AFINITOR®
NOVARTIS
Inhibidor de protein quinasa.
Composición.
Cada comprimido de Afinitor 2,5 mg contiene: Everolimus 2,5 mg. Excipientes: butilhidroxitolueno 0,05 mg, estearato de magnesio 0,63 mg, lactosa monohidratada 2,45 mg, hidroxipropilmetilcelulosa 22,5 mg, crospovidona 25,0 mg, lactosa anhidra 71, 88 mg c.s. Cada comprimido de Afinitor® 5 mg contiene: Everolimus 5 mg. Excipientes: butilhidroxitolueno 0,10 mg, estearato de magnesio 1,25 mg, lactosa monohidratada 4,90 mg, hidroxipropilmetilcelulosa 45 mg, crospovidona 50 mg, lactosa anhidra 143, 78 mg c.s. Cada comprimido de Afinitor® 10 mg contiene: Everolimus 10 mg. Excipientes: butilhidroxitolueno 0,20 mg, estearato de magnesio 2,50 mg, lactosa monohidratada 9,80 mg, hidroxipropilmetilcelulosa 90 mg, crospovidona 100 mg, lactosa anhidra 287,50 mg c.s.
Farmacología.
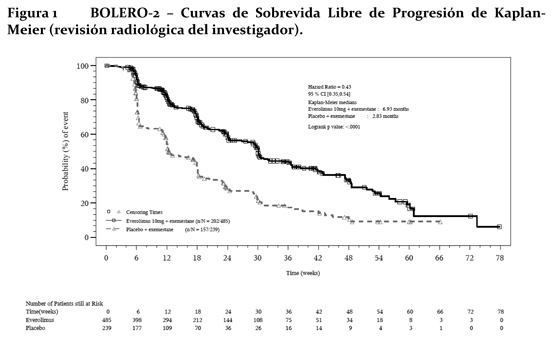
Mecanismo de acción (MA): Everolimus es un inhibidor de la transducción de señales que actúa sobre mTOR (el blanco de rapamicina en los mamíferos) y más concretamente sobre mTORC1 (complejo 1 de la diana de rapamicina en los mamíferos). mTOR es una serina-treonina quinasa clave que desempeña una función central en la regulación del crecimiento, la proliferación y la sobrevida celulares. La regulación de la transducción de señales a través de mTORC1 es compleja, pues depende de mitógenos, de factores de crecimiento y de la disponibilidad de energía y de nutrientes. mTORC1 es un regulador esencial de la síntesis general de proteínas en las últimas etapas del sistema de PI3K/AKT; la regulación de este sistema falla en la mayoría de las neoplasias malignas humanas. La activación del vía del mTOR es un cambio adaptativo clave llevando a la resistencia endocrina en el cáncer de mama. Varias vías de transducción de señales son activadas para escapar al efecto de la terapia endocrina. Otra vía es la vía PI3K/Akt/mTOR, la cual es activada principalmente en células de cáncer de mama con remoción de terapia estrogénica a largo plazo y resistente a inhibidores de aromatasa (AI). En células de cáncer de mama, la resistencia a AI debido a la activación de Akt puede ser revertida por la administración con everolimus. Dos de los principales reguladores de la transducción de señales a través de mTORC1 son los supresores tumorales TSC1 (hamartina, cuyo déficit es la causa de la esclerosis tuberosa de tipo 1) y TSC2 (tuberina, cuyo déficit es la causa de la esclerosis tuberosa de tipo 2) que interactúan entre sí y forman un complejo. La pérdida o la inactivación de TSC1 o de TSC2 da lugar a altas concentraciones de Rheb-GTP, una GTPasa de la familia RAS que interactúa con el complejo mTORC1 para activarlo. La activación de mTORC1 pone en marcha una cascada de transducción de señales mediada por quinasas que incluye la activación de la S6K1. Un sustrato del complejo 1 de mTOR (mTORC1), S6K1, fosforila el dominio 1 de activación de los receptores de estrógeno, que es el responsable de la activación de los receptores independiente del ligando. En el síndrome de esclerosis tuberosa, que es un trastorno genético, mutaciones que inactivan los genes de dichas proteínas (TSC1 o TSC2) provocan la aparición de hamartomas en todo el organismo. Propiedades farmacodinámicas: Everolimus es un inhibidor selectivo de mTOR (el blanco de rapamicina en los mamíferos) y más específicamente del complejo de transducción de señales mTOR-Raptor (mTORC1). mTOR es una serina-treonina-quinasa clave en la cascada de transducción de señales de PI3K/AKT; la regulación de este sistema falla en la mayoría de las neoplasias malignas humanas. Everolimus ejerce su actividad a través del receptor intracelular FKBP12 con el que interactúa con elevada afinidad. El complejo formado por la proteína FKBP12 y everolimus se fija a mTORC1 e inhibe su capacidad transductora de señales. La transducción de señales a través de mTORC1 se efectúa por medio de la modulación de la fosforilación de efectores consecutivos en la serie, entre cuyos componentes mejor caracterizados figuran los reguladores de la traducción de proteínas S6K1 (quinasa 1 de la proteína ribosómica S6) y 4E-BP (proteína de fijación al factor de elongación 4E en los eucariontes). El desmantelamiento de la función de S6K1 y de 4E-BP1, como resultado de la inhibición de mTORC1, interfiere la traducción de los ARNm codificadores de proteínas que son esenciales para la regulación del ciclo celular, la glucólisis y la adaptación a condiciones de baja concentración de oxígeno (hipoxia). Ello inhibe el crecimiento del tumor y la expresión de factores que se inducen en condiciones de hipoxia (como el factor de transcripción HIF-1); la inhibición de tales factores reduce la expresión de otros factores implicados en la potenciación de procesos angiógenicos tumorales (como el factor de crecimiento del endotelio vascular VEGF). Everolimus es un inhibidor potente del crecimiento y la proliferación de células tumorales, células endoteliales, fibroblastos y de las células del músculo liso de los vasos sanguíneos. En consonancia con la función reguladora primaria que ejerce mTORC1, se ha visto que everolimus reduce la proliferación de células tumorales, la glucólisis y la angiogénesis en los tumores sólidos in vivo y de esa forma ofrece dos modos independientes de inhibición del crecimiento tumoral: una actividad antineoplásica directa en las células y una inhibición del estroma tumoral. En un modelo neuronal murino del TSC en el que TSC1 estaba desactivado en la mayor parte de las neuronas durante el desarrollo cortical, everolimus elevó la mediana de la sobrevida de 33 días a más de 100 días, y también mejoraron notablemente la conducta, el fenotipo y el aumento de peso. Se observó penetración encefálica, acumulación a lo largo del tiempo con el tratamiento repetido y reducción efectiva de las concentraciones de la proteína ribosómica S6 fosforilada, que es un marcador eferente de mTORC1. El tratamiento mejoró las anomalías de los neurofilamentos, la mielinización y el aumento de tamaño de las células, si bien persistieron los signos de displasia neuronal y sólo se observaron modificaciones moderadas de la densidad y la longitud de las espinas dendríticas. Ratones tratados con everolimus sólo durante 23 días (entre el 7° y el 30° día de vida) mostraron una mejora persistente del fenotipo, con una mediana de sobrevida de 78 días. En resumen, everolimus es muy activo en este modelo neuronal del TSC, con efectos beneficiosos atribuibles aparentemente a los efectos sobre la vía de transducción de señales de mTORC1 y AKT y, en consecuencia, sobre el tamaño celular y la mielinización. Ensayos clínicos: Cáncer de mama avanzado con receptores hormonales positivos: BOLERO-2 (Estudio CRAD001Y2301), un estudio aleatorio, doble ciego, multicéntrico de Fase III de Afinitor® + Exemestane vs. Placebo + Exemestane fue conducido en mujeres posmenopáusicas con cáncer de mama avanzado con receptores estrogénicos positivos, HER-2-neu/neutro con recurrencia o progresión seguido de terapia previa con letrozol o anastrozol. Las pacientes fueron aleatorizadas en una relación de 2:1 para recibir Everolimus/Placebo (10mg día), en adición a exemestane (25mg día). La aleatorización fue estratificada por sensibilidad documentada a la terapia hormonal previa (+ vs. -) y por la presencia de metástasis visceral (+ vs. -). La sensibilidad a la terapia hormonal previa fue definida como (1) beneficio clínico documentado (respuesta completa [CR], respuesta parcial [PR], enfermedad estable ≥-24 semanas) a por lo menos una terapia hormonal previa en un estadio avanzado o (2) por lo menos 24 meses de terapia hormonal adyuvante previa a la recurrencia. El punto final primario para el estudio fue la sobrevida libre de progresión (PFS) evaluada por el Criterio de Evaluación de Respuesta en Tumores Sólidos (RECIST), basado en las evaluaciones de los investigadores (radiología local). El análisis de soporte de PFS fue basado en una revisión radiológica central independiente. Los puntos finales secundarios incluyeron sobrevida global (OS), tasa de respuesta global (ORR), tasa de beneficio clínico (CBR), cambio en la calidad de vida (QoL) y deterioro de ECOG PS. Los puntos finales adicionales incluyeron cambios en los marcadores óseos a las 6 y 12 semanas. Un total de 724 pacientes fueron aleatorizados en una relación de 2:1 la rama de la combinación everolimus (10mg diarios) + exemestane (25mg diarios) (n=485) o rama de placebo + exemestane (25mg diarios) (n=239). Los dos grupos de tratamiento fueron balanceados con respecto a la demografía basal de las características de la enfermedad y a la historia de uso previo de antineoplásico. La mediana de la edad de los pacientes fue 61 años (rango 20 a 93) y 75% eran caucásicos. Los resultados de eficacia fueron obtenidos de un análisis intermedio después que se observaron 359 eventos locales de PFS y 217 eventos centrales de PFS. Pacientes en la rama de tratamiento placebo + exemestane no cruzaron al brazo de everolimus en el momento de la progresión. El estudio demostró un beneficio clínico estadísticamente significativo de everolimus + exemestane sobre placebo + exemestane por una prolongación 2.4 veces mayor en la mediana de la PFS (mediana: 6.93 meses vs. 2.83 meses), resultando en una reducción del riesgo de progresión o muerte del 57% (PFS HR 0.43; 95% CI: 0.35, 0.54; test Log Rank, valor p < 0.0001 por evaluación de investigador local) (ver tabla 5 y figura No. 1). Los análisis de PFS basados en evaluación radiológica central independiente fueron de soporte y mostraron una prolongación 2.6 veces mayor en la mediana de la sobrevida libre de progresión (10.58 meses vs. 4.14 meses), resultando en una reducción del riesgo progresión o muerte del 64% (PFS HR: 0.36; 95% CI: 0.27, 0.47; test Log Rank, valor p < 0.0001) (ver tabla 6 y figura No. 2). La respuesta objetiva de la evaluación del investigador basada en RECIST se observó en 9.5% de los pacientes (95% CI: 7.0, 12.4) en el brazo de tratamiento de everolimus + exemestane vs. 0.4% (95% CI: 0.0-2.3) en el brazo de placebo + exemestane (p < 0.0001 para la comparación entre ramas). La tasa de beneficio clínico para everolimus + exemestane fue de 33.4% vs. 18.0% en el brazo control; p < 0.0001 (ver tabla 1).
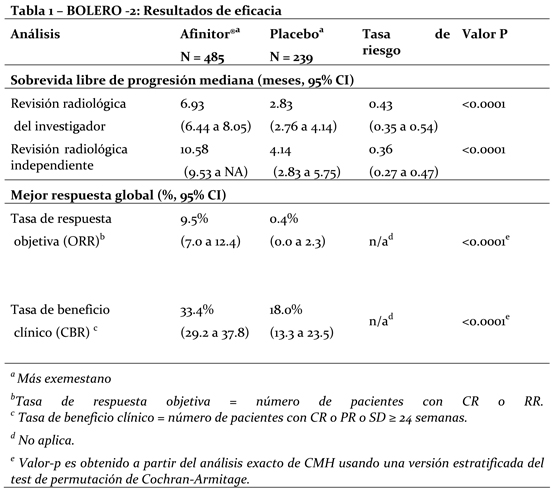
Los datos de sobrevida global (OS) no permiten al momento del análisis de PFS contribuir a una interpretación final de los mismos. 83 muertes fueron reportadas en el análisis interino, representando 10.6% y 13.0% de los pacientes muertos en las ramas de tratamiento de everolimus + exemestane y placebo + exemestane, respectivamente.
Tasas a los 9 meses de PFS fue de 40% de pacientes recibiendo everolimus + exemestane comparado con 15% en la rama de placebo + exemestane en una mediana de seguimiento de 7.6 meses.
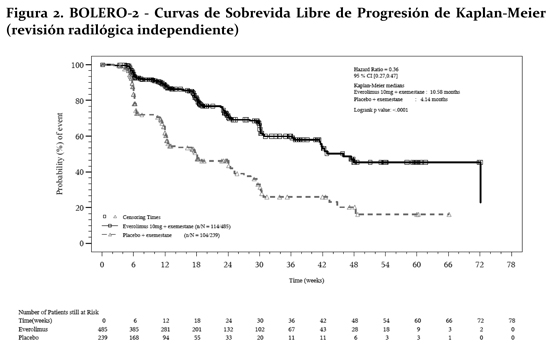
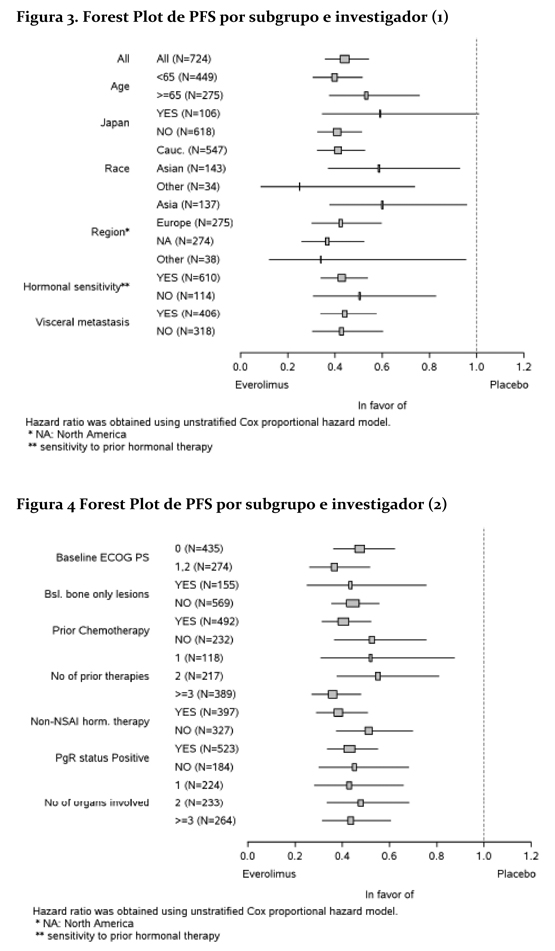
El efecto en el tratamiento del PFS estimado fue soportado por un análisis planeado de los subgrupos del PFS por evaluación del investigador. Para todos los subgrupos analizados, un efecto del tratamiento positivo fue visto con everolimus + exemestane con un Hazard ratio vs. placebo + exemestane entre 0.25 y 0.60 (tabla 2, figuras 3 y 4). Los análisis de subgrupos demostraron un efecto de tratamiento homogéneo y constante independientemente de la sensibilidad a la terapia hormonal previa y a la presencia de metástasis visceral, y entre los subgrupos de mayor prognóstico y demográficos.
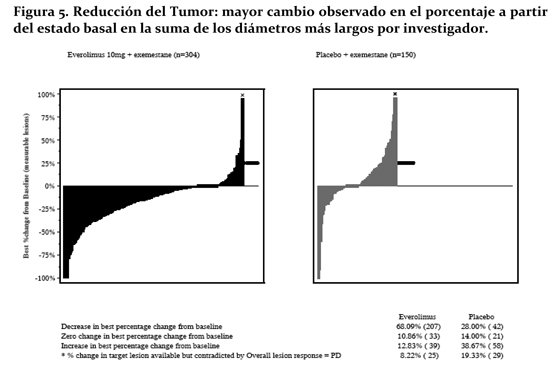
La reducción tumoral fue evidente a partir de la correspondiente cascada de Plot. Los resultados indican que el 68.1% de las pacientes en el brazo de everolimus + exemestane experimentaron una reducción tumoral vs. 28% para placebo + exemestane (Figura No. 5).
Las diferencias clínicamente o estadísticamente significativas no fueron observadas entre los dos brazos de tratamiento en términos de deterioro de ECOG PS (≥1) y las medianas de los tiempos hasta el deterioro (≥5%) de los puntajes de dominio de QLQ-C30. Efectos en tejido óseo: No hay datos a largo plazo del efecto de everolimus en el tejido óseo. Los datos comparativos obtenidos de BOLERO-2 mostraron una mejoría en los marcadores séricos óseo durante las primeras 12 semanas de tratamiento, demostrando una mejoría a nivel óseo. Tumores neuroendocrinos avanzados de origen pancreático: El estudio RADIANT-3 (CRAD001C2324), un ensayo de Fase III, aleatorizado, doble ciego y multicéntrico, de comparación entre Afinitor® más el mejor cuidado de soporte (MCS) y el placebo más el mismo MCS en pacientes con tumores neuroendocrinos pancreáticos avanzados (pNET), demostró un beneficio clínico estadísticamente significativo de Afinitor® en comparación con el placebo debido a una prolongación 2,4 veces mayor de la mediana sobrevida libre de progresión (SLP) (11,04 meses frente a 4,6 meses), que produjo una reducción de riesgo igual al 65% en la SLP (Hazard ratio [HR]: 0,35; IC 95%:0,27-0,45; p < 0,0001) (ver Tabla 1 de la Figura 1). En el estudio RADIANT-3 participaron pacientes con pNET avanzados que habían sufrido progresión tumoral en los 12 meses precedentes. Se estratificó a los pacientes según si habían recibido o no quimioterapia citotóxica previa y según al estado funcional de la OMS (0 contra 1 y 2). Como parte del tratamiento complementario óptimo se admitió el uso de análogos de la somastatina. El criterio principal de valoración del ensayo fue la sobrevida libre de progresión (SLP) evaluada a través de los RECIST (criterios de evaluación de la respuesta aplicables a tumores sólidos). Cuando se tenían indicios de progresión por estudios radiológicos, el investigador podía revelar a los pacientes el tratamiento recibido, entonces los del grupo del placebo tenían la posibilidad de recibir Afinitor® sin enmascaramiento. Los criterios de valoración secundarios fueron la seguridad, la tasa de respuesta objetiva (ORR de su sigla en inglés), ya sean respuestas completas (RC) o bien parciales (RP), la duración de la respuesta y la sobrevida global (SG). En total, 410 pacientes fueron repartidos aleatoriamente en dos grupos (1:1) para recibir Afinitor® en dosis de 10 mg/día (n=207) o placebo (n=203). Los datos demográficos estaban bien equilibrados (la edad mediana fue de 58 años, el 55% era varones y el 78,5% de raza blanca).
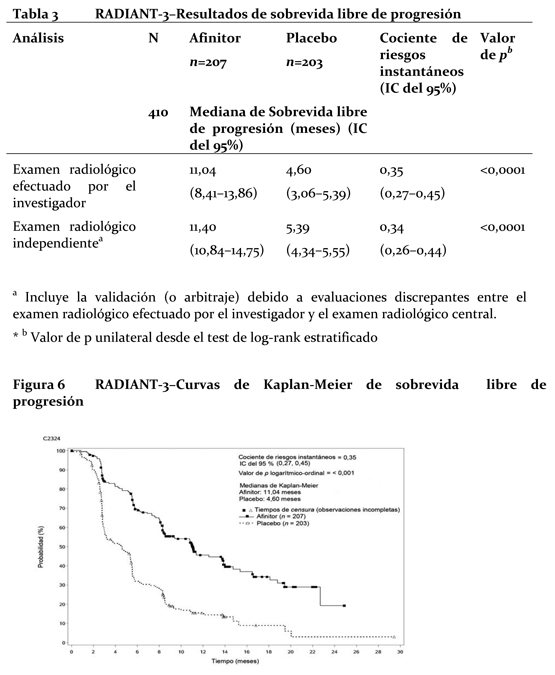
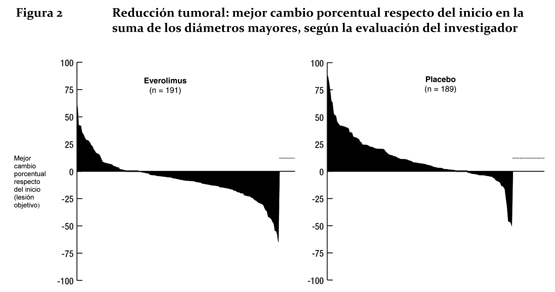
Las tasas de sobrevida libre de progresión a los 18 meses fueron del 34,2% en el grupo de Afinitor® y del 8,9% en el del placebo. La tasa de respuesta objetiva por evaluación del investigador fue de 4,8 % en el grupo de everolimus en comparación con 2,0 % en el grupo de placebo. La reducción tumoral también fue evidente en la gráfica de cascada correspondiente. Los resultados indican que 64,4 % de los pacientes del grupo de everolimus experimentaron reducción tumoral en comparación con 20,6 % del grupo de placebo (Figura 2).

Los resultados de la sobrevida global (OS) aun no están listos y no se observó ninguna diferencia estadísticamente significativa en OS (HR = 0,99 (CI de 95 %, 0,68 a 1,43) en un análisis actualizado). El cruzamiento de > 72 % de los pacientes del grupo de placebo al de Afinitor® abierto después de la progresión de la enfermedad probablemente generó confusión en la detección de cualquier diferencia en OS relacionada con el tratamiento. Carcinoma de células renales en estadio avanzado: El estudio RECORD-1 (CRAD001C2240) fue un estudio de Fase III, internacional, multicéntrico, aleatorizado, con doble ciego y comparativo de Afinitor® (10 mg/día) con placebo (más un tratamiento óptimo de apoyo en cualquiera de los dos casos) en pacientes con carcinoma renal metastásico con progresión tumoral al tratamiento previo con un VEGFR-TKI (inhibidor del dominio con actividad tirosina-quinasa del receptor del factor de crecimiento del endotelio vascular), como sunitinib, sorafenib (o ambos fármacos). También se admitieron pacientes que habían recibido tratamiento previo con bevacizumab e interferón a. Se clasificó a los pacientes en estratos o grupos según el riesgo pronóstico del MSKCC (Memorial Sloan-Kettering Cancer Center) (grupos de riesgo elevado, moderado o reducido) y el tratamiento antineoplásico anterior (1 o 2 VEGFR-TKI previos). El criterio principal de valoración fue la sobrevida libre de progresión, documentada según los criterios RECIST (Response Evaluation Criteria in Solid Tumours) y evaluada mediante un examen centralizado, independiente y ciego. Los criterios secundarios de valoración fueron la seguridad, la tasa objetiva de respuesta tumoral, la sobrevida global, los síntomas relacionados con la enfermedad y la calidad de vida. Cuando los estudios radiológicos indicaban que había progresión, el investigador podía revelar a los pacientes el tratamiento recibido, entonces los del grupo del placebo podían recibir Afinitor® (10 mg/día) sin ser tratamiento ciego. El Comité Independiente de Vigilancia de Datos (CIVD) recomendó la finalización de este ensayo en el momento de realizar el segundo análisis interino por haberse cumplido el criterio principal de valoración. Unos 416 pacientes en total fueron repartidos aleatoriamente en dos grupos para recibir Afinitor® (n=277) o placebo (n=139). Las características demográficas estaban debidamente equilibradas (edad mediana de los grupos de sujetos 61 años [entre 27 y 85 años], sexo [masculino en un 77%], raza blanca en un 88%, 74% habían recibido al menos tratamiento previo con VEGFR-TKI. Los resultados del análisis interino preplanificado indicaron que Afinitor® era superior al placebo en lo referente al criterio principal de valoración de sobrevida libre de progresión, al producir una reducción estadísticamente significativa del 67% en el riesgo de progresión o muerte.
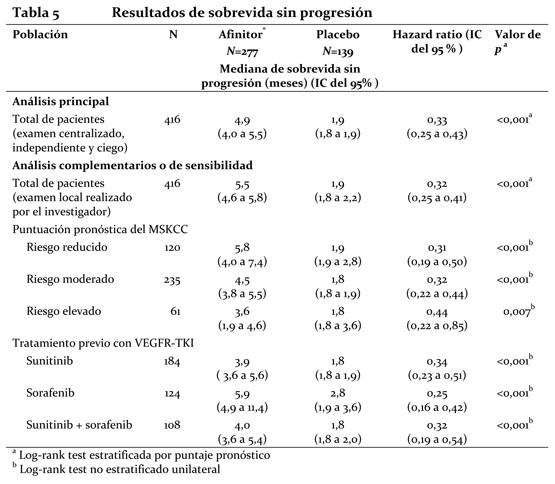
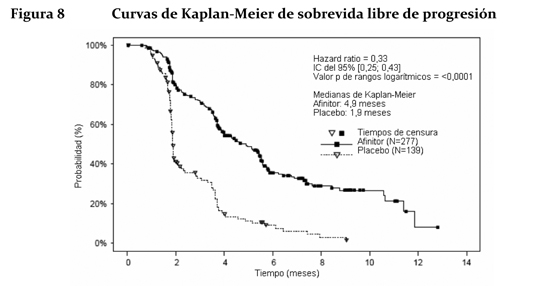
La sobrevida libre de progresión a 6 meses fue del 36% con Afinitor® comparada con 9% en la rama placebo. Se observaron respuestas tumorales, objetivas y confirmadas en 5 pacientes (2%) del grupo de Afinitor® y en ninguno del grupo del placebo. Por lo tanto, la sobrevida libre de progresión es un reflejo de lo que ocurre en la población cuya enfermedad se ha vuelto estable (que viene a ser el 67% del grupo tratado con Afinitor®). No se observó ninguna diferencia estadísticamente significativa con relación al tratamiento en la sobrevida global, pero sí una tendencia a favor de Afinitor® (HR 0,82; 95% CI: 0,57 a 1,17; p=0,137). El cambio por Afinitor® al romper el ciego tras comprobarse la progresión de la enfermedad en el grupo del placebo funcionó como confundidor para la detección de cualquier posible diferencia vinculada al tratamiento en la sobrevida global. La valoración de los síntomas relacionados con la enfermedad reveló una fuerte tendencia hacia una mejor calidad de vida en el grupo de Afinitor® (HR 0,75: 95%; CI: 0,53 a 1,06; p=0,053). ASCG: Se llevó a cabo un estudio de Fase II prospectivo y sin enmascaramiento para evaluar la seguridad y la eficacia de Afinitor® en pacientes con diagnóstico de de ASCG. Para la inclusión en el estudio se exigían pruebas radiológicas seriadas de crecimiento del ASCG. El criterio principal de valoración de la eficacia fue la variación del volumen del ASCG durante la fase principal de tratamiento de 6 meses, evaluada mediante un examen centralizado e independiente de las radiografías. Tras dicha fase, los pacientes podían pasar a la fase de extensión del tratamiento, en la que se evaluaba el volumen del ASCG cada 6 meses. En conjunto, recibieron tratamiento con Afinitor® 28 pacientes, cuya mediana de edad era de 11 años (intervalo: 3-34 años) y de los que el 61 % eran varones y el 86 % eran de raza blanca. Trece pacientes (46 %) presentaban un ASCG secundario de menor tamaño, que en 12 de ellos se situaba en el ventrículo contralateral. Afinitor® se asoció a una reducción clínicamente importante y estadísticamente significativa del volumen del ASCG primario a los 6 meses respecto al volumen inicial (p < 0,001). La disminución del volumen del tumor fue más rápida durante los tres primeros meses de tratamiento, y la remisión se mantuvo en las evaluaciones ulteriores (ver la tabla 3). Ningún paciente presentó lesiones nuevas, empeoramiento de la hidrocefalia ni aumento de la presión intracraneal, y en ninguno hubo que recurrir a la resección quirúrgica ni a otros tratamientos del ASCG.
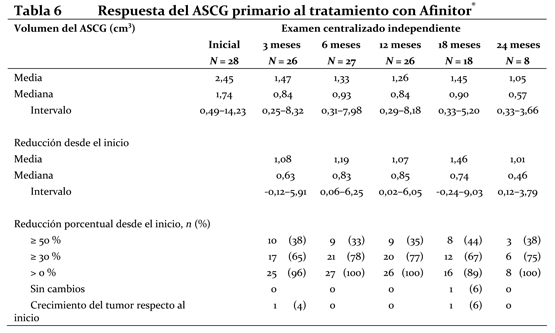
El análisis principal se fundaba en: La variación del volumen del ASCG primario según la evaluación del investigador local (p < 0,001): en el 75 % de los pacientes se redujo al menos un 30 % y en el 39 % se redujo al menos un 50 %. La variación del volumen total del ASCG según la revisión centralizada independiente (p < 0,001) o la evaluación del investigador local (p < 0,001). Un paciente satisfizo el criterio preestablecido de éxito del tratamiento (reducción del volumen del ASCG superior al 75 %) y se le retiró temporalmente el tratamiento del ensayo, pero en los tres meses siguientes se constató que el ASCG había vuelto a crecer y se reanudó el tratamiento. Everolimus se asoció a una reducción clínicamente significativa de la frecuencia general de convulsiones a los 6 meses respecto al inicio (mediana -1,0; p = 0,022). Propiedades Farmacocinéticas: Absorción: En los pacientes con tumores sólidos avanzados, las concentraciones pico de everolimus se alcanzan 1 o 2 horas después de la administración de dosis orales de entre 5 y 70 mg de fármaco en ayunas o tras un refrigerio exento de grasas. La Cmáx es proporcional a la dosis en el intervalo de 5 a 10 mg, tanto en las pautas diarias como en las semanales. A partir de los 20 mg/semana, el aumento de Cmáx aumenta de forma menos proporcional a la dosis, a pesar de que el ABC (área bajo la curva) es proporcional a la dosis en el intervalo de 5 a 70 mg. Efecto de los alimentos: En sujetos sanos, las comidas ricas en grasas redujeron la exposición sistémica a Afinitor® (10 mg)(valorada según el ABC) en un 22% y la concentración plasmática máxima en un 54%; las comidas pobres en grasas redujeron el ABC un 32% y la Cmáx en un 42%. Sin embargo, los alimentos, no tuvieron ningún efecto aparente sobre la curva de concentración - tiempo en la fase posterior a la absorción. Distribución: El cociente sangre/plasma de everolimus, que es concentración dependiente en el intervalo de 5 a 5000 ng/mL, varía entre el 17% y el 73%. La cantidad de everolimus confinada en el plasma es de un 20% en los pacientes con cáncer que toman Afinitor® en dosis de 10 mg/día. La unión a proteínas plasmáticas es de un 74% en los sujetos sanos y los pacientes con insuficiencia hepática moderada. En ratas usadas como modelo, everolimus administrado por vía intravenosa atravesó la barrera hematoencefálica de forma dependiente a la dosis, pero no proporcional a ella, lo cual es un signo de saturación de la bomba de expulsión en dicha barrera. La penetración encefálica de everolimus se pudo comprobar asimismo en ratas que habían recibido dosis orales de everolimus. Metabolismo: Everolimus es un sustrato del CYP3A4 y de la glucoproteína P (PgP). Luego de la administración oral, es el componente principal en la circulación sanguínea. Se han detectado seis importantes metabolitos de everolimus en la sangre humana: tres metabolitos monohidroxilados, dos productos con anillos abiertos por hidrólisis y un conjugado fosfatidilcolínico de everolimus. Estos metabolitos se habían identificado en las especies animales de los estudios de toxicidad, y su actividad era casi cien veces menor que la del propio everolimus. Por consiguiente, se considera que la mayor parte de la actividad farmacológica corresponde al compuesto inalterado. Excreción: No se han efectuado estudios de excreción específicos en pacientes oncológicos, pero se tienen datos procedentes de trasplantes. Tras la administración de una dosis única de everolimus radioactivo combinado con ciclosporina, el 80% de la radioactividad se recuperó en las heces y el 5%, en la orina. No se detectó compuesto inalterado en la orina ni en las heces. Farmacocinética en el estado de equilibrio: Tras la administración diaria o semanal de everolimus a pacientes con tumores sólidos avanzados, el ABC0-t de equilibrio fue proporcional a la dosis tanto en la gama de concentraciones de 5 a 10 mg con la pauta diaria como en la de 5 a 70 mg con la semanal. Con la pauta diaria, el estado de equilibrio se alcanzó en dos semanas. La Cmáx es proporcional a la dosis en el intervalo de 5 a 10 mg en las pautas diaria y semanal, pero cuando las dosis son iguales o superiores a 20 mg/semana, la Cmáx aumenta de forma menos proporcional a la dosis y se detecta 1 o 2 horas (tmáx) después de la administración. En el estado de equilibrio y con la pauta diaria, se observó una correlación significativa entre el ABC0-t y la concentración mínima anterior a la dosis. La vida media de eliminación de everolimus es aproximadamente igual a 30 horas. Pacientes con insuficiencia hepática: La seguridad, tolerabilidad y farmacocinética de Afinitor® fue evaluada en un estudio de dosis única de Everolimus en 34 pacientes con función hepática deteriorada versus pacientes con función hepática normal. Comparado con sujetos normales, hubo un incremento de 1.6, 3.3 y 3.6 veces en la exposición (ABC(0-inf)) para sujetos con insuficiencia hepática leve (Child-Pugh A), moderada (Child-Pugh B), y severa (Child-Pugh C), respectivamente. Las simulaciones de farmacocinética a dosis múltiple avalaron las recomendaciones de dosificación en sujetos con insuficiencia hepática basándose en su estado de Child-Pugh status. El ajuste de dosis es recomendado en pacientes con insuficiencia hepática (ver Dosificacion). Pacientes con insuficiencia renal: En un análisis farmacocinético poblacional de 170 pacientes con cáncer avanzado no se detectó ningún efecto significativo de la depuración de creatinina (25 - 178 mL/min) sobre la depuración oral de everolimus (CL/F). En los receptores de un trasplante, la insuficiencia renal (depuración de creatinina: 11 - 107 mL/min) posterior al trasplante no alteró la farmacocinética de everolimus. Pacientes pediátricos: No está indicado el uso de Afinitor® en niños con cáncer (ver Dosificación). En pacientes con diagnóstico de ASCG, las concentraciones mínimas intraindividuales del estado de equilibrio eran proporcionales a la dosis, con dosis diarias de entre 1,5 y 14,6 mg/m2 (ver Dosificación). Pacientes de edad avanzada: En un análisis farmacocinético de una población de pacientes con cáncer no se apreció un efecto significativo de la edad (27-85 años) en la depuración oral de everolimus (CL/F: entre 4,8 y 54,5 litros/hora). Origen étnico: La depuración oral de everolimus (CL/F) es semejante en pacientes japoneses o de raza blanca con diagnóstico de cáncer y función hepática similar. Según un análisis farmacocinético poblacional, la depuración oral (CL/F) es un 20% mayor, en promedio, en los receptores de trasplante de raza negra. Relación entre la exposición y la respuesta: Tras la administración diaria de 5 o 10 mg de everolimus, se observó una moderada correlación entre la disminución de la fosforilación de 4E-BP1 (P4E-BP1) en el tejido tumoral y la Cmín sanguínea media de dicho fármaco en el estado de equilibrio. Datos adicionales indican que la inhibición de la fosforilación de la quinasa S6 es muy sensible a la inhibición de mTOR por parte de everolimus. La inhibición de la fosforilación de F-4G fue completa en todas las concentraciones mínimas (Cmín) que siguieron a las dosis de 10 mg diarios. En pacientes con ASCG, las concentraciones mínimas más altas de everolimus parecen asociarse a mayores reducciones del volumen del tumor. Sin embargo, se han observado respuestas con concentraciones mínimas de apenas 2 ng/ml; por ello, una vez lograda una eficacia aceptable, puede que no sea necesario aumentar más la dosis (ver Dosificación). La sobrevida libre de progresión tiende a prolongarse conforme aumentaba la Cmín (definido como (área bajo la curva Cmín-tiempo desde el comienzo del estudio al tiempo del evento)/(tiempo desde el comienzo del estudio al evento)) en los pacientes con tumores neuroendocrinos pancreáticos avanzados (pNET, razón de riesgos: 0,73; IC 95%: 0,50-1,08) y en pacientes con tumores carcinoides avanzados (razón de riesgos: 0.66; 95% IC; 0.40 a 1.08). La Cmín de everolimus afectó a la probabilidad de reducción del tamaño del tumor (p < 0,001), con hazard ratio de 1,62 y 1,46, respectivamente, cuando se modificó la exposición de 5 ng/mL a 10 ng/mL en los pacientes con pNET avanzados. Datos de toxicidad preclínica: La toxicidad preclínica de everolimus se estudió en ratones, ratas, cerdos de raza Minipig, monos y conejos. Los órganos más afectados fueron los del aparato reproductor femenino y masculino de diversas especies (degeneración tubular testicular, reducción del contenido de espermatozoides en los epidídimos y atrofia uterina), así como los pulmones de los ratones y las ratas (aumento de macrófagos alveolares) y los ojos sólo de las ratas (opacidades en la línea de sutura anterior del cristalino). Se observaron alteraciones renales mínimas en las ratas (incremento de lipofuscina en el epitelio tubular vinculada a la edad del animal) y los ratones (agravamiento de lesiones subyacentes). No hubo signos de toxicidad renal en los monos o los cerdos Minipig. Everolimus pareció exacerbar espontáneamente las enfermedades subyacentes (miocarditis crónica en las ratas, infección del plasma y del corazón por el virus de Coxsackie en los monos, infestación del tubo digestivo por coccidios en los cerdos Minipig, lesiones cutáneas en los ratones y los monos). Estos efectos se observaron generalmente con una exposición sistémica incluida dentro del intervalo terapéutico o superior al mismo, salvo en las ratas, en las que ocurrieron con una exposición inferior al intervalo terapéutico debido a la elevada distribución en los tejidos. En un estudio sobre fecundidad de ratas macho se observó una alteración de la morfología testicular con la dosis de 0,5 mg/kg o mayor, así como una reducción de la motilidad de los espermatozoides, del número de cabezas de los espermatozoides y de las concentraciones plasmáticas de testosterona con la dosis de 5 mg/kg, que se sitúa dentro del rango de exposición terapéutico (52 ng.h/ml y 414 ng.h/ml, respectivamente, comparado con 560 ng.h/ml a una exposición humana de 10 mg/día y deterioró la fecundidad animal. Hubo signos de reversibilidad. Everolimus no alteró la fecundidad de las hembras, pero atravesó la barrera placentaria y fue tóxico para el producto de la concepción. Causó embriotoxicidad y fetotoxicidad por debajo del nivel terapéutico de exposición sistémica en las ratas, lo cual se manifestó como mortalidad y un menor peso fetal. Con 0,3 y 0,9 mg/kg se observó una mayor incidencia de malformaciones y anomalías óseas (por ejemplo, hendidura esternal). En los conejos, el aumento de resorciones tardías fue una señal de embriotoxicidad. En estudios de toxicidad en ratas jóvenes con dosis de apenas 0,15 mg/kg/día, las manifestaciones de toxicidad sistémica comprendieron la reducción del aumento ponderal y del consumo de alimentos, y el retraso en el logro de algunos hitos del desarrollo con todas las dosis; se observó una recuperación total o parcial tras suspender la administración. Con la posible excepción del efecto observado específicamente en el cristalino de las ratas (al que resultaron más sensibles los animales jóvenes), parece que no existe una diferencia significativa entre la sensibilidad de los animales jóvenes a los efectos adversos del everolimus y la de los animales adultos con dosis diarias de entre 0,5 y 5 mg/kg. No se observaron signos de toxicidad en monos jóvenes con dosis de hasta 0,5 mg/kg/día durante 4 semanas. Los estudios de genotoxicidad en los que se investigaron variables pertinentes de genotoxicidad no evidenciaron actividad clastógena ni mutágena alguna. La administración de everolimus a ratones y ratas durante dos años tampoco reveló poder cancerígeno alguno, incluso cuando se usaron las dosis más elevadas, que son unas 3,9 y 0,2 veces mayores que la exposición clínica prevista para una dosis de 10 mg.
Indicaciones.
Afinitor® está indicado: Para el tratamiento de pacientes con carcinoma de células renales avanzado, en los que la enfermedad ha progresado durante o después del tratamiento con una terapia dirigida al factor de crecimiento del endotelio vascular (VEGF). Para el tratamiento de pacientes con astrocitoma subependimario de células gigantes (ASCG) asociado con esclerosis tuberosa (ET) que requiere intervención terapéutica, pero no es candidato para la resección quirúrgica curativa. La efectividad de Afinitor® se basa en un análisis del cambio del volumen de SEGA. No se ha demostrado beneficio clínico tal como la mejora en los síntomas relacionados. Para el tratamiento de pacientes con tumores neuroendocrinos avanzados de origen pancreático (PNET) progresados en pacientes con enfermedad irresecable, localmente avanzada o metastásica. La seguridad y eficacia de Afinitor® en el tratamiento de pacientes con tumores carcinoides no han sido establecidas. Para el tratamiento de mujeres posmenopáusicas con cáncer de mama avanzado con receptores hormonales positivos, en combinación con inhibidor de la aromatasa, después de una terapia endocrina previa. Pacientes con complejo de esclerosis tuberosa (tuberous sclerosis complex, TSC) con angiomiolipoma renal que no requiere cirugía inmediata.
Dosificación.
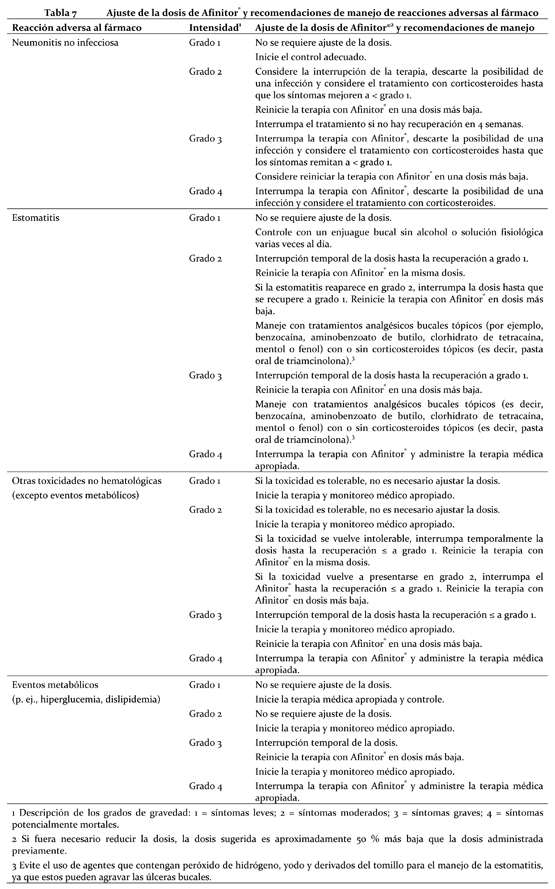
Afinitor® debe administrarse por vía oral una vez al día, a la misma hora todos los días, consistentemente con o sin alimentos (ver Farmacocinética). Los comprimidos deben ser deglutidos enteros con un vaso con agua. No se deben masticar ni triturar. Si el paciente no puede ingerir los comprimidos de Afinitor®, estos deben dispersarse por completo en un vaso de agua (que contenga unos 30 mL) agitando con suavidad el contenido, inmediatamente antes de beberlo. Seguidamente debe enjuagarse el vaso con el mismo volumen de agua y el paciente debe beber toda esta agua para tener la seguridad de que se administra la dosis completa. (Ver Farmacocinética). Es necesario continuar con el tratamiento mientras se observen beneficios clínicos y no surjan reacciones adversas intolerables. Población destinataria en general: Adultos: Posología en cáncer de mama avanzado con receptores hormonales positivos, tumores neuroendocrinos avanzados de origen pancreático y en el carcinoma de células renales avanzado: La dosis recomendada de Afinitor® es de 10 mg una vez al día. El tratamiento con Afinitor® ha de instaurarlo un médico experimentado en el uso de terapias antineoplásicas. Posología en casos de complejo de esclerosis tuberosa con angiomiolipoma renal: Un médico con experiencia en el tratamiento de pacientes con TSC debe iniciar el tratamiento con Afinitor®. La dosis recomendada de Afinitor® es de 10 mg, tomada una vez al día. Reacciones adversas: el tratamiento de presuntas reacciones adversas graves o intolerables en el caso de pacientes que requieren ajustes de dosis en el cáncer de mama avanzado positivo para receptores hormonales; tumores neuroendocrinos avanzados de origen pancreático; carcinoma de células renales avanzado y complejo de esclerosis tuberosa (TSC) con angiomiolipoma renal puede exigir la reducción momentánea de la dosis de Everolimus (Afinitor®) o la interrupción del tratamiento con Afinitor®. Si fuera necesario reducir la dosis, la dosis sugerida es aproximadamente 50 % más baja que la dosis diaria administrada previamente (ver Advertencias y Precauciones). En la Tabla 7 se resumen las recomendaciones de reducción de la dosis, interrupción o discontinuación de Afinitor® en el manejo de las ADR. También se proporcionan las recomendaciones generales de manejo que sean necesarias. El criterio clínico del médico tratante debe orientar el plan de manejo de cada paciente en función de la evaluación individual de beneficio-riesgo.
Inhibidores moderados del CYP3A4 o de la glucoproteína P (PgP): Usar con cautela en caso de administrar Afinitor® combinado con inhibidores moderados del CYP3A4 o de la PgP. Si el paciente requiere la coadministración de estas medicaciones, se debe reducir la dosis de Afinitor® a aproximadamente al 50 % de la dosis previamente administrada. En los casos de reducción de la dosis por debajo de la concentración más baja de Afinitor® disponible, deberá considerarse la administración de la dosis día por medio. Posiblemente sea necesario reducir aún más la dosis para manejar las reacciones adversas (ver Precauciones). Inductores potentes del CYP3A4: Debe evitarse el uso concomitante de inductores potentes del CYP3A4. Si el paciente requiere el uso de estas medicaciones, debe considerarse incrementar la dosis de Afinitor® de 10 mg a 20 mg diarios (basado en datos farmacocinéticos), usando incrementos de 5 mg por vez. La dosis de Afinitor® es predictiva para ajustar el ABC al rango observado sin inductores. Sin embargo, no hay datos clínicos con la dosis ajustada en pacientes que están recibiendo inductores potentes de CYP3A4. Si el uso de inductores potentes del CYP3A4 es discontinuado, la dosis de Afinitor® debe regresar a la prescripta antes de la iniciación de dicho inductor (ver Precauciones e Interacciones). Pacientes adultos y pediátricos: Posología en astrocitoma subependimario de células gigantes (ASCG) asociado a esclerosis tuberosa (ET): El tratamiento con Afinitor® debe instaurarlo un médico experimentado en el tratamiento de pacientes con ET que tenga acceso a servicios de determinación de las concentraciones sanguíneas de everolimus. En los pacientes en tratamiento por ASCG deben determinarse las concentraciones sanguíneas de everolimus (ver Determinación de las concentraciones sanguíneas de everolimus). Puede que haya que ajustar la dosis para obtener un efecto terapéutico óptimo. Las dosis toleradas y eficaces varían de unos pacientes a otros. La administración concurrente de tratamiento antiepiléptico puede afectar al metabolismo del everolimus y contribuir a esta diversidad (véase Interacciones). La administración de la dosis se personaliza en función del área de superficie corporal (Body Surface Area, BSA, en m2) mediante la fórmula de Dubois, donde el peso (weight, W) se expresa en kilogramos y la altura (height, H), en centímetros:
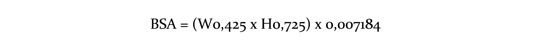
La dosis diaria inicial recomendada de Afinitor® para el tratamiento de pacientes con TSC y SEGA es de 4,5 mg/m2, redondeada a
la concentración más cerca a de comprimidos de Afinitor®. Asimismo, se pueden combinar diferentes concentraciones de los comprimidos de Afinitor® para lograr la dosis deseada. Unas dos semanas después de iniciar el tratamiento se deben determinar las concentraciones mínimas de everolimus en sangre. Se debe ajustar la dosis para lograr concentraciones mínimas de entre 3 y 15 ng/ml. Si las concentraciones son inferiores a 3ng/ml, se puede aumentar la dosis diaria en 2,5 mg cada 2 semanas según la tolerabilidad (ver Farmacología). Se debe evaluar el volumen del ASCG unos tres meses después de iniciar el tratamiento con Afinitor® y ajustar ulteriormente la dosis en función de las variaciones del volumen tumoral, la concentración mínima correspondiente y la tolerabilidad (ver Farmacología). Modificaciones de la dosis en casos de TSC con SEGA: El tratamiento de las reacciones adversas graves o intolerables puede exigir la reducción momentánea de la dosis o la interrupción del tratamiento con Afinitor® (ver Advertencias y Precauciones). Si fuera necesario reducir la dosis, la dosis sugerida es aproximadamente 50 % más baja que la dosis administrada previamente. En casos de reducción de la dosis por debajo de la concentración más baja disponible, deberá considerarse la administración de la dosis día por medio. Inhibidores moderados del CYP3A4 o de la glucoproteína P (PgP): Se debe ejercer cautela a la hora de administrar Afinitor® con inhibidores moderados del CYP3A4 o de la PgP. Si fuera necesario coadministrar un inhibidor moderado del CYP3A4 o de la PgP, reduzca la dosis diaria de Afinitor® en un 50 % aproximadamente. Tal vez sea necesario reducir aún más la dosis para mitigar las reacciones adversas (ver Advertencias, Precauciones e Interacciones). Se deben determinar las concentraciones mínimas de everolimus unas dos semanas después de la adición de un inhibidor moderado del CYP3A4 o de la PgP. Si se suspende la administración del inhibidor moderado, se debe volver a administrar la dosis de everolimus (Afinitor®) que se administraba antes de instaurar dicho inhibidor y determinar de nuevo la concentración mínima de everolimus unas dos semanas después (ver Advertencias y Precauciones). Inductores potentes del CYP3A4: Se debe evitar el uso concomitante de inductores potentes del CYP3A4. Los pacientes que estén recibiendo también inductores potentes del CYP3A4 (p. ej., fármacos antiepilépticos inductores de enzimas) pueden necesitar una dosis mayor de Afinitor® para lograr concentraciones mínimas de entre 5 y 15 ng/ml. Si las concentraciones son inferiores a 5 ng/ml, puede aumentarse la dosis diaria en 2,5 mg cada 2 semanas, comprobando la concentración mínima y evaluando la tolerabilidad antes de aumentarla. Si se suspende la administración del inductor potente, se debe volver a administrar la dosis de Afinitor® que se administraba antes de instaurar dicho inductor y determinar de nuevo la concentración mínima de everolimus unas dos semanas después (ver Advertencias y Precauciones). Determinación de las concentraciones sanguíneas de everolimus en los pacientes tratados por ASCG: Es preciso determinar las concentraciones sanguíneas de everolimus en los pacientes tratados por ASCG utilizando un método bioanalítico validado de cromatografía de líquidos acoplada a espectrometría de masas (LC-MS). Se medirán las concentraciones mínimas unas dos semanas después de la dosis inicial, después de cada cambio de dosis y después de iniciar o modificar la coadministración de inductores o inhibidores del CYP3A4 (ver Advertencias y Precauciones) o después de cualquier cambio en el estado hepático (Child-Pugh). Se debe ajustar la dosis con el objetivo de alcanzar concentraciones mínimas de everolimus de entre 3 y 15 ng/ml, según la tolerabilidad. La dosis se puede incrementar a fin de lograr una concentración mínima más alta dentro del rango objetivo, para obtener eficacia óptima, si no hay problemas de tolerabilidad (ver Características farmacológicas / Propiedades). Posología en poblaciones especiales: Población pediátrica: Afinitor® no ha sido estudiado en pacientes con SEGA < 3 años de edad o con BSA < 0,58 m2. El uso de Afinitor no está recomendado en pacientes pediátricos con cáncer. No se recomienda el uso de Afinitor® en pacientes pediátricos con TSC que tienen angiomiolipoma renal sin SEGA. Las recomendaciones de administración de la dosis para los pacientes pediátricos con TSC y SEGA coinciden con aquellas para la población adulta correspondiente, excepto por los pacientes con disfunción hepática. El uso de Afinitor® no se recomienda en pacientes < 18 años de edad con TSC y SEGA que también padecen de disfunción hepática. Pacientes de edad avanzada (de 65 años como mínimo): En estos pacientes no es necesario ajustar la dosis (ver Farmacología). Pacientes con insuficiencia renal: En estos pacientes no es necesario ajustar la dosis (ver Farmacología). Pacientes con insuficiencia hepática: Pacientes con cáncer de mama avanzado con receptores hormonales positivos, tumores neuroendocrinos avanzados de origen pancreático, carcinoma de células renales avanzado y TSC con angiomiolipoma renal. Insuficiencia hepática leve (Child-Pugh A) - dosis recomendada de 7.5 mg diarios. Insuficiencia hepática moderada (Child-Pugh B) - dosis recomendada de 2.5 mg diarios. Insuficiencia hepática severa (Child-Pugh C) - no recomendado. Si los beneficios deseados superan los riesgos, no se debe exceder de una dosis de 2.5 mg diarios. Deben realizarse ajustes de dosis si el estado hepático del paciente (Child-Pugh Clase C) cambia durante el tratamiento. Astrocitoma subependimario de células gigantes (ASCG) asociado con esclerosis tuberosa (ET). Pacientes ≥ 18 años de edad: Disfunción hepática leve (clase A de Child-Pugh): 75 % de la dosis calculada en función del BSA (redondeada a la concentración más cercana). Disfunción hepática moderada (clase B de Child-Pugh): 25 % de la dosis calculada en función del BSA (redondeada a la concentración más cercana). Disfunción hepática grave (clase C de Child-Pugh): no se recomienda. Las concentraciones mínimas en sangre total del everolimus se deben evaluar aproximadamente 2 semanas después de iniciar el tratamiento o luego de cualquier cambio en el estado hepático (clase de Child-Pugh). La administración de la dosis se debe ajustar para lograr concentraciones mínimas de 3 a 15 ng/ml. La dosis se puede incrementar a fin de lograr una concentración mínima más alta dentro del rango objetivo, para obtener eficacia óptima, si no hay problemas de tolerabilidad. Si las concentraciones están por debajo de 3 ng/ml, la dosis diaria se puede incrementar en 2,5 mg, si no hay problemas de tolerabilidad. Pacientes de < 18 años de edad: El uso de Afinitor® no se recomienda en pacientes < 18 años de edad con TSC y SEGA que también padecen de disfunción hepática.
Contraindicaciones.
Hipersensibilidad al principio activo, a otros derivados de la rapamicina o a cualquiera de los excipientes. (Ver Advertencias y Precauciones)
Reacciones adversas.
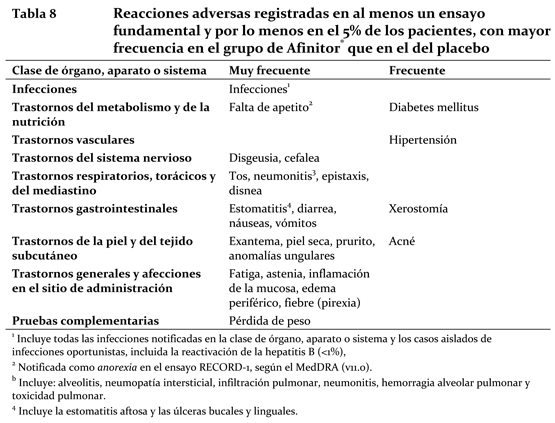
Cáncer de mama avanzado con receptores hormonales positivos, tumores neuroendocrinos avanzados de origen pancreático, carcinoma de células renales avanzado y carcinoma de células renales avanzado: Resumen del perfil de seguridad: La información sobre reacciones adversas se basa principalmente en datos procedentes de cuatro ensayos aleatorizados de Fase III, con doble enmascaramiento y placebo controlados: BOLERO-2 (CRAD001Y2301): Afinitor® en combinación con exemestane en el tratamiento de mujeres posmenopáusicas con cáncer de mama metastásico o avanzado localmente con receptores de estrógeno positivos que son refractarias a letrozol o anastrozol. De los datos obtenidos de la fecha de corte de análisis interino (11-Febrero-2011), la duración mediana del tratamiento fue 14.6 semanas para pacientes recibiendo Afinitor® y 12.0 semanas para aquellos pacientes recibiendo placebo más exemestane. RADIANT-3 (CRAD001C2324): Afinitor® más un tratamiento complementario óptimo en pacientes con tumores neuroendocrinos pancreáticos avanzados. La duración mediana del tratamiento enmascarado del estudio fue de 37,8 semanas en los pacientes que recibieron Afinitor® y de 16,1 semanas en los del grupo del placebo. RECORD-1 (CRAD001C2240): Afinitor® más un tratamiento complementario óptimo en pacientes con carcinoma de células renales avanzado. La duración mediana del tratamiento enmascarado de estudio fue de 141 días en los pacientes que recibieron Afinitor® y de 60 días en los del grupo del placebo. Las reacciones adversas más frecuentes (con una incidencia igual o superior al 10% en al menos un ensayo Fase III y, a juicio del investigador, presuntamente relacionadas con el tratamiento) fueron (en orden decreciente): estomatitis, exantema, diarrea, fatiga, infecciones, astenia, náuseas, edema periférico, falta de apetito, cefalea, disgeusia, epistaxis, inflamación de la mucosa, neumonitis, pérdida de peso, vómitos, prurito, tos, disnea, piel seca, anomalías ungulares y fiebre (pirexia). Las reacciones adversas de grado 3 o 4 más frecuentes (frecuencia igual o superior al 2% en al menos un ensayo fundamental) fueron: estomatitis, fatiga, diarrea, infecciones, neumonitis y diabetes mellitus. Resumen tabulado de reacciones adversas obtenidas de los estudios clínicos: La Tabla 8 presenta la categoría de frecuencia de las reacciones adversas notificadas con una incidencia igual o superior al 5% en los pacientes que recibieron Afinitor® (10 mg/día) en al menos uno de los ensayos fundamentales; todos los términos incluidos se basan en el mayor porcentaje registrado en un ensayo fundamental. Las reacciones adversas se enumeran con arreglo a la clase de órgano, aparato o sistema del MedDRA. Dentro de cada clase de órgano, aparato o sistema se presentan las reacciones adversas por orden decreciente de frecuencia. Además, para clasificar cada reacción adversa en la correspondiente categoría de frecuencia, se ha seguido la convención siguiente (CIOMS III): muy frecuente (≥1/10); frecuente (≥1/100, < 1/10); infrecuente (≥1/1000, < 1/100); rara (≥1/10 000, < 1/1000); muy rara ( < 1/10 000), incluidas las notificaciones aisladas.
A continuación se detallan otras reacciones adversas importantes cuya frecuencia fue mayor en el grupo de Afinitor® que en el del placebo, aunque inferior al 5%, en al menos un ensayo fundamental. Todos los términos incluidos se basan en el mayor porcentaje registrado en un ensayo fundamental. Trastornos de la sangre y del sistema linfático: Infrecuente: aplasia exclusivamente eritrocítica (eritroblastopenia) ( < 1%). Trastornos del metabolismo y de la nutrición: Frecuente: deshidratación (2,5%), agravamiento de la diabetes mellitus existente (1,1%). Infrecuente: diabetes mellitus de nueva aparición ( < 1%). Trastornos psiquiátricos: Frecuente: insomnio (3,3%). Trastornos del sistema nervioso: Infrecuente: ageusia ( < 1%). Desórdenes Vasculares: Frecuente: Hemorragia (4.7%, mayormente grado 1 en varios sitios). Infrecuente: trombosis venosa profunda ( < 1%). Trastornos cardíacos: Infrecuente: insuficiencia cardíaca congestiva ( < 1%). Trastornos respiratorios, torácicos y del mediastino: Frecuente: embolia pulmonar (1,5%), hemoptisis (1,1%). Infrecuente: síndrome de dificultad respiratoria aguda (distrés respiratorio agudo) ( < 1%). Trastornos gastrointestinales: Frecuente: dolor bucal (3,7%), dolor abdominal (3,6%), dispepsia (2,9%), disfagia (2,6%). Trastornos de la piel y del tejido subcutáneo: Frecuente: eritrodisestesia palmoplantar (4,7%), eritema (3,7%). Trastornos osteomusculares y del tejido conectivo: Frecuente: artralgia (2,8%). Trastornos renales y urinarios: Frecuente: proteinuria (2,5%), insuficiencia renal (2,3%, incluida la insuficiencia renal aguda), micción diurna elevada (1,8%). Trastornos generales y afecciones en el sitio de administración: Frecuente: dolor torácico (1,1%). Infrecuente: cicatrización deficiente de heridas ( < 1%). Se observaron anomalías de laboratorio importantes en al menos un ensayo pivote con mayor frecuencia en el grupo de Afinitor® que en placebo. En los cuatro ensayos de Fase III la mayoría de las anomalías de laboratorio importantes observadas se registraron con una frecuencia ≥10% (enumeradas por orden decreciente de frecuencia): Cifras reducidas de parámetros hematológicos tales como hemoglobina, linfocitos, trombocitos y neutrófilos (o colectivamente como pancitopenia). Cifras elevadas de parámetros bioquímicos, como colesterol, triglicéridos, glucosa, aspartato-transaminasa, creatinina, alanina-aminotransferasas y bilirrubina. Cifras reducidas de parámetros bioquímicos, como fosfato y potasio. La mayoría de las anomalías eran leves (grado 1) o moderadas (grado 2). Las anomalías de grado 4 abarcaron reducciones en las cifras de linfocitos (2,2%), hemoglobina (2%) y potasio (2%), neutrófilos, trombocitos y fosfato (cada cifra < 1%) e incrementos de creatinina (1%), colesterol, AST, ALT, bilirrubina y glucosa (cada cifra < 1%) y hemoglobina (2%). Complejo de esclerosis tuberosa (TSC) con angiomiolipoma renal: Resumen del perfil de seguridad: Los datos descritos abajo se basan en un ensayo aleatorizado, doble ciego y controlado de Fase III de Afinitor® (n = 79) en comparación con el placebo (n = 39) en pacientes con TSC que tienen angiomiolipoma o linfangioleiomiomatosis (LAM) esporádica con angiomiolipoma. La mediana de duración del tratamiento del estudio a ciego fue de 38,1 semanas (intervalo: 2 a 105) en pacientes que recibieron Afinitor® y 34,0 semanas (intervalo: 9 a 112) en los que recibieron el placebo. La exposición total fue de 67,7 pacientes-años en pacientes que recibieron Afinitor® y 29,9 pacientes-años en los que recibieron el placebo. La mediana de edad de los pacientes fue de 31,0 años (intervalo: 18,0 a 61,0). A la fecha límite, 91,1 % de los pacientes del grupo de Afinitor® y 66,7 % de los pacientes del grupo de placebo continuaban en el estudio. No fue evidente ninguna diferencia en la proporción de pacientes que interrumpieron el fármaco del estudio a causa de las ADR (2,5 % con Afinitor®, en comparación con 2,6 % con el placebo). Las ADR más frecuentes (incidencia ≥ 10 % y presuntamente relacionadas con el tratamiento, según el investigador) fueron estomatitis, hipercolesterolemia, acné, fatiga, anemia, aumento de la lactato deshidrogenasa en sangre, leucopenia y náuseas. Las ADR de grados 3 y 4 más frecuentes (incidencia ≥ 2 % y presuntamente relacionadas con el tratamiento, según el investigador) fueron la estomatitis y amenorrea. Se informó una sola muerte en el grupo de Afinitor® como resultado del estado epiléptico de un paciente con antecedentes de ataques convulsivos severos; no hubo presunción de relación con el fármaco, según el investigador del estudio. Resumen tabulado de reacciones adversas al fármaco derivadas de ensayos clínicos: En la Tabla 9 se presentan las categorías por frecuencia de las ADR informadas con incidencia ≥ 5 % en los pacientes tratados con 10 mg/día de Afinitor® en este ensayo de Fase III. Las reacciones adversas a un medicamento de este estudio de Fase III se enumeran según la clase de órgano, aparato o sistema afectados de MedDRA. Dentro de cada clase de órgano, aparato o sistema, las ADR se clasifican según su frecuencia, de modo que aparecen primero las de mayor frecuencia. Además, la categoría de frecuencia correspondiente a cada ADR se basa en la convención siguiente (CIOMS III): muy frecuente (≥ 1/10); frecuente (≥ 1/100 a < 1/10); poco frecuente (≥ 1/1000 a < 1/100), infrecuente (≥ 1/10.000 a < 1/1000) y muy infrecuente ( < 1/10.000).

A continuación se enumeran otras ADR que se han presentado con mayor frecuencia en el caso de Afinitor® que en el del placebo, pero con una incidencia < 5 % y consideradas clínicamente relevantes. Trastornos del sistema inmunitario: Frecuente: Hipersensibilidad (1,3 %). Trastornos del sistema nervioso: Frecuente: Disgeusia (3,8 %) y ageusia (1,3 %). Trastornos respiratorios, torácicos y mediastínicos: Frecuente: Epistaxis (3,8 %) y neumonitis (1,3 %). Trastornos del aparato urinario: Frecuente: Insuficiencia renal aguda (1,3 %). Trastornos del aparato reproductor y las mamas: Frecuente: Menorragia (3,8 %), hemorragia vaginal (3,8 %), atraso de la menstruación (1,3 %). Anomalías en los análisis de laboratorio clínicamente relevantes: Los informes de la mayoría de las anomalías en los análisis de laboratorio clínicamente relevantes tuvieron una incidencia ≥ 10 % (se enumeran en orden de frecuencia decreciente): Disminuciones de parámetros hematológicos como hemoglobina, leucocitos, neutrófilos, linfocitos y plaquetas. Aumento de parámetros químicos clínicos como colesterol, triglicéridos, aspartato y alanina transaminasas, glucosa y bilirrubina. Disminución de parámetros químicos clínicos como fosfatos en sangre. Casi todas las anomalías en los análisis de laboratorio fueron leves (grado 1) o moderadas (grado 2). Las anomalías en los análisis de laboratorio de grados 3 y 4 más frecuentes fueron reducciones de fosfatos (5,1 %), fibrinógeno (2,5 %), linfocitos y neutrófilos (1,3 % en cada caso), y aumentos de fosfatasa alcalina, potasio, aspartato transaminasas y alanina transaminasas (1,3 % en cada caso). Ensayo de Fase III en pacientes con TSC y SEGA: Resumen del perfil de seguridad: Los datos que se describen a continuación se basan en un ensayo de Fase III aleatorizado, doble ciego, controlado, de Afinitor® (n = 78) en comparación con placebo (n = 39) en pacientes con TSC y SEGA, independientemente de la edad. La mediana de la edad de los pacientes fue de 9,5 años (intervalo: 0,8 a 26,6). La mediana de la duración del tratamiento del estudio a ciego fue de 9,6 meses (intervalo: 5,5 a 18,1) en los pacientes tratados con Afinitor®, y de 8,3 meses (intervalo: 3,2 a 18,3) en los que recibieron placebo. La exposición total fue de 66,5 pacientes-años en los pacientes tratados con Afinitor®, y de 30,8 pacientes-años en los que recibieron placebo. A la fecha de corte de datos, 97,4 % de los pacientes del grupo de Afinitor® y 79,5 % de los del grupo de placebo continuaban en el ensayo. Ningún paciente interrumpió el fármaco del estudio debido a ADR. La ADR más frecuente (incidencia ≥ 10 % y sospecha de relación con el tratamiento, según el investigador) fue la estomatitis. Las ADR de grado 3 más frecuentes (incidencia ≥ 2 % y sospecha de relación con el tratamiento, según el investigador) fueron: estomatitis, neutropenia y gastroenteritis viral. No se informaron ADR de grado 4. Resumen tabulado de reacciones adversas de los estudios clínicos: La tabla 10 resume las reacciones adversas surgidas durante el tratamiento que se notificaron con una frecuencia ≥5%. Las reacciones adversas se enumeran en relación a la clase de órgano, aparato o sistema afectado del MedDRA. Dentro de cada clase de órgano, aparato o sistema, se presentan las reacciones adversas por orden decreciente de frecuencia.

A continuación se enumeran otras ADR del ensayo de Fase III que se han presentado con mayor frecuencia en el caso de Afinitor® que en el de placebo, pero con incidencia < 5 % y consideradas clínicamente relevantes. Infecciones e infestaciones: Frecuente: Otitis media (3,8 %), gastroenteritis viral (2,6 %). Trastornos sanguíneos y del sistema linfático: Frecuente: Anemia (2,6 %). Trastornos psiquiátricos: Frecuente: Agresividad (1,3 %), insomnio (1,3 %). Trastornos del sistema nervioso: Frecuente: Convulsiones (2,6 %). Trastornos respiratorios, torácicos y mediastínicos: Frecuente: Epistaxis (1,3 %), neumonitis (1,3 %). Trastornos gastrointestinales: Frecuente: Dolor oral (3,8 %). Trastornos del aparato reproductor y las mamas: Frecuente: Amenorrea (2,6 %), irregularidad menstrual (1,3 %). Trastornos generales y afecciones del lugar de administración: Frecuente: Trastornos del andar (1,3 %). Exploraciones complementarias: Frecuente: Aumento de triglicéridos en sangre (2,6 %). Anomalías en los análisis de laboratorio clínicamente relevantes: Las anomalías en los análisis de laboratorio clínicamente relevantes informadas con una incidencia ≥ 10 % (enumeradas por orden de frecuencia decreciente): Los parámetros hematológicos incluyeron: aumento del tiempo de tromboplastina parcial, neutropenia y anemia. Los parámetros químicos clínicos incluyeron: hipercolesterolemia, aumento de la aspartato aminotransferasa (AST), hipertrigliceridemia, aumento de la alanina aminotransferasa (ALT), hipofosfatemia e hipopotasiemia. Casi todas las anomalías en los análisis de laboratorio fueron leves (grado 1) o moderadas (grado 2). La anomalía en los análisis de laboratorio de grado 3 más frecuente (incidencia ≥ 2 % y que se presentó con más frecuencia con Afinitor® que con el placebo) fue la neutropenia. Ensayo de Fase II en pacientes con TSC y SEGA: El estudio CRAD001C2485 es un ensayo de fase II, no aleatorizado, de etiqueta abierta, monocéntrico, de Afinitor® (n = 28) en pacientes de ≥ 3 años de edad con TSC y SEGA. La mediana de edad de los pacientes fue de 11 años (intervalo: 3 a 34). La mediana de la duración del tratamiento fue de 34,2 meses (intervalo: 4,7 a 47,1). La exposición total fue de 75,6 pacientes-años. Hasta la fecha de corte, 89,3 % de los pacientes continuaban en el ensayo. Reacciones adversas al fármaco: Se informaron las siguientes ADR adicionales clínicamente relevantes durante el ensayo de fase II (Nota: El protocolo estipuló que todas las infecciones fueran ADR clasificadas): Muy frecuentes: Sinusitis, celulitis, gastroenteritis, faringitis, otitis externa, infección cutánea, tiña (micosis), infección gástrica, infección de las vías urinarias, forúnculo, nasofaringitis, diarrea, dermatitis acneiforme, acné, conjuntivitis e hipertrigliceridemia. Frecuentes: Infección, absceso en extremidad, bronquitis viral, agitación, inflamación faríngea, gastritis, vómitos, proteinuria, disminución de la inmunoglobulina G en sangre. Otras ADR clínicamente relevantes de grado 3 informadas en el ensayo de Fase II (incidencia ≥ 2 %) fueron: neutropenia y casos aislados de sinusitis, neumonía, extremidad con absceso y bronquitis viral. Se informaron las siguientes ADR clínicamente relevantes en una categoría de frecuencia más alta en el ensayo de Fase II que en el ensayo de Fase III controlado (aumento de frecuente a muy frecuente): Muy frecuentes: Infección de las vías respiratorias altas, otitis media, neumonía, tos, pirexia, incremento de colesterol en sangre, incremento de triglicéridos en sangre y disminución del recuento de neutrófilos. Anomalías en los análisis de laboratorio clínicamente relevantes: Se informaron las siguientes anomalías en los análisis de laboratorio clínicamente relevantes adicionales, con una incidencia ≥ 10 %, en el ensayo de Fase II: Los parámetros hematológicos incluyeron leucopenia, trombocitopenia y linfopenia. Los parámetros químicos clínicos incluyeron aumento de la fosfatasa alcalina en sangre, hiperglucemia, hipercreatinemia e hipoglucemia. Las anomalías en los análisis de laboratorio clínicamente relevantes de grado 3 adicionales informadas en el ensayo de Fase II fueron aumento de la fosfatasa alcalina en sangre y aumento de la aspartato aminotransferasa. Las anomalías en los análisis de laboratorio clínicamente relevantes de grado 4 fueron neutropenia y linfopenia. Descripción de reacciones adversas seleccionadas: En ensayos clínicos, everolimus se ha asociado a casos graves de reactivación de hepatitis B, incluidos desenlaces mortales. La reactivación de infecciones es un evento esperado durante períodos de inmunosupresión. En ensayos clínicos y en informes espontáneos posteriores a la comercialización, el everolimus se ha relacionado con episodios de insuficiencia renal (lo que incluye episodios mortales) y proteinuria. Se recomienda el control de la función renal (véase Advertencias y Precauciones). En los ensayos clínicos e informes posteriores a la comercialización, everolimus ha sido asociado con casos de amenorrea (incluso amenorrea secundaria).
Precauciones.
Interacciones farmacológicas: Debe evitarse la coadministración de inhibidores potentes del CYP3A4 y de la PgP (ver Interacciones). Debe usarse con cautela en caso de administrarse en combinación con inhibidores moderados del CYP3A4 o de la PgP. Llegado el caso de que Afinitor® deba administrarse concurrentemente con un inhibidor moderado del CYP3A4 o de la PgP, se deberá monitorear atentamente la aparición de efectos indeseados en el paciente y reducir la dosis si fuera necesario. (Ver Dosificación) Debe evitarse la coadministración de inductores potentes del CYP3A4 o de la PgP (ver Interacciones). Llegado el caso de que sea necesario administrar Afinitor® con un inductor potente del CYP3A4 o de la PgP concurrentemente, se deberá vigilar atentamente la respuesta clínica del paciente. Considerar el incremento de la dosis de Afinitor® cuando se coadministra con inductores potentes del CYP3A4 o de la PgP si no hay otra alternativa terapeútica (ver Interacciones y Dosificación). Debe tenerse cuidado cuando Afinitor® es administrado en combinación con sustratos orales del CYP3A4 con una ventana terapéutica estrecha debido al potencial de producir interacciones. Si Afinitor® se administra con sustratos orales del CYP3A4 con una ventana terapéutica estrecha, el paciente debe ser monitoreado para efectos adversos descriptos en la información del producto del sustrato de CYP3A4 administrado oralmente (ver Interacciones). Insuficiencia hepática: La exposición a everolimus fue incrementada en pacientes con insuficiencia hepática leve (Child-Pugh A), moderada (Child-Pugh B) y grave (clase C de Child-Pugh) (ver Farmacología). Afinitor® no está recomendado para su uso en pacientes con disfunción hepática grave (clase C de Child-Pugh) para el tratamiento del cáncer de mama avanzado positivo para receptores hormonales en mujeres posmenopáusicas, o en pacientes con tumores neuroendocrinos avanzados de origen pancreático o carcinoma de células renales avanzado con insuficiencia hepática severa (Child-Pugh C) o TSC con angiomiolipoma renal, salvo que el beneficio potencial supere el riesgo (ver Dosificación y Farmacología). Afinitor® no está recomendado para el uso de pacientes < 18 años de edad con TSC y SEGA que tienen insuficiencia hepática (Child-Pugh A,B o C) o en pacientes ≥ 18 años con disfunción hepática grave (clase C de Child-Pugh) (ver Dosificación y Farmacología). Vacunas: Durante el tratamiento con Afinitor® es preciso evitar la aplicación de vacunas atenuadas (elaboradas con microbios vivos), así como el contacto íntimo con personas que han recibido tales vacunas (ver Interacciones). Interacciones: El everolimus es un sustrato del CYP3A4 y asimismo un sustrato e inhibidor moderado de la bomba de expulsión de fármacos conocida como PgP. Por consiguiente, los fármacos que afectan al CYP3A4 o la PgP pueden alterar la absorción y la eliminación posterior de everolimus. In vitro, el everolimus es un inhibidor competitivo del CYP3A4 y un inhibidor mixto del CYP2D6. Agentes que pueden aumentar las concentraciones sanguíneas de everolimus: Las sustancias que inhiben la actividad del CYP3A4 (y que por eso mismo reducen el metabolismo de everolimus) pueden incrementar las concentraciones sanguíneas de everolimus. Los inhibidores de la PgP (capaces de reducir la expulsión de everolimus de las células intestinales) pueden aumentar las concentraciones sanguíneas de everolimus. Debe evitarse el tratamiento concurrente con inhibidores potentes del CYP3A4 o de la PgP (como, por ejemplo: ketoconazol, itraconazol, voriconazol, ritonavir, claritromicina y telitromicina). Se apreció un significativo incremento de exposición a everolimus (la Cmáx y el ABC aumentaron unas 3,9 y 15 veces, respectivamente) en sujetos sanos que habían recibido everolimus junto con ketoconazol (inhibidor potente del CYP3A4 e inhibidor de la PgP). El tratamiento simultáneo con inhibidores moderados del CYP3A4 (incluyendo, sin que esta mención sea limitativa, eritromicina, verapamilo, ciclosporina, fluconazol, diltiazem, amprenavir, fosamprenavir o aprepitant) y de la PgP requiere cautela. Se debe reducir la dosis de Afinitor® si se administraran concomitantemente inhibidores moderados del CYP3A4 / PgP (ver Dosificación). Hubo un aumento de exposición a everolimus en sujetos sanos que habían recibido everolimus junto con: eritromicina (inhibidor moderado del CYP3A4 e inhibidor de la PgP; Cmáx 2,0 veces mayor y ABC 4,4 veces mayor). Verapamilo (inhibidor moderado del CYP3A4 e inhibidor de la PgP; Cmáx 2,3 veces mayor y ABC 3,5 veces mayor). Ciclosporina (sustrato del CYP3A4 e inhibidor de la PgP; Cmáx 1,8 vez mayor y ABC 2,7 veces mayor). Algunos antimicóticos, como el fluconazol, y bloqueantes de los canales de calcio, como el diltiazem, son inhibidores moderados del CYP3A4 y de la PgP que pueden aumentar las concentraciones sanguíneas de everolimus. Durante el tratamiento con Afinitor® se debe evitar el consumo de pomelo, jugo de pomelo, fruta estrella, naranja de Sevilla y cualquier otro alimento que pueda alterar la actividad de la PgP y del citocromo P450. No se apreciaron diferencias en la Cmín de everolimus después de un tratamiento con la dosis diaria de 10 o 5 mg en presencia o ausencia de sustratos del CYP3A4 o de la PgP. La coadministración de inhibidores débiles el CYP3A4, con o sin inhibidores de la PgP, no produjo efectos evidentes en la Cmín de everolimus después de un tratamiento con la dosis diaria de 10 o 5 mg. Agentes que pueden reducir las concentraciones sanguíneas de everolimus: Las sustancias inductoras del CYP3A4 o de la PgP pueden reducir las concentraciones sanguíneas de everolimus mediante un aumento del metabolismo de everolimus o la expulsión de everolimus de las células intestinales. Debe evitarse el tratamiento simultáneo con inductores potentes del CYP3A4 o de la PgP. Si el uso concurrente de inductores potentes del CYP3A4 o de la PgP es requerido (por ejemplo: rifampicina y rifabutina) puede ser necesario ajustar la dosis (ver Dosificación y Precauciones). El pretratamiento de sujetos sanos con dosis múltiples de 600 mg de rifampicina (un inductor de CYP3A4 y de la PgP) al día durante 8 días y, luego, con una dosis única de everolimus prácticamente triplica la depuración de everolimus y reduce la Cmáx en un 58%, así como el ABC, en un 63%. Entre los inductores potentes del CYP3A4 que pueden aumentar el metabolismo de everolimus y reducir sus concentraciones sanguíneas figuran asimismo la hierba de San Juan (Hypericum perforatum), corticoides (como la dexametasona, prednisona, prednisolona), anticonvulsivantes (como la carbamazepina, el fenobarbital y la fenitoína) y antirretrovíricos (como el efavirenz y la nevirapina). Agentes cuyas concentraciones sanguíneas pueden verse alteradas por everolimus: Los estudios en sujetos sanos indican que no existen interacciones farmacocinéticas clínicamente significativas entre Afinitor® y dos inhibidores de la HMG-CoA reductasa como la atorvastatina (un sustrato del CYP3A4) y la provastatina (que no es sustrato del CYP3A4), y los análisis de farmacocinética poblacional tampoco arrojaron indicios de que la simvastatina (un sustrato del CYP3A4) afecte de alguna forma la depuración de everolimus (Afinitor®). In vitro, el everolimus es un inhibidor competitivo del metabolismo de la ciclosporina (un sustrato del CYP3A4) y un inhibidor mixto del dextrometorfano (un sustrato del CYP2D6). Tras la administración de una dosis oral de 10 mg al día o de 70 mg a la semana, la concentración máxima media de everolimus en el estado de equilibrio es más de 12 o 36 veces menor que los valores de Ki de la inhibición in vitro. Por consiguiente, no cabe esperar que everolimus afecte el metabolismo de los sustratos de CYP3A4 o CYP2D6. Un estudio con pacientes sanos demostró que la coadministración de una dosis oral de midazolam con everolimus resultó en un 25% de incremento en la Cmáx de midazolam y un 30% de incremento en ABC (0-inf) de midazolam mientras que la proporción metabólica de ABC (0-ibf) (1-hidroximidazolam/midazolam) y el t½ terminal de midazolam no fueron afectados. Esto sugiere que la exposición incrementada a midazolam se debe al efecto de everolimus en el sistema gastrointestinal cuando ambos medicamentos se administran al mismo tiempo. Por ello, everolimus puede afectar la biodisponibilidad de medicamentos orales que son sustratos del CYP3A4. Se considera poco probable que everolimus afecte la exposición de otros sustratos de la CYP3A4. Es una rareza que everolimus afecte la exposición de otros sustratos CYP3A4 que sean administrados por otras vías no orales, como ser endovenosa, subcutánea y administración transdérmica (ver Precauciones). La coadministración de everolimus y una formulación de depósito de octreotida aumenta la Cmín de octeotrida con un cociente de medias geométricas (everolimus/placebo) de 1,47 (IC del 90%: 1,32-1,64), que probablemente no afecte de forma clínicamente significativa a la eficacia de everolimus en los pacientes con tumores neuroendocrinos avanzados. La coadministración de everolimus y exemestane incrementa la Cmín y C2h de exemestane en 45% y 71%, respectivamente. Sin embargo, los correspondientes niveles de estradiol en un nivel estable (4 semanas) no fueron diferentes entre los dos brazos de tratamiento. No se observó incremento en las reacciones adversas relacionadas a exemestane en pacientes con cáncer de mama avanzado con receptores hormonales positivos recibiendo la combinación. No es probable que el incremento en los niveles de exemestane tenga un impacto sobre la eficacia o seguridad. Vacunas: Los inmunodepresores pueden alterar la respuesta a una vacuna y ésta puede ser menos eficaz durante el tratamiento con Afinitor®. Es necesario evitar el uso de vacunas atenuadas durante el tratamiento con Afinitor® (ver Precauciones). Dichas vacunas son, por ejemplo: la antigripal intranasal, la antisarampionosa, la antiparotidítica, la antirrubeólica, la antipoliomielítica oral, la BCG (antituberculosa), la antiamarílica, la vacuna contra la varicela y la vacuna antitifoidea elaborada con la cepa TY21a de S. Typhi. Embarazo y Lactancia: Embarazo: No se dispone de datos suficientes sobre el uso de Afinitor® en mujeres embarazadas. Los estudios en animales han evidenciado efectos tóxicos durante la reproducción tales como embriotoxicidad y fetotoxicidad (ver Datos sobre toxicidad preclínica). Se desconoce el riesgo para el ser humano. Afinitor® no debe administrarse a mujeres embarazadas, a menos que los posibles beneficios justifiquen el riesgo para el feto. Lactancia: No se sabe si el everolimus pasa a la leche humana. En los estudios con animales, tanto el everolimus como sus metabolitos pasaban fácilmente a la leche de las ratas lactantes. Por consiguiente, las mujeres que toman Afinitor® no deben amamantar. Mujeres en edad de procrear: Debe aconsejarse a las mujeres en edad fértil que utilicen métodos anticonceptivos altamente eficaces durante el tratamiento con Afinitor® y en las 8 semanas siguientes a la finalización de la terapia. Fertilidad: Se desconoce el potencial de everolimus de causar esterilidad en pacientes de ambos sexos. Sin embargo, se ha observado amenorrea (incluso amenorrea secundaria). A juzgar por los hallazgos preclínicos, el tratamiento con Afinitor® puede comprometer la fecundidad masculina. La fertilidad femenina no se vio afectada (ver Datos sobre toxicidad preclínica). Efectos sobre la capacidad de conducir vehículos y utilizar máquinas: No se han estudiado los efectos sobre la capacidad de conducir vehículos y utilizar máquinas.
Advertencias.
Neumonitis no infecciosa: La neumonitis no infecciosa es un efecto de la clase farmacológica de los derivados de la rapamicina. También fueron descriptos casos de neumonitis no infecciosa (incluida la enfermedad intersticial pulmonar) en pacientes tratados con Afinitor® (ver Reacciones adversas). Algunos de estos casos fueron severos y en raras ocasiones se observaron casos fatales. Se debe considerar el diagnóstico de neumonitis no infecciosa en los pacientes que presentan signos y síntomas respiratorios inespecíficos, como hipoxia, derrame pleural, tos o disnea, en quienes se han descartado las causas infecciosas o neoplásicas y otras causas no farmacológicas por medio de estudios apropiados. Se ha de aconsejar al paciente que comunique de inmediato cualquier síntoma respiratorio, ya sea nuevo o agravado. Los pacientes en quienes se descubren signos radiológicos indicativos de neumonitis no infecciosa, pero con escasos síntomas o ninguno, pueden continuar su tratamiento con Afinitor® sin modificar la dosis. Si los síntomas son moderados, debe considerarse la posibilidad de interrumpir el tratamiento hasta que mejoren. Puede ser necesario el uso de corticoesteroides. La administración de Afinitor® se puede reanudar con una dosis diaria aproximadamente 50 % menor que la dosis administrada previamente. En los casos en que los síntomas de neumonitis no infecciosa sean graves (grado 3 o 4), la terapia con Afinitor® se deberá interrumpir, y el uso de corticosteroides podría estar indicado hasta que los síntomas clínicos se resuelvan. En los casos de neumonitis no infecciosa de grado 3, la administración de Afinitor® se puede reiniciar en una dosis diaria aproximadamente 50 % inferior a la dosis previamente administrada, según las circunstancias clínicas de la persona (véase Dosificación). Infecciones: Afinitor® tiene propiedades inmunodepresoras y puede hacer que los pacientes sean más propensos a contraer infecciones bacterianas, fúngicas, virales o por protozoarios, incluyendo las atribuidas a patógenos oportunistas (ver Reacciones adversas). En pacientes tratados con Afinitor® se han descripto infecciones locales y generales, incluidas las neumonías, otras infecciones bacterianas y micosis invasoras, como la aspergilosis o la candidiasis e infecciones virales incluyendo la reactivación de hepatitis B. Algunas de estas infecciones han sido graves (p. ej.: produjeron insuficiencia respiratoria o hepática) y en ocasiones llevaron a la muerte del paciente. Los médicos y los pacientes deben ser conscientes del mayor riesgo de infección asociado a Afinitor®. Deben tratarse las infecciones preexistentes antes de iniciar el tratamiento c
on Afinitor®. Durante el tratamiento con Afinitor® debe evaluarse permanentemente la aparición de síntomas y signos de infección y, si se diagnostica una infección, debe establecerse un tratamiento adecuado sin demora y estudiar la posibilidad de suspender temporal o definitivamente la administración de Afinitor®. Si se realiza el diagnóstico de una infección fúngica sistémica invasiva, debe discontinuarse Afinitor® y tratarla con la terapia antifúngica apropiada. Reacciones de hipersensibilidad: Con el uso de everolimus, se han observado reacciones de hipersensibilidad manifestadas por síntomas, incluyendo: anafilaxia, disnea, crisis vasomotoras, dolor torácico o angioedema (p. ej.: edema de las vías respiratorias o de la lengua, con o sin falla respiratoria), entre otros (ver Contraindicaciones). Ulceras bucales: Se han observado úlceras, estomatitis y mucositis bucales en pacientes tratados con Afinitor® (ver Reacciones adversas). En dichas situaciones se recomiendan los tratamientos tópicos, pero no los colutorios a base de alcohol o peróxido, que pueden exacerbar la afección. No se deben utilizar fungicidas, salvo si se ha diagnosticado una micosis (ver Interacciones). Insuficiencia Renal: Se ha descripto casos de falla renal (incluyendo falla renal aguda), algunos con resultados fatales, en pacientes tratados con Afinitor® (ver Reacciones adversas y Vigilancia y análisis de laboratorio). Vigilancia y análisis de laboratorio: Función renal: Se han notificado elevaciones de creatinina sérica y proteinuria, usualmente leves, y proteinuria en los ensayos clínicos (ver Reacciones adversas). Se recomienda la vigilancia de la función renal, que incluye la determinación de urea (BUN), proteína urinaria o de creatinina sérica, antes del inicio del tratamiento con Afinitor® y periódicamente durante el mismo. Glucemia: Se ha comunicado hiperglucemia en los ensayos clínicos (ver Reacciones adversas). Se aconseja la vigilancia de la glucemia en ayunas antes de comenzar el tratamiento con Afinitor® y periódicamente durante el mismo. La glucemia debe estar perfectamente regulada antes de instaurar un tratamiento con Afinitor® en el paciente. Parámetros hematológicos: Se han registrado cifras reducidas de hemoglobina, linfocitos, neutrófilos y trombocitos en los ensayos clínicos (ver Reacciones adversas). Se aconseja la supervisión del hemograma completo antes de comenzar el tratamiento con Afinitor® y periódicamente durante el mismo.
Conservación.
No conservar a más de 30°C; conservar en el envase original para proteger el producto de la luz y la humedad.
Sobredosificación.
En los estudios en animales, everolimus reveló tener un bajo potencial de toxicidad aguda. No se observó letalidad ni toxicidad grave en las ratas o ratones que recibieron dosis orales únicas de 2000 mg/Kg (ensayo límite). Se conocen muy pocos casos de sobredosis en seres humanos. Se han administrado dosis únicas de hasta 70 mg con una aceptable tolerabilidad aguda. En todos los casos de sobredosis se deben tomar medidas generales de apoyo. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital A. Posadas: (011) 4654-6648/4658-7777.
Información al paciente.
Lea todo el prospecto detenidamente antes de empezar a tomar el medicamento. Conserve este prospecto, ya que puede tener que volver a leerlo. Si tiene alguna duda, consulte a su médico o farmacéutico. Este medicamento se le ha recetado a usted y no debe dárselo a otras personas, ni lo use para otra enfermedad. Si considera que alguno de los efectos adversos la afecta gravemente o si aprecia cualquier efecto adverso no mencionado en este prospecto, informe a su médico o farmacéutico. Contenido del prospecto: 1. Qué es Afinitor® y para qué se utiliza. 2. Antes de tomar Afinitor®. 3. Cómo tomar Afinitor®. 4. Posibles efectos adversos. 1. QUE ES AFINITOR® Y PARA QUE SE UTILIZA: Qué es Afinitor®: Afinitor® es un medicamento contra el cáncer que previene que crezcan ciertas células en el cuerpo. Puede ser utilizado para tratar cáncer avanzado y también crecimientos no oncológicos asociados con un desorden genético llamado esclerosis tuberosa o complejo de esclerosis tuberosa en niños y adultos. Para qué se utiliza Afinitor®: Su médico puede prescribirle Afinitor® para el tratamiento de: Tumores neuroendocrinos avanzados (NET) localizados en el páncreas. Cáncer de riñón avanzado (Carcinoma de células renales avanzado (RCC)). Astrocitoma subependimario de células gigantes (SEGA, tumor de cerebro específico) asociado con esclerosis tuberosa (ET). Cáncer de mama avanzado con receptores hormonales positivos, en combinación con un inhibidor de la aromatasa, después de terapia endócrina previa en mujeres postmenopáusicas. Complejo de esclerosis tuberosa (TSC) con angiomiolipoma renal (un tumor de los riñones), que no requiere cirugía inmediata. Cómo actúa Afinitor®: La sustancia activa en Afinitor® es everolimus. Tratamiento de cáncer de mama avanzado con receptores hormonales positivos: El crecimiento de este tipo de cáncer de mama es estimulado por los estrógenos que son hormonas sexuales femeninas. Los inhibidores de la aromatasa reducen la cantidad de estrógeno y pueden enlentecer el crecimiento del cáncer de mama. La administración de Afinitor® junto con un inhibidor de la aromatasa puede prevenir que las células de cáncer sean resistentes a la terapia hormonal, consecuentemente reduce el crecimiento del cáncer de mama y retrasa las recurrencias de esta enfermedad. Tratamiento de tumores neuroendocrinos avanzados: Los tumores neuroendocrinos son tumores raros los cuales puede encontrarse en diferentes partes del cuerpo. Afinitor® es utilizado para controlar estos tumores localizados en el páncreas. Tratamiento de cáncer de riñón avanzado: Afinitor® detiene el desarrollo de nuevas células de cáncer y corta el suministro de sangre al cáncer. Esto disminuye el crecimiento y la diseminación del cáncer de riñón. Tratamiento de pacientes con astrocitoma subpendimario de células gigantes asociado a esclerosis tuberosa: Afinitor® reduce el tamaño de los tumores cerebrales que son causados por un desorden genético llamado esclerosis tuberosa. Esto puede prevenir que los tumores causen problemas mientras crecen, como por ejemplo hidrocefalia (acumulación excesiva de fluido dentro del cerebro). Tratamiento del TSC con angiomiolipoma renal: Afinitor® reduce el tamaño del angiomiolipoma renal relacionado con un trastorno genético llamado complejo de esclerosis tuberosa (TSC). Esto podría disminuir el riesgo de que el o los tumores causen complicaciones hemorrágicas y podría ayudar a la conservación de la función renal. Monitoreo durante el tratamiento con Afinitor®: Durante el tratamiento se le realizarán exámenes de sangre regularmente. Estos estudios controlarán la cantidad de células sanguíneas (glóbulos blancos, glóbulos rojos y plaquetas) en su organismo para evaluar si Afinitor® está produciendo algún efecto no deseado en este tipo de células. Además se realizarán pruebas sanguíneas para evaluar la función de sus riñones (nivel de creatinina), la función hepática (nivel de transaminasas) y los niveles de azúcar en sangre, ya que estos parámetros también pueden estar afectados por Afinitor®. Si usted recibe Afinitor® para el tratamiento de astrocitoma subpendimario de células gigantes asociado a esclerosis tuberosa, son necesarios exámenes de sangre regulares para medir cuando Afinitor® hay en su sangre ya que esto ayudará a su médico a decidir cuanto Afinitor® usted necesita tomar. Ante cualquier duda acerca de Afinitor®, o de la razón por la cual se le indicó esta medicación, consulte a su médico. 2. ANTES DE TOMAR AFINITOR®. Siga cuidadosamente todas las instrucciones del médico. Pueden ser diferentes de la información general contenida en este prospecto. Tratamiento de cáncer de mama avanzado con receptores hormonales positivos, tumores neuroendocrinos avanzados o cáncer de riñón avanzado: Afinitor® le será recetado únicamente por un médico con experiencia en el tratamiento del cáncer. Tratamiento del TSC con angiomiolipoma renal: Afinitor® será recetado únicamente para usted por un médico con experiencia en el tratamiento de pacientes con TSC. Tratamiento de pacientes con astrocitoma subpendimario de células gigantes asociado a esclerosis tuberosa: Afinitor® le será recetado únicamente por un médico con experiencia en el tratamiento de pacientes con astrocitoma subpendimario de células gigantes asociado a esclerosis tuberosa, requiere tener acceso acceso a análisis de sangre para permitir medir cuanto Afinitor® hay en su sangre. No tome Afinitor®: si es alérgico (hipersensible) a everolimus, a sustancias relacionadas con everolimus como sirolimus (rapamicina) o temsirolimus, o a cualquiera de los demás componentes de Afinitor®. Si usted se encuentra en cualquiera de estas situaciones, dígaselo al médico y no tome Afinitor®. Si cree que puede ser alérgico, consulte a su médico. Tenga especial cuidado con Afinitor®: Si algo de lo siguiente aplica a usted informe a su médico antes de tomar Afinitor®: si tiene problemas en el hígado o si ha tenido alguna enfermedad que puede haber afectado su hígado. En este caso, su médico puede prescribirle una dosis diferente de Afinitor®. Si tiene diabetes (alto nivel de azúcar en la sangre). si tiene alguna infección. Puede ser necesario realizar un tratamiento para controlar la infección antes de iniciar la terapia con Afinitor®. Si previamente tuvo Hepatitis B, dado que puede reactivarse durante el tratamiento con Afinitor® (ver Posibles efectos adversos). Si está tomando otros medicamentos (ver Uso de otros medicamentos). Si está embarazada, si sospecha que puede estar embarazada, o si existe alguna posibilidad de haber quedado embarazada durante el tratamiento con Afinitor® (ver Embarazo). Si está amamantando (ver Lactancia). Qué debe saber durante el tratamiento con Afinitor®: Problemas pulmonares o respiratorios: Los pacientes pueden experimentar problemas pulmonares o respiratorios, como neumonitis, embolia pulmonar o síndrome de dificultad respiratoria aguda. Informe inmediatamente a su médico si experimenta síntomas pulmonares/respiratorios nuevos o que se agravan, como tos, dolor del pecho o falta de aire, ya que los problemas respiratorios o pulmonares graves podrían tener consecuencias mortales. Su médico podría tener que reducirle la dosis de Afinitor® o interrumpir de modo temporal o definitivo su tratamiento con Afinitor® y agregar otro medicamento para ayudarle a sobrellevar este efecto secundario. Infección: Afinitor® puede hacer más probable que contraiga una infección (como neumonía, infección de las vías urinarias, infección micótica o infección viral, incluida la reactivación de la hepatitis B). Algunas infecciones podrían ser graves y tener consecuencias potencialmente mortales. Informe de inmediato a su médico si experimenta síntomas de infección. Ulceras bucales: Los pacientes podrían experimentar úlceras y lesiones bucales. Informe a su médico si experimenta dolor o malestar en la boca, o si tiene úlceras abiertas en la boca. Es posible que el médico tenga que reducirle la dosis de Afinitor® o interrumpir de modo temporal o definitivo su tratamiento con Afinitor®. Podría necesitar tratamiento con un enjuague o gel bucal. Algunos enjuagues y geles bucales pueden agravar las úlceras, así que no pruebe ningún producto sin consultar primero a su médico. Reacciones alérgicas: Si durante el tratamiento con Afinitor® desarrolla síntomas como hinchazón de las vías aéreas o de la lengua y/o dificultad para respirar, estos pueden ser signos de una reacción alérgica seria. En ese caso contactar a su médico inmediatamente. Desórdenes hepáticos: se ha observado falla hepática en algunos pacientes recibiendo Afinitor®. Su doctor monitoreará las funciones hepáticas durante su tratamiento con Afinitor®. Vacunación: Si necesita recibir alguna vacuna durante el tratamiento con Afinitor®, debe consultar a su médico antes de la administración de la misma. Uso de otros medicamentos: Afinitor® puede afectar la forma como actúan otros medicamentos. Puede ser necesario modificar la dosis de Afinitor®. Informe a su médico o farmacéutico si está utilizando, o ha utilizado recientemente otros medicamentos, incluso los adquiridos sin receta. Esto incluye en particular: Algunos medicamentos utilizados para el tratamiento de infecciones. Esto incluye medicamentos para el tratamiento de infecciones por hongos (antifúngicos como el ketoconazol, itraconazol y fluconazol), o medicamentos para el tratamiento de infecciones bacterianas (antibióticos como claritromicina, telitromicina o eritromicina). Algunos medicamentos utilizados para el tratamiento de la tuberculosis como la rifampicina o rifabutina. La hierba de San Juan -un producto a base de hierbas usado para el tratamiento de la depresión y otras condiciones (también conocido como Hypercum Perforatum). Algunos corticosteroides como la dexametasona, prednisona y prednisolona. Medicamentos para controlar las convulsiones o epilepsia (antiepilépticos como la fenitoína, carbamazepina, fenobarbital). Algunos medicamentos usados para el tratamiento del SIDA (VIH) como el ritonavir, amprenavir, fosamprenavir, efavirenz, nevirapina. Algunos medicamentos usados para el tratamiento de condiciones cardíacas o de hipertensión arterial (bloqueantes de los canales de calcio como el verapamilo y el diltiazem). Ciclosporina, un medicamento utilizad para controlar el rechazo de órgano en pacientes transplantados. Aprepitant, un medicamento utilizado para la prevención de náuseas y vómitos. Midazolam, un medicamento utilizado para tratar convulsiones severas, o utilizado como sedante antes o durante la cirugía o un procedimiento médico. Deberá evitarse el uso de estos medicamentos durante el tratamiento con Afinitor®. Si está tomando alguno de ellos, su médico puede prescribirle un medicamento diferente para impedir la aparición de efectos colaterales adicionales causados por la combinación de estos medicamentos con Afinitor®. Para pacientes con astrocitoma subpendimario de células gigantes asociado a esclerosis tuberosa los cuales están tomando medicamentos anticonvulsivantes, un cambio en la dosis de esta medicación puede requerir un cambio en la dosis de Afinitor®. Mientras esté recibiendo el tratamiento con Afinitor® no debe iniciar una terapia con un nuevo medicamento sin consultar previamente con el médico que le indicó Afinitor®. Esto incluye medicamentos de venta libre y derivados de hierbas o medicinas alternativas. Toma de Afinitor® con los alimentos y bebidas: Debe tomar Afinitor® a la misma hora cada día, siempre consistentemente con las comidas o bien consistentemente siempre en ayuno. No coma pomelo ni tome jugo de pomelo, tamarindo o naranja de sevilla mientras esté tomando Afinitor® ya que puede aumentar la cantidad de Afinitor® en la sangre, posiblemente a un nivel perjudicial. Personas de edad avanzada (de 65 años o mayores): Si usted tiene 65 o más años de edad, puede recibir Afinitor® en la misma dosis que la población adulta más joven. Niños y adolescentes: Para las siguientes indicaciones Afinitor® no está indicado en niños u adolescentes: Tratamiento de cáncer de mama avanzado con receptores hormonales positivos, tumores neuroendocrinos o de cáncer de riñón avanzado. En cambio para el tratamiento de pacientes con astrocitoma subpendimario de células gigantes asociado a esclerosis tuberosa Afinitor® puede ser utilizado en niños a partir de 3 años de edad y adolescentes lo cuales tienen una función hepática normal. Tratamiento del TSC con angiomiolipoma renal: Afinitor® no debe ser usado en niños o adolescentes con TSC que tienen angiomiolipoma renal en ausencia de SEGA. Embarazo y lactancia: Consulte a su médico antes de iniciar el tratamiento con cualquier medicamento. Afinitor® puede ser prejudicial para el feto y para el bebé durante la lactancia. No se recomienda el tratamiento con Afinitor® durante el embarazo. Informe a su médico si está embarazada o piensa que puede estarlo. Su médico evaluará con usted el riesgo potencial de recibir Afinitor® durante el embarazo. No debe amamantar durante el tratamiento con Afinitor®. Informe a su médico si está amamantando. Mujeres en edad de procrear: Las mujeres que pueden quedar embarazadas deben utilizar un método anticonceptivo altamente efectivo durante el tratamiento con Afinitor® (como comprimidos o preservativos) y continuarlo por 8 semanas después de suspender el tratamiento. Si, a pesar de estas medidas, cree que puede estar embarazada, consulte con su médico antes de tomar más Afinitor®. Fertilidad: Afinitor® puede comprometer la fertilidad del hombre. Se ha observado ausencia de períodos menstruales (amenorrea) en algunas mujeres tratadas con Afinitor®. 3. COMO TOMAR AFINITOR®: Siga exactamente las instrucciones de administración de Afinitor® indicadas por su médico. No tome dosis mayores de la que su medico le indicó. Cantidad de Afinitor® que debe tomar: Su médico le dirá exactamente cuántos comprimidos de Afinitor® tomará. No cambie la dosis sin hablar primero con el médico. Si Afinitor® le causa ciertos efectos secundarios (p. ej., problemas pulmonares o de respiración, úlceras bucales), es posible que el médico tenga que reducirle la dosis de Afinitor®; o incluso interrumpir temporal o definitivamente su tratamiento con Afinitor®. No deje de tomar Afinitor® a menos que su médico se lo indique. Tratamiento de cáncer de mama avanzado con receptores hormonales positivos, tumores neuroendocrinos avanzados o de cáncer de riñón avanzado: La dosis usual de 10 mg, una vez al día. Su médico podría recomendarle una dosis más alta o más baja con base en sus necesidades de tratamiento personales, p. ej., si tiene problemas hepáticos o si toma ciertos medicamentos adicionales. Tratamiento del TSC con angiomiolipoma renal: La dosis usual es de 10 mg, tomada una vez al día. Su médico podría recomendarle una dosis más alta o más baja con base en sus necesidades de tratamiento personales, p. ej., si tiene problemas hepáticos o si toma ciertos medicamentos adicionales. Tratamiento de pacientes con astrocitoma subpendimario de células gigantes asociado a esclerosis tuberosa: Su médico determinará la dosis de Afinitor® dependiendo de su tamaño corporal, el estado de su hígadoy que medicinas este tomando. Exámenes de sangre son necesarios durante el tratamiento con Afinitor® para medir la cantidad de Afinitor® en sangre y encontrar la mejor dosis diaria para usted. Siga cuidadosamente las instrucciones de su médico acerca de cuánto Afinitor® tomar. Afinitor® no debe utilizarse en niños y adolescentes (menos de 18 años de edad) con problemas hepáticos. Cuándo tomar Afinitor®: Tome Afinitor® una vez al día, a la misma hora. Es importante recibir Afinitor® a la misma hora todos los días para que se mantenga constante la cantidad del medicamento en sangre. Cómo debe tomar Afinitor®: Los comprimidos de Afinitor® deben ser ingeridos por vía oral. Usted debe tomar Afinitor® todos los días en el mismo horario, siempre con comida o siempre sin comida. Trague el(los) comprimido(s) enteros con un vaso con agua. Los comprimidos no deben masticarse ni triturarse. Si usted no puede ingerir los comprimidos, puede verterlos en un vaso de agua: Coloque el o los comprimidos en un vaso con agua (conteniendo aproximadamente 30 mL). Mezclar suavemente el contenido hasta que los comprimidos se disuelvan y tomar inmediatamente. Enjuague el vaso con la misma cantidad de agua (aproximadamente 30 mL) y tome el contenido entero para asegurar la toma completa de la dosis de Afinitor®. Duración del tratamiento con Afinitor®: Continúe el tratamiento con Afinitor® por el tiempo que le indique su médico. Si toma más Afinitor® del que debiera: Si ha tomado más Afinitor®de lo que corresponde o si alguien tomó sus comprimidos de forma accidental, consulte a su médico o acuda al hospital inmediatamente. Presente el envase y este prospecto para que el médico conozca qué es lo que ha tomado. Puede ser necesario tratamiento urgente. Si olvidó tomar Afinitor®: Si olvidó tomar Afinitor®, puede recibir esa dosis hasta 6 horas después del horario habitual. Si recuerda haber olvidado la dosis diaria después de 6 horas del horario en que normalmente toma Afinitor®, no tome la dosis por ese día. Al día siguiente, tome el comprimido en el horario habitual. No tome una dosis doble para compensar las dosis olvidadas. 4. POSIBLES EFECTOS ADVERSOS: Al igual que todos los medicamentos, Afinitor® puede producir efectos adversos, aunque no todas las personas los sufran. Afinitor® puede afectar los resultados de algunos análisis de sangre. Algunos efectos adversos pueden ser serios: Se ha informado de reacciones alérgicas (reacciones de hipersensibilidad, que incluyen la hinchazón debajo de la piel y/o de las vías respiratorias o la lengua). Las reacciones alérgicas graves deben recibir atención médica inmediata. Informe de inmediato a su médico o enfermera si experimenta síntomas que indican una reacción alérgica, como sibilancias, dificultad para respirar o hinchazón de la cara, cuello y garganta, erupciones de la piel y urticaria o fiebre después de tomar Afinitor®. Problemas de pulmón o respiratorios (por ej. Neumonitis, embolismo pulmonar o síndrome de stress respiratorio (deterioro de la función respiratoria)): dígale inmediatamente a su médico si presenta aparición o empeoramiento de síntomas respiratorios o pulmonares tales como tos, dolor de pecho o dificultad respiratoria dado que pueden tener consecuencias fatales. Su médico puede requerir cambiar la dosis indicada de Afinitor® o agregarle otra medicación para controlar los efectos adversos. Infecciones: Afinitor® puede predisponerlo a contraer infecciones (como neumonía o una infección urinaria). Dígale a su medico inmediatamente si tiene fiebre o escalofríos o cualquier otro signo o síntoma de infección. Puede necesitar tratamiento urgente. Desórdenes hepáticos: se ha observado falla hepática en algunos pacientes que recibían Afinitor®. Su doctor monitoreará la función hepática durante el tratamiento con Afinitor®. Reactivación de la Hepatitis B: se ha observado reactivación de la Hepatitis B en algunos pacientes recibiendo Afinitor®. Informe a su médico si usted experimenta síntomas de hepatitis B durante el tratamiento con Afinitor®: el primer síntoma puede ser no específico, incluyendo fiebre, picazón, dolor e inflamación de articulaciones. Otros síntomas pueden incluir fatiga, pérdida del apetito, náuseas, ictericia (piel amarilla) y dolor en la parte superior del abdomen. Materia fecal pálida u orina oscura también pueden ser síntomas de hepatitis. Embolismo Pulmonar: se observó embolismo pulmonar en pacientes recibiendo Afinitor®. El embolismo pulmonar es una condición que ocurre cuando uno o más arterias en sus pulmones se tapan. Los síntomas pueden ser repentinos, falta de aire, dolor de pecho o tos con sangre. Informe a su médico inmediatamente si usted experimenta alguno de estos síntomas durante el tratamiento con Afinitor®. Tratamiento de cáncer de mama avanzado con receptores hormonales positivos, de tumores neuroendocrinos avanzados y cáncer de riñón avanzado: Algunos efectos adversos son muy frecuentes: Estos efectos secundarios pueden afectar a más de 1 de cada 10 pacientes. problemas de pulmón y respiración (neumonitis). Infecciones. Ulceras en la boca: Afinitor® puede producir úlceras bucales y ampollas. Informe a su médico si presenta dolor o molestias en la boca o presenta ampollas en la boca, puesto que puede necesitar tratamiento con un enjuague o gel. Algunos enjuagues y geles pueden empeorar las úlceras, por lo que deberá comprobar primero con su médico antes de utilizar alguno de ellos. Hinchazón de los brazos, manos, pies, tobillos u otras partes del cuerpo (signos de edema). Sensación de mareo (náuseas), vómitos, diarrea. Pérdida de apetito. Debilidad o cansancio. Erupción. Piel seca. Alteración del sentido del gusto. Picazón. Sangrado de la nariz (epistaxis). Cefalea (dolor de cabeza). Tos. Disnea (falta de aire). Inflamación de las mucosas. Fiebre. Pérdida de peso. Desórdenes en la uñas. Si cualquiera de estos efectos lo afecta severamente, dígaselo a su médico. Algunos efectos adversos son frecuentes: Estos efectos secundarios pueden afectar entre 1 y 10 de cada 100 pacientes. Erupción y dolor en las palmas de la mano o plantas del pie (síndrome de mano-pie). Sequedad de boca. Dolor abdominal. Dificultad para tragar (disfagia). Problemas para dormir (insomnio). Presión arterial alta (hipertensión). Dolor en el pecho. Ardor en el estómago o indigestión (dispepsia). Falla hepática. Orinar más frecuentemente de lo habitual (poliuria). Deshidratación. Emporamiento de la diabetes. Tos con flema con sangre (hemoptisis). Altos niveles de azúcar en sangre. Embolismo pulmonar: (una condición que ocurre cuando una o más arterias en sus pulmones están bloqueadas. Los síntomas pueden ser repentina falta de aire, dolor de pecho o tos con sangre). - Acné. - Enrojecimiento de la piel (eritema). Dolor de las articulaciones. Proteínas en la orina. Sangrado (hemorragia), por ejemplo en la pared del intestino. Si cualquiera de estos efectos lo afecta severamente, dígaselo a su médico. Algunos efectos adversos son poco frecuentes: Estos efectos secundarios pueden afectar entre 1 y 10 de cada 1000 pacientes. Tipo de anemia llamada aplasia pura de células rojas. Aparición de diabetes mellitus. Pérdida del sentido del gusto. Síntomas de insuficiencia cardíaca como respiración jadeante, dificultad para respirar al estar acostado, hinchazón de los pies o las piernas. - falta de aire o respiración acelerada (síndrome de estrés respiratorio agudo). Problemas de cicatrización de las heridas. - Bloqueo u obstrucción de los vasos sanguíneos (venas) en las piernas (trombosis venosa profunda). Los síntomas pueden ser hinchazón y/o dolor en las piernas, usualmente su pantorrilla (enrojecimiento o piel caliente en el área afectada). Se ha observado ausencia de períodos menstruales (amenorrea) en algunas mujeres tratadas con Afinitor®. Si considera que alguno de los efectos adversos que sufre es grave, informe a su médico. Tratamiento del TSC con angiomiolipoma renal: Algunos efectos secundarios son muy frecuentes. Estos efectos secundarios podrían afectar a más de 1 de cada 10 pacientes. Nivel bajo de glóbulos rojos (anemia). Nivel bajo de glóbulos blancos (leucopenia). Nivel alto de colesterol en la sangre (hipercolesterolemia). Ulceras bucales: Afinitor® puede causar úlceras e inflamación bucales. Informe a su médico si experimenta dolor o malestar en la boca, o si tiene úlceras abiertas en la boca. Podría necesitar tratamiento con un enjuague o gel bucal. Algunos enjuagues y geles bucales pueden agravar las úlceras, así que no pruebe ningún producto sin consultar primero a su médico. Malestar estomacal, como sentir ganas de vomitar (náuseas). Acné. Sensación de cansancio. Nivel alto en la sangre de una enzima denominada lactato deshidrogenasa sanguínea, la cual proporciona información acerca de la salud de ciertos órganos. Si alguno de estos efectos secundarios lo afecta gravemente, informe a su médico. Algunos efectos secundarios son frecuentes. Estos efectos secundarios podrían ocurrir entre 1 y 10 de cada 100 pacientes. Infección de las vías urinarias. Inflamación de los senos y las fosas nasales (sinusitis). Los síntomas podrían incluir cefalea, sensación de presión en el área de los ojos, la nariz o las mejillas. Infección de las vías respiratorias superiores. Nivel bajo de plaquetas (trombocitopenia). Reacciones alérgicas (hipersensibilidad). Nivel bajo de fosfatos en la sangre (hipofosfatemia). Nivel alto de lípidos en la sangre (hiperlipidemia). Disminución del apetito. Nivel bajo de hierro (deficiencia de hierro). Cefalea. Alteraciones del gusto (disgeusia). Pérdida del gusto (ageusia). Tos. Hemorragia nasal (epistaxis). Problemas pulmonares o respiratorios. Diarrea. Vómitos. Dolor abdominal. Cantidad excesiva de gases en los intestinos (flatulencia). Un trastorno inflamatorio de la piel, caracterizado por enrojecimiento, comezón y quistes llenos de líquido que supuren, que pueden presentar escamas, costras o endurecimiento (dermatitis acneiforme). Sequedad de la piel. Protuberancias pequeñas y sólidas de color rosa o rojo en la piel (pápula). Insuficiencia renal. Trastornos menstruales como ausencia de períodos (amenorrea), períodos irregulares o atrasados, períodos con sangrado abundante (menorragia) o sangrado vaginal. Si alguno de estos efectos secundarios lo afecta gravemente, informe a su médico. Tratamiento del TSC con SEGA: Algunos efectos secundarios son muy frecuentes. Estos efectos secundarios podrían afectar a más de 1 de cada 10 pacientes. Infecciones como inflamación de los senos y las fosas nasales (sinusitis), infección del oído medio o externo, infección gástrica, dolor de garganta y secreción nasal, infecciones de la piel, tiña (una infección micótica de la piel), infecciones de los folículos pilosos, infección de las vías urinarias, conjuntivitis, infección de las vías respiratorias altas, neumonía. Niveles bajos de glóbulos blancos (un tipo de células de la sangre que combaten las infecciones; su médico los evaluará periódicamente). Nivel alto de colesterol en la sangre. Niveles altos de grasas en la sangre (triglicéridos altos). Tos. Ulceras bucales: Afinitor® puede causar úlceras e inflamación bucales. Informe a su médico si experimenta dolor o malestar en la boca, o si tiene úlceras abiertas en la boca. Podría necesitar tratamiento con un enjuague o gel bucal. Algunos enjuagues y geles bucales pueden hacer que empeoren las úlceras, de modo que no pruebe ningún producto sin consultar primero a su médico. Diarrea. Una erupción de la piel parecida al acné. Acné. Fiebre. Si alguno de estos lo afecta gravemente, informe a su médico. Algunos efectos secundarios son frecuentes: Estos efectos secundarios podrían ocurrir entre 1 y 10 de cada 100 pacientes: Absceso en alguna extremidad. Bronquitis viral. Bajos niveles de eritrocitos (anemia). Agresividad. Incapacidad para dormir. Agitación. Ataques (convulsiones). Sangrados nasales. Inflamación de la garganta. Trastorno pulmonar o respiratorio (como la falta de aire, tos seca o dificultad para respirar). Inflamación del recubrimiento interno del estómago (gastritis). Vómitos. Erupción cutánea. Proteínas en la orina. Trastornos menstruales como ausencia de períodos (amenorrea) o períodos irregulares. Sensación de cansancio. Irritabilidad. Dificultad para caminar (trastornos de la marcha). Disminución de los niveles de anticuerpos en la sangre (pregunte a su médico). Aumento de las lipoproteínas de baja densidad en la sangre. Si alguno de estos efectos secundarios lo afecta gravemente, informe a su médico. Si nota cualquier otro efecto secundario no mencionado en este folleto, informe a su médico o farmacéutico. Si cualquiera de estos efectos lo afecta severamente, dígaselo a su médico. Si usted nota cualquier otro efecto adverso no mencionado en este prospecto, por favor infórmelo a su médico.
Presentación.
Envases conteniendo 28, 30, 56, 60, 90 y 120 comprimidos.
Revisión.
27/03/2012.