clínicos aleatorizados (véase Reacciones adversas). Se deben monitorizar los pacientes con signos y síntomas de hemorragia en el SNC e interrumpir el tratamiento con Avastin en casos de hemorragia intracraneal. No existe información sobre el perfil de seguridad de Avastin en pacientes con diátesis hemorrágica congénita, coagulopatía adquirida o en aquellos que estaban recibiendo dosis completas de anticoagulantes para el tratamiento del tromboembolismo antes del inicio de la terapia con Avastin, ya que estos pacientes fueron excluidos de los ensayos clínicos. Por lo tanto, en ellos se debe tener precaución antes de iniciar la terapia. Sin embargo, los pacientes que desarrollaron trombosis venosa durante el tratamiento aparentemente no tuvieron una mayor incidencia de hemorragia de Grado 3 o superior cuando fueron tratados con dosis completas de warfarina concomitantemente con Avastin (NCI-CTCAE v.3). Hemorragia pulmonar/Hemoptisis: Los pacientes con cáncer de pulmón no microcítico tratados con Avastin pueden tener riesgo de hemorragia pulmonar/hemoptisis grave, en algunos casos mortal. Los pacientes con hemorragia pulmonar
/hemoptisis reciente ( > 2,5 ml de sangre roja) no deben ser tratados con Avastin. Insuficiencia Cardíaca Congestiva (ICC) (véase Reacciones adversas): En los ensayos clínicos se notificaron reacciones relacionadas con ICC. Los acontecimientos oscilaron desde la disminución asintomática en la fracción de eyección del ventrículo izquierdo hasta la ICC sintomática, requiriendo tratamiento u hospitalización. Se debe tener precaución cuando se trate con Avastin a pacientes con afección cardiovascular clínicamente significativa, como por ejemplo enfermedad arterial coronaria preexistente, o insuficiencia cardíaca congestiva preexistente. La mayoría de los pacientes que desarrollaron ICC tenía cáncer de mama metastásico y había recibido previamente tratamiento con antracicilinas, radioterapia sobre la pared torácica izquierda o tenía otros factores de riesgo para el desarrollo de ICC. En los pacientes del ensayo clínico AVF3694g tratados con antraciclinas y que no habían recibido antraciclinas anteriormente, no se observó aumento en la incidencia de ICC de cualquier Grado en el grupo tratado con antraciclina más bevacizumab en comparación con el que recibió sólo antraciclinas. Las reacciones de ICC de Grado 3 o superiores fueron algo más frecuentes entre los pacientes tratados con bevacizumab en combinación con quimioterapia que en aquellos a los que se administró quimioterapia. Estos datos concuerdan con los resultados en pacientes de otros ensayos en cáncer de mama metastásico que no recibieron tratamiento concomitante con antraciclinas (NCI-CTCAE v. 3) (véase Reacciones adversas). Neutropenia e infecciones (véase Reacciones adversas): En los pacientes tratados con algunos regímenes de quimioterapia mielotóxica junto con Avastin se ha observado un aumento de la incidencia de neutropenia grave, neutropenia febril o infección asociada o no con neutropenia grave (incluyendo casos mortales), en comparación con aquellos tratados sólo con quimioterapia. Esta situación se ha comprobado principalmente en tratamientos basados en la combinación con platino o taxanos en el tratamiento del CPNM, en CMm y en asociación con paclitaxel y topotecán en carcinoma persistente, recurrente o metastásico del cuello uterino. Reacciones de hipersensibilidad / reacciones a la infusión (véase Reacciones adversas): Existe el riesgo de que los pacientes presenten reacciones a la infusión o reacciones de hipersensibilidad. Se recomienda una observación estrecha del paciente durante y después de la administración de bevacizumab, al igual que con cualquier otra infusión de un anticuerpo monoclonal humanizado. Si apareciera una reacción, debe interrumpirse la infusión y administrarse los tratamientos médicos adecuados. No se considera necesaria la premedicación en forma sistemática. Osteonecrosis del maxilar (ONM) (véase Reacciones adversas): Se han notificado casos de ONM en pacientes oncológicos tratados con Avastin, la mayoría de los cuales habían recibido tratamiento previo o concomitante con bisfosfonatos por vía intravenosa y en estos casos la ONM es un riesgo identificado. Se debe proceder con precaución cuando se administran simultánea o secuencialmente Avastin y bisfosfonatos por vía intravenosa. Los procesos dentales invasivos también están identificados como un factor de riesgo. Antes de comenzar el tratamiento con Avastin se debe considerar llevar a cabo un examen dental y una apropiada odontología preventiva. En aquellos pacientes que hayan recibido previamente o que estén recibiendo bisfosfonatos por vía intravenosa deben evitarse los procesos dentales invasivos siempre que sea posible. Población pediátrica: Avastin no está aprobado para su uso en pacientes pediátricos. En los informes publicados, se han observado casos de osteonecrosis en otros sitios diferentes de la mandíbula en pacientes menores de 18 años de edad expuestos a Avastin. Trastornos oculares: Durante la etapa poscomercialización, se han notificado eventos adversos después del uso no registrado de Avastin por vía intravítrea para el tratamiento de desórdenes oculares. Los eventos fueron: pérdida permanente de la visión, endoftalmitis (infecciosa y estéril), inflamación intraocular, desprendimiento de retina, aumento de la presión intraocular, hemorragia conjuntival, hemorragia vítrea o hemorragia retinal, partículas flotantes en el vítreo, hiperemia ocular, molestia o dolor ocular. Insuficiencia ovárica/Fertilidad: Avastin puede afectar la fertilidad femenina (véanse Fertilidad, embarazo y lactancia y Reacciones adversas). Por lo tanto, antes de comenzar el tratamiento con Avastin se debe consultar con las mujeres en edad fértil las estrategias de preservación de la fertilidad. Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de Avastin sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Sin embargo, se ha notificado somnolencia y síncope con el uso de Avastin (véase Reacciones adversas, Tabla 1. Si los pacientes experimentan síntomas que afectan a su visión o concentración, o su capacidad de reacción, deben ser advertidos de no conducir y utilizar máquinas hasta que estas manifestaciones desaparezcan. Fertilidad, embarazo y lactancia: Mujeres en edad fértil: Las mujeres en edad fértil deben utilizar métodos anticonceptivos efectivos durante el tratamiento (y hasta 6 meses después del mismo). Embarazo: No existen ensayos clínicos con datos sobre el tratamiento con Avastin en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad reproductiva, incluyendo malformaciones (véase Farmacología; Datos preclínicos sobre seguridad). Dado que se conoce que las IgGs atraviesan la placenta, se espera que Avastin inhiba la angiogénesis en el feto, y, por lo tanto, se sospecha que provoca defectos congénitos graves si se administra durante el embarazo. En la etapa poscomercialización, se han observado casos de anomalías fetales en las mujeres tratadas con bevacizumab solo o en combinación con agentes quimioterápicos embriotóxicos conocidos (véase Reacciones adversas). Avastin está contraindicado durante el embarazo (véase Contraindicaciones). Lactancia: Se desconoce si bevacizumab se excreta en la leche materna. Dado que la IgG materna se excreta en la leche y que bevacizumab puede afectar negativamente al crecimiento y desarrollo del niño (véase Farmacología; Datos preclínicos sobre seguridad), se debe interrumpir la lactancia materna durante la terapia y durante por lo menos los 6 meses posteriores a la administración de la última dosis de Avastin. Fertilidad: Estudios de toxicidad de dosis repetidas en animales han demostrado que bevacizumab podría tener un efecto adverso sobre la fertilidad femenina (véase Farmacología; Datos preclínicos sobre seguridad). Un subestudio con mujeres premenopáusicas de un ensayo en Fase III para el tratamiento adyuvante de pacientes con cáncer de colon, mostró una mayor incidencia de nuevos casos de insuficiencia ovárica en el grupo de bevacizumab comparado con el grupo control. En la mayoría de las pacientes, después de discontinuar bevacizumab se recuperó la función ovárica. Se desconoce el efecto a largo plazo del tratamiento con bevacizumab en la fertilidad.
Interacciones.
Efecto de agentes antineoplásicos en la farmacocinética de bevacizumab: No se han observado interacciones farmacocinéticas clínicamente relevantes en la farmacocinética de bevacizumab con la administración concomitante de quimioterapia según los resultados del análisis de farmacocinética poblacional. En los pacientes tratados con Avastin en monoterapia no se registraron diferencias estadísticamente significativas ni clínicamente relevantes en el clearance de bevacizumab en comparación con los que recibieron Avastin en combinación con interferón alfa-2a, erlotinib o quimioterapias (IFL, 5-FU/LV, carboplatino/paclitaxel, capecitabina, doxorrubicina o cisplatino/gemcitabina). Efecto de bevacizumab en la farmacocinética de otros agentes antineoplásicos: No se observaron interacciones clínicamente relevantes de bevacizumab en la farmacocinética de la administración concomitante de interferón alfa 2-a, erlotinib (y su metabolito activo OSI-429), o quimioterapia con irinotecán (y su metabolito activo SN38), capecitabina, oxaliplatino (que se determinó midiendo los niveles de platino libre y total), y cisplatino. No se pudieron extraer conclusiones del efecto de bevacizumab en la farmacocinética de gemcitabina. Combinación de bevacizumab y maleato de sunitinib: En dos ensayos clínicos de carcinoma de células renales metastásico, se notificó anemia hemolítica microangiopática (MAHA) en 7 de 19 pacientes tratados con la combinación de bevacizumab (10 mg/kg cada dos semanas) y maleato de sunitinib (50 mg diarios). MAHA es un trastorno hemolítico que se puede presentar con fragmentación de glóbulos rojos, anemia y trombocitopenia. Además, en algunos pacientes se observó hipertensión (incluyendo crisis hipertensiva), creatinina elevada y síntomas neurológicos. Todos estos acontecimientos fueron reversibles después del retiro de bevacizumab y maleato de sunitinib (véanse Precauciones; Hipertensión; Proteinuria y Síndrome de Encefalopatía Posterior Reversible). Combinación con tratamientos basados en platinos o taxanos (véanse Precauciones; y Reacciones adversas): Se ha observado un aumento en las tasas de neutropenia grave, neutropenia febril, o infección con o sin neutropenia grave (incluyendo algunos casos mortales), principalmente en pacientes tratados con terapias basadas en platinos o taxanos en el tratamiento del CPNM o CMm. Radioterapia: El estudio BO21990, Fase III, aleatorizado, doble-ciego, controlado con placebo, evaluó la seguridad y eficacia de la administración simultánea de quimioterapia (temozolomida), radioterapia y Avastin, en 921 pacientes con diagnóstico reciente de glioblastoma. En este estudio no se informó ningún nuevo evento adverso asociado con Avastin. En otras indicaciones no se han establecido la seguridad y la eficacia de la administración concomitante de radioterapia y Avastin. Anticuerpos monoclonales dirigidos al EGFR en combinación con diferentes regímenes de bevacizumab: No se han realizado estudios de interacción. Para el tratamiento del CCRm los anticuerpos monoclonales dirigidos al EGFR no se deben administrar en combinación con regímenes de quimioterapia que contengan bevacizumab. Los resultados de dos estudios aleatorizados, Fase III, PACCE y CAIRO-2 en pacientes con CCRm, sugieren que el uso de anticuerpos monoclonales anti-EGFR, panitumumab y cetuximab respectivamente, en combinación con bevacizumab más quimioterapia, se asocia con un descenso de la sobrevida libre de progresión y/o de la sobrevida global y con un incremento de la toxicidad, si se compara con bevacizumab solo más regímenes de quimioterapia.
Incompatibilidades.
Este medicamento no debe mezclarse con otros fármacos, excepto los mencionados en "Precauciones especiales de eliminación y otras manipulaciones". Se ha observado que el perfil de degradación de bevacizumab depende de la concentración cuando se diluye con soluciones de glucosa (5%).
Conservación.
Los viales deben conservarse en heladera entre 2°C a 8°C. Conservar el vial en el embalaje exterior para proteger su contenido de la luz. No congelar. No agitar. Para las condiciones de conservación del medicamento después de su dilución, véase "Período de validez". Precauciones especiales de eliminación y otras manipulaciones: Avastin debe ser preparado por un profesional de la salud empleando técnicas asépticas para asegurar la esterilidad de la disolución preparada. Se deberá extraer la cantidad necesaria de bevacizumab y diluir con solución inyectable de 9 mg/ml de cloruro sódico (0,9%) hasta el volumen requerido para la administración. La concentración de la solución final de bevacizumab debe mantenerse dentro del intervalo de 1,4 - 16,5 mg/ml. Los medicamentos de uso parenteral deben comprobarse visualmente antes de su administración para detectar la posible existencia de partículas o decoloración. Avastin es de uso único, debido a que el producto no contiene conservantes. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local. No se han observado incompatibilidades entre Avastin y el equipo de infusión o las bolsas de cloruro de polivinilo o poliolefina. Este medicamento no debe ser utilizado después de la fecha de vencimiento indicada en el envase. Este medicamento debe ser usado exclusivamente bajo prescripción y vigilancia médica y no puede repetirse sin nueva receta médica. Mantenga los medicamentos fuera del alcance de los niños. Período de validez: Medicamento diluido: Se ha demostrado la estabilidad química y física en uso entre 2° C y 30° C durante 48 horas una vez diluido con una solución inyectable de 9 mg/ml (0,9%) de cloruro sódico. Desde el punto de vista microbiológico, el producto debe ser usado inmediatamente. Si no se utiliza en forma inmediata, el tiempo y las condiciones de almacenamiento hasta su empleo serán responsabilidad del usuario y normalmente no deberían ser superiores a 24 horas entre 2°C y 8°C, a menos que la dilución se haya realizado bajo condiciones asépticas controladas y validadas.
Sobredosificación.
La dosis más alta ensayada en seres humanos (20 mg/kg de peso corporal, por vía intravenosa, cada 2 semanas) se asoció con migraña grave en varios pacientes. Ante la eventualidad de una sobredosificación concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Dr. Ricardo Gutiérrez: 4962-6666/2247; Policlínico Dr. G. A. Posadas: 4654-6648; 4658-7777; Hospital General de Niños Dr. Pedro de Elizalde: 4300-2115; 4363-2100/2200 Interno 6217.
Presentación.
Vial de 4 ml con 100 mg (25mg/ml): envase con 1. Vial de 16 ml con 400 mg (25mg/ml): envase con 1.
Revisión.
Noviembre 2016. NI(erlotinib)+RI+EMA+ ANMAT 1° rcp+CDS: 33.0C+34.0S.