Actemra® s.c.
ROCHE
Tocilizumab.
Agente inmunosupresor, inhibidor de interleucina.
Composición.
Cada jeringa prellenada de 1 ml contiene 162 mg en 0,9 ml (180 mg/ml) de tocilizumab (anticuerpo monoclonal IgG1 recombinante humanizado antirreceptor de interleucina-6 (IL-6) humana, producido en células de ovario de hámster chino mediante tecnología de ADN recombinante). Cada jeringa prellenada contiene 162 mg/0,9 ml de tocilizumab en un excipiente compuesto por: polisorbato 80: 0,18 mg, L-arginina 0,132 mg, clorhidrato de L-arginina 18,8 mg, L-metionina 4,03 mg, L-histidina 1,40 mg, clorhidrato de L-histidina monohidratado 1,90 mg y agua para inyectables c.s.
Farmacología.
Código ATC: L04AC07. Grupo farmacoterapéutico: Agentes inmunosupresores, inhibidores de interleucina. Propiedades farmacodinámicas: Mecanismo de acción: Tocilizumab se une específicamente a los receptores de IL-6 tanto solubles como unidos a membranas (IL-6Rs e IL-6Rm). Se ha demostrado que tocilizumab inhibe la señalización mediada por IL-6Rs e IL-6Rm. La IL-6 es una citocina proinflamatoria pleiotrópica producida por diversos tipos celulares, incluidas células T y B, los monocitos y los fibroblastos. La IL-6 participa en diferentes procesos fisiológicos como la activación de los linfocitos T, la inducción de secreción de inmunoglobulina, la inducción de síntesis hepática de proteínas de la fase aguda y la estimulación de la hemopoyesis. Se ha implicado a la IL-6 en la patogenia de enfermedades, tales como afecciones inflamatorias, osteoporosis y neoplasias. Efectos farmacodinámicos: En ensayos clínicos con tocilizumab, se observaron reducciones rápidas de la proteína C reactiva (PCR), la velocidad de sedimentación globular (VSG) y el amiloide A sérico (AAS). En forma coherente con el efecto sobre los reactantes de la fase aguda, el tratamiento con tocilizumab se asoció con reducción del recuento de plaquetas dentro del rango normal. Se observaron aumentos de los niveles de hemoglobina, debidos a la reducción por tocilizumab de los efectos impulsados por la IL-6 sobre la producción de hepcidina para incrementar la disponibilidad del hierro. En pacientes tratados con tocilizumab, se registraron disminuciones de los niveles de PCR dentro de los valores normales ya a la segunda semana y las reducciones se mantuvieron mientras duraba el tratamiento. En voluntarios sanos a los que se administró tocilizumab en dosis de 2 a 28 mg/kg por vía intravenosa y 81 a 162 mg por vía subcutánea, el recuento absoluto de neutrófilos descendió a su nivel más bajo transcurridos de 2 a 5 días después de la administración. Posteriormente los niveles de neutrófilos se recuperaron hasta el valor basal de una manera dosis-dependiente. Los pacientes con artritis reumatoidea demostraron un descenso comparable a los voluntarios sanos, en el recuento absoluto de neutrófilos después de la administración de tocilizumab (véase Reacciones adversas). Uso intravenoso: Eficacia clínica y seguridad: Se ha evaluado la eficacia de tocilizumab para aliviar los signos y síntomas de artritis reumatoidea en cinco ensayos aleatorizados, doble-ciego, multicéntricos. Los ensayos I - V incluyeron a pacientes ≥ 18 años de edad con artritis reumatoidea activa diagnosticada según los criterios del American College of Rheumatology (ACR) y que tenían por lo menos ocho articulaciones doloridas y seis inflamadas al inicio. En el ensayo I, tocilizumab se administró por vía intravenosa cada cuatro semanas como monoterapia y en los estudios II, III y V, por la misma vía cada cuatro semanas en combinación con MTX frente a placebo y MTX. En el estudio IV, tocilizumab se administró por vía intravenosa cada 4 semanas en combinación con otros FAMEs frente a placebo y otros FAMEs. El objetivo primario de los cinco estudios fue la proporción de pacientes que alcanzó respuesta ACR20 a la semana 24. En el ensayo I se evaluaron 673 pacientes que no habían sido tratados con MTX en los seis meses previos a la aleatorización y que no habían suspendido el tratamiento previo con MTX como consecuencia de efectos tóxicos clínicamente importantes o falta de respuesta. La mayoría (67%) no había recibido nunca MTX. Se administraron dosis de 8 mg/kg de tocilizumab cada cuatro semanas como monoterapia. El grupo de comparación recibió MTX semanal (dosis ajustada desde 7,5 mg a un máximo de 20 mg por semana durante un período de ocho semanas). En el ensayo II, un estudio de dos años con análisis planificados en las semanas 24, 52 y 104, se evaluaron 1.196 pacientes con una respuesta clínica inadecuada a MTX. Se administraron dosis de 4 u 8 mg/kg de tocilizumab o placebo cada cuatro semanas como terapia ciega durante 52 semanas en combinación con MTX estable (de 10 mg a 25 mg semanales). Después de 52 semanas todos los pacientes pudieron recibir tratamiento abierto con tocilizumab 8 mg/kg. De los pacientes que completaron el estudio, el 86% de los que inicialmente fueron asignados al grupo placebo + MTX recibió tratamiento abierto con tocilizumab 8 mg/kg en el segundo año. El objetivo primario en la semana 24 fue la proporción de pacientes que alcanzaron respuesta ACR20. Entre las semanas 52 y 104, los objetivos co-primarios fueron la prevención del daño articular y la mejora de la función física. En el ensayo III se evaluaron 623 pacientes con una respuesta clínica inadecuada a MTX. Se administraron dosis de 4 u 8 mg/kg de tocilizumab o placebo cada cuatro semanas, en combinación con MTX estable (de 10 mg a 25 mg semanales). En el ensayo IV se estudiaron 1.220 pacientes con una respuesta inadecuada a un tratamiento reumatológico instaurado, con uno o más FAMEs. Se administraron dosis de 8 mg/kg de tocilizumab o placebo cada cuatro semanas, en combinación con FAMEs estables. En el ensayo V se evaluaron 499 pacientes con una respuesta clínica inadecuada o intolerancia a uno o más tratamientos antagonistas del TNF. Esta terapia se suspendió antes de la aleatorización. Se administraron dosis de 4 u 8 mg/kg de tocilizumab o placebo cada cuatro semanas, en combinación con MTX estable (de 10 mg a 25 mg semanales). El criterio de valoración primario para los estudios III-V fue la proporción de pacientes que alcanzaron una respuesta ACR20 a la semana 24. El porcentaje de pacientes que logró respuestas ACR20, 50 y 70 en los estudios I a V se muestra en la Tabla 1. Respuesta clínica: En todos los ensayos, los pacientes tratados con tocilizumab 8 mg/kg tuvieron tasas de respuesta en ACR20, 50 y 70 significativamente mayores a los 6 meses comparados con control (Tabla 1). En el estudio I, se demostró la superioridad de tocilizumab 8 mg/kg frente al comparador activo MTX. El efecto del tratamiento fue similar en los pacientes independientemente del factor reumatoide, edad, sexo, raza, número de tratamientos previos o estado de la enfermedad. El tiempo hasta al inicio de la acción fue rápido (ya a la semana 2) y la magnitud de la respuesta siguió mejorando con la duración del tratamiento. En los ensayos de extensión abiertos que están en marcha, I-V, se han registrado respuestas duraderas continuadas durante más de 3 años. En pacientes tratados con tocilizumab 8 mg/kg, se observaron mejorías significativas en todos los componentes individuales de la respuesta ACR, incluidos: recuentos de articulaciones doloridas e inflamadas; evaluación global por los pacientes y los médicos; puntuaciones del índice de discapacidad; evaluación del dolor y PCR en comparación con los pacientes que recibieron placebo más MTX / u otros FAMEs en todos los estudios. Los pacientes de los estudios I-V tenían un nivel medio de actividad de la enfermedad DAS28 de 6,5-6,8 al inicio. Se observó una reducción significativa (mejoría media), del nivel inicial DAS28 de 3,1-3,4 en los tratados con tocilizumab frente a los controles (1,3-2,1). La proporción de pacientes que alcanzó una remisión clínica DAS28 (DAS28 < 2,6), a las 24 semanas, fue significativamente mayor en los que recibieron tocilizumab (28-34%), comparado con los pacientes control (1-12%). En el ensayo II, un 65% logró un DAS28 < de 2,6 a la semana 104, comparado con un 48% que lo alcanzó en la semana 52 y un 33% en la semana 24. En un análisis conjunto de los estudios II, III y IV, la proporción de pacientes que alcanzó una respuesta ACR20, 50 y 70 fue significativamente mayor (59% frente al 50%, 37% frente a 27%, 18% frente a 11%, respectivamente) en el grupo de tocilizumab 8 mg/kg más FAMEs frente al grupo tratado con tocilizumab 4 mg/kg más FAMEs (p < 0,03). En forma similar, la proporción de pacientes que alcanzó remisión en el DAS28 (DAS28 < 2,6) fue significativamente mayor (31% frente al 16%, respectivamente) en aquéllos que recibieron tocilizumab 8 mg/kg más FAMEs que en los tratados con tocilizumab 4 mg/kg más FAMEs (p < 0,0001).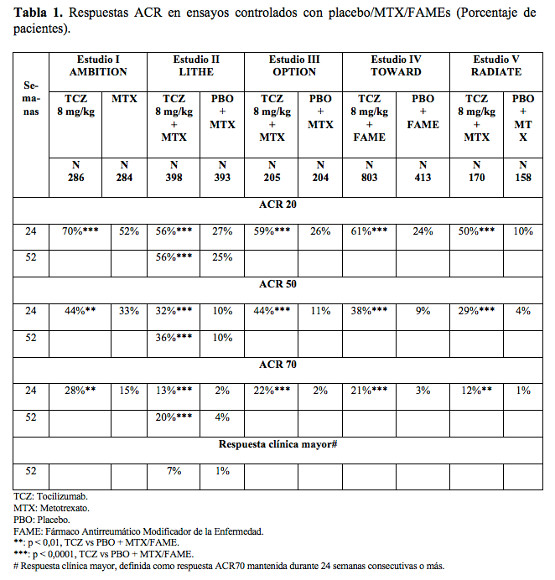
Respuesta clínica mayor: Después de dos años de tratamiento con tocilizumab y metotrexato, el 14% de los pacientes alcanzó una respuesta clínica mayor (mantenimiento de la respuesta ACR70 durante 24 semanas o más). Respuesta radiográfica: En el ensayo II, en pacientes con una respuesta inadecuada a MTX, se evaluó radiográficamente la inhibición del daño articular estructural y se expresó como un cambio en la escala de Sharp modificada y sus componentes, la puntuación de erosión y la del estrechamiento del espacio articular. Se demostró inhibición del daño estructural articular con una progresión radiográfica significativamente menor en los pacientes que recibieron tocilizumab en comparación con el control (Tabla 2). En la extensión abierta del estudio II la inhibición de la progresión del daño estructural en pacientes tratados con tocilizumab más MTX se mantuvo en el segundo año del tratamiento. En la semana 104 el cambio medio desde la basal en el índice total Sharp-Genant fue significativamente menor en los pacientes del grupo tocilizumab 8 mg/kg más MTX (p < 0,0001) que en los del grupo placebo más MTX.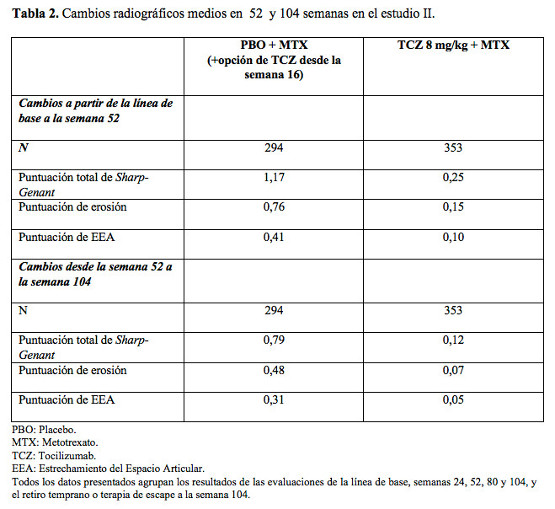
Después de un año de tratamiento con tocilizumab más MTX, el 85% de los pacientes (n = 348) no presentó progresión del daño estructural, definido por un cambio en el índice total de Sharp de cero o menos, comparado con el 67% de aquéllos tratados con placebo más MTX (n = 290) (p ≤0,0001). Estas cifras se mantuvieron coherentes después de dos años de tratamiento (83%; n = 353). El noventa y tres por ciento (93%; n = 271) de los pacientes no manifestó progresión entre las semanas 52 y 104. Resultados relacionados con la salud y la calidad de vida: Los pacientes tratados con tocilizumab comunicaron una mejora en todos los resultados notificados: Cuestionario de evaluación de la salud, Índice de Discapacidad (HAQ-DI), Formulario breve 36 (SF-36) y Evaluación funcional del tratamiento de enfermedades crónicas. Se observaron mejoras estadísticamente significativas en las puntuaciones de HAQ-DI en pacientes tratados con Actemra en comparación con los que recibieron FAMEs. En la semana 24, la proporción de pacientes tratados con 8 mg/kg de tocilizumab que mostraron una mejoría importante en HAQ-DI (definida como una disminución de la puntuación total individual de > 0,25) fue significativamente más elevada que la de los pacientes que recibieron placebo + MTX/FAMES en todos los estudios. Durante el período abierto del estudio II, la mejora de la función física se ha mantenido hasta los 2 años. En el Estudio II, los cambios en PCS, MCS y FACIT-Fatiga a la semana 52 fueron 10,1, 5,4 y 8,4 respectivamente, en el grupo de 8 mg/kg de TCZ + MTX comparado con 5,6, 3,8 y 5,5, en el grupo placebo + MTX, respectivamente. En la semana 52, el cambio medio en HAQ-DI fue de -0,58 en el grupo de tocilizumab 8 mg/kg más MTX comparado con -0,39 del grupo placebo más MTX. El cambio medio en HAQ-DI se mantuvo en la semana 104 en el grupo tocilizumab 8 mg/kg más MTX (-0,61). Niveles de hemoglobina: Se comprobaron mejoras estadísticamente significativas en los niveles de hemoglobina con tocilizumab en comparación con los FAMEs (p < 0,0001) en la semana 24. Los niveles medios de hemoglobina aumentaron en la semana 2 y permanecieron dentro del intervalo normal hasta la semana 24. Un marcado descenso en los niveles promedio de los reactantes de fase aguda PCR, ERS y amiloide sérico A ocurrió rápidamente luego de la administración de tocilizumab. Junto con este efecto sobre los reactantes de fase aguda el tratamiento con tocilizumab se asoció con una reducción en el recuento de plaquetas, pero dentro del rango considerado normal. Tocilizumab versus adalimumab en monoterapia: En el ensayo (WA19924), doble-ciego de 24 semanas, que comparó tocilizumab en monoterapia con adalimumab en monoterapia, se evaluó a 326 pacientes con AR que eran intolerantes a MTX o donde el tratamiento continuado con MTX se consideraba inapropiado (incluyendo respondedores inadecuados a MTX). Los pacientes en el grupo de tocilizumab recibieron una infusión intravenosa (i.v.) de tocilizumab (8 mg/kg) cada 4 semanas y una inyección subcutánea (s.c.) de placebo cada 2 semanas. A los pacientes en el grupo de adalimumab se les administró una inyección subcutánea de adalimumab (40 mg) cada 2 semanas más una infusión intravenosa de placebo cada 4 semanas. Se observó un efecto de tratamiento superior, estadísticamente significativo de tocilizumab sobre adalimumab, en el control de la actividad de la enfermedad, desde el valor basal a la semana 24, para la variable primaria cambio en DAS28 y para todas las variables secundarias (Tabla 3).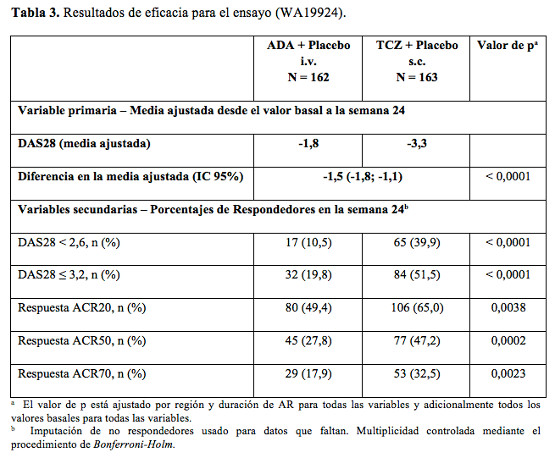
El perfil clínico global de eventos adversos fue similar entre tocilizumab y adalimumab. La proporción de pacientes con reacciones adversas graves fue equilibrada entre los grupos de tratamiento (tocilizumab 11,7% comparado con adalimumab 9,9%). Las reacciones adversas medicamentosas en el grupo de tocilizumab concordaron con el perfil de seguridad conocido de tocilizumab y la frecuencia de las reacciones adversas notificadas fue similar en comparación con la Tabla 5. Se informó en el grupo de tocilizumab una mayor incidencia de infecciones e infestaciones (48% comparado con 42%), sin diferencias en las infecciones graves (3,1%). Ambos tratamientos en estudio indujeron el mismo patrón de cambios en los parámetros de seguridad del laboratorio (disminución en neutrófilos y recuento de plaquetas, aumento en ALT, AST y lípidos); sin embargo, la magnitud de las modificaciones y la frecuencia de fuertes anomalías fueron superiores con tocilizumab en comparación con adalimumab. Cuatro pacientes (2,5%) en el grupo de tocilizumab y 2 (1,2%) en el grupo de adalimumab experimentaron una disminución en el recuento de neutrófilos de Grados 3 o 4 según los Criterios de Toxicidad Común (CTC). Once pacientes (6,8%) en el grupo de tocilizumab y 5 (3,1%) en el de adalimumab tuvieron un incremento de las ALT de Grado 2 o superior según CTC. El aumento medio de LDL desde el valor basal fue 0,64 mmol/litro (25 mg/dl) para pacientes en el grupo de tocilizumab y 0,19 mmol/litro (7 mg/dl) para los del grupo de adalimumab. La seguridad observada en el grupo de tocilizumab concordó con el perfil de seguridad conocido de tocilizumab y no se observaron reacciones adversas al medicamento nuevas o inesperadas (véase Tabla 5). Uso subcutáneo: Eficacia clínica: Se ha evaluado la eficacia de tocilizumab administrado por vía subcutánea para aliviar los signos y síntomas de la artritis reumatoidea y la respuesta radiográfica, en dos ensayos aleatorizados, doble-ciego, controlados, multicéntricos. Para el estudio I (SC-I), los pacientes tenían que ser > 18 años de edad con artritis reumatoidea activa de moderada a grave, diagnosticada según los criterios del American College of Rheumatology (ACR), con al menos 4 articulaciones doloridas y 4 inflamadas al inicio del estudio. Todos los pacientes recibieron anteriormente FAMEs no biológicos. Para el estudio II (SC-II), los pacientes tenían que ser > 18 años de edad con artritis reumatoidea activa de moderada a grave, diagnosticada según los criterios del American College of Rheumatology (ACR), con al menos 8 articulaciones doloridas y 6 inflamadas al comienzo del estudio. El cambio de 8 mg/kg vía intravenosa una vez cada 4 semanas a 162 mg vía subcutánea una vez por semana altera la exposición en el paciente. La medida varía con el peso corporal del paciente (aumentando en aquéllos de bajo peso corporal y disminuyendo en los de alto peso corporal) pero el resultado clínico coincide con el observado en pacientes tratados con tocilizumab por vía intravenosa. Respuesta clínica: En el ensayo SC-I se evaluaron pacientes con artritis reumatoidea activa de moderada a grave, que presentaron respuesta clínica inadecuada a las terapias reumatológicas existentes, incluidos uno o más FAMEs, de los cuales el 20% presentaba antecedentes de respuesta inadecuada a por lo menos un inhibidor del TNF. En este ensayo, 1.262 pacientes fueron aleatorizados en una proporción de 1:1 para recibir 162 mg de tocilizumab por vía subcutánea, 1 vez por semana, u 8 mg/kg de tocilizumab intravenoso, cada cuatro semanas en combinación con FAMEs no biológicos. La variable principal en el estudio fue la diferencia en la proporción de pacientes que alcanzó una respuesta ACR20 en la semana 24. Los resultados del ensayo SC-I se detallan en la Tabla 4.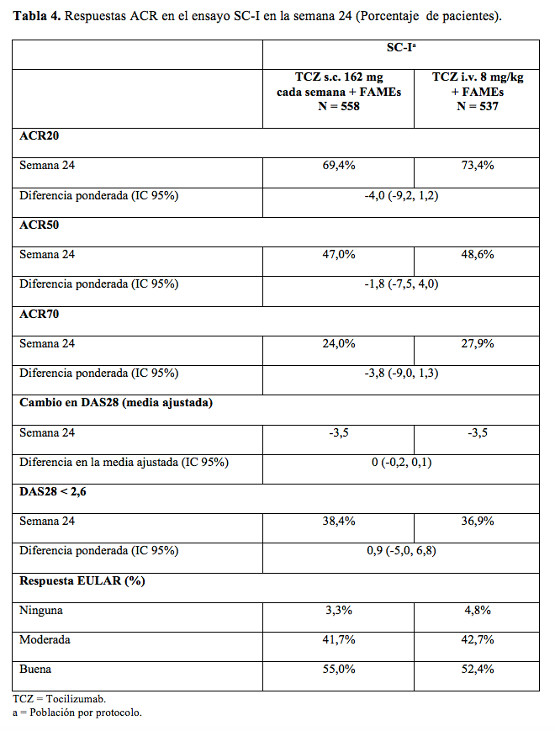
Los pacientes del ensayo SC-I presentaron un nivel medio de actividad de la enfermedad (DAS28) al inicio de 6,6 y 6,7 en los grupos que recibieron el tratamiento por vía subcutánea e intravenosa, respectivamente. En la semana 24, se observó una reducción significativa en DAS28 desde el inicio (mejoría media) de 3,5 en ambos grupos de tratamiento, y una proporción similar de pacientes había alcanzado remisión clínica DAS28 (DAS28 < 2,6) en los grupos tratados por vía subcutánea (38,4%) e intravenosa (36,9%). Respuesta radiográfica: La respuesta radiográfica de tocilizumab por vía subcutánea fue evaluada en un ensayo doble-ciego, controlado, multicéntrico, en pacientes con artritis reumatoidea activa (SC-II). En este ensayo se evaluó a pacientes con artritis reumatoidea activa, de moderada a grave, que manifestaron una respuesta inadecuada a los tratamientos reumatológicos existentes, incluidos uno o más FAMEs, de los cuales aproximadamente el 20% tenía antecedentes de respuesta inadecuada a por lo menos un inhibidor del TNF. Los pacientes debían ser > 18 años, con artritis reumatoidea activa diagnosticada de acuerdo con los criterios ACR y por lo menos 8 articulaciones doloridas y 6 inflamadas al inicio. En el ensayo, 656 pacientes fueron aleatorizados en una proporción de 2:1 para recibir 162 mg de tocilizumab por vía subcutánea, cada dos semanas o placebo, en combinación con FAMEs no biológicos. En el ensayo SC-II, se evaluó radiográficamente la inhibición del daño articular estructural y se expresó como un cambio desde el inicio en la escala total de Sharp promedio modificada por van der Heijde. En la semana 24, se observó inhibición del daño estructural, con una progresión radiográfica significativamente menor en pacientes que recibieron tocilizumab subcutáneo en comparación con placebo (Sharp-van der Heijde promedio de 0,62 comparada con 1,23, p=0,0149 [van Elteren]). Estos resultados están alineados con los observados en pacientes tratados con tocilizumab intravenoso. En el ensayo SC-II en la semana 24 en pacientes tratados con tocilizumab subcutáneo cada dos semanas en comparación con placebo, se obtuvieron resultados de ACR20 60,9%, ACR50 39,8%, ACR70 19,7% en el grupo de tocilizumab y ACR20 31,5%, ACR50 12,3% y ACR70 5,0% en el de placebo. La media del DAS28 al inicio del estudio era de 6,7 en tocilizumab subcutáneo y de 6,6 en el de placebo. En la semana 24 hubo una reducción significativa del DAS28 de 3,1 en tocilizumab subcutáneo y de 1,7 en placebo, se observaron valores de DAS28 menores de 2,6 en el 32% de los pacientes con tocilizumab en el grupo subcutáneo y en el 4% en el de placebo. Resultados relacionados con la salud y la calidad de vida: En el estudio SC-I, la reducción promedio en HAQ-DI desde el inicio hasta la semana 24 fue de 0,6 en ambos grupos subcutáneo e intravenoso. La proporción de pacientes que alcanzó una mejoría clínicamente significativa en el HAQ-DI en la semana 24 (cambio desde el inicio ≥0,3 unidades) también fue similar en el grupo subcutáneo (65,2%) en comparación con el grupo intravenoso (67,4%), con una diferencia ponderada en las proporciones de -2,3% (IC 95% - 8,1, 3,4). Para SF-36, el cambio promedio observado desde el inicio hasta la semana 24 en el índice del componente mental y del físico fue de 6,22 y 9,49 para el grupo subcutáneo y de 6,54 y 9,65 para el grupo intravenoso, respectivamente. En el estudio SC-II, el descenso medio en HAQ-DI desde el inicio hasta la semana 24, fue significativamente mayor en pacientes tratados con tocilizumab subcutáneo cada dos semanas (0,4) frente a placebo (0,3). La proporción de pacientes que consiguieron mejorías en HAQ-DI clínicamente significativas en la semana 24 (cambios desde el inicio ≥3 unidades) fue mayor en tocilizumab subcutáneo cada dos semanas (58%) frente a placebo (46,8%). SF-36 (cambio medio en los valores del componente mental y físico) fue significativamente mayor en el grupo de tocilizumab subcutáneo (6,5 y 5,3) frente a placebo (3,8 y 2,9). La Agencia Europea de Medicamentos ha aplazado la obligación de presentar las conclusiones de los ensayos clínicos realizados con Actemra en uno o más grupos de población pediátrica en el tratamiento de la artritis idiopática crónica (incluyendo artritis reumatoidea, espondilitis anquilosante, artritis psoriásica y artritis idiopática juvenil (véase Posología y formas de administración, para más información sobre uso pediátrico). Propiedades farmacocinéticas: Uso intravenoso: Absorción: Se determinó la farmacocinética de tocilizumab usando un análisis de farmacocinética poblacional en una base de datos compuesta por 3.552 pacientes con artritis reumatoidea tratados con una infusión de una hora de 4 u 8 mg/kg de tocilizumab cada 4 semanas durante 24 semanas o con 162 mg de tocilizumab administrados por vía subcutánea una vez por semana o cada dos semanas durante 24 semanas. Los siguientes parámetros son válidos para una dosis de 8 mg/kg de tocilizumab administrados cada 4 semanas: los valores medios previstos (± DE) en equilibrio fueron de área bajo la curva (ABC) = 38.000 ± 13.000 h•mg/ml, concentración mínima (Cmín) = 15,9 ± 13,1 mg/ml y concentración máxima (Cmáx) = 182 ± 50,4 mg/ml de tocilizumab. Los cocientes de acumulación para ABC y Cmáx fueron pequeños, de 1,32 y 1,09, respectivamente. Este cociente de acumulación fue mayor para la Cmín (2,49), lo que era esperado de acuerdo con la contribución del clearance no lineal en concentraciones menores. Se alcanzó el equilibrio después de la primera administración para la Cmáx y después de 8 y 20 semanas para el ABC y la Cmín, respectivamente. El ABC, la Cmín y la Cmáx de tocilizumab aumentaron con el incremento del peso corporal. Para un peso corporal ≥100 kg, los valores medios previstos (±DE), en equilibrio de ABC, Cmin y Cmáx de tocilizumab fueron de 50.000 ± 16.800 mg•h/ml, 24,4 ± 17,5 mg/ml, y 226 ± 50,3 mg/ml, respectivamente, los cuales son mayores que los valores de exposición media de los pacientes (es decir, cualquier peso corporal) indicados anteriormente. La curva dosis-respuesta para tocilizumab se aplana a mayor exposición, resultando en una menor ganancia de eficacia por cada incremento en la concentración de tocilizumab, de manera que no se demostró mayor eficacia clínicamente significativa en pacientes tratados con > 800 mg de tocilizumab. Por lo tanto, no se recomiendan dosis de tocilizumab superiores a 800 mg en infusión en pacientes que pesen ≥ de 100 kg (véase Posología y formas de administración). Los siguientes parámetros son válidos para una dosis de 4 mg/kg de tocilizumab administrada cada 4 semanas. Los valores medios pronosticados (± DE) en estado de equilibrio de ABC (área bajo la curva), concentración mínima (Cmín) y concentración máxima (Cmáx) de tocilizumab fueron de 13.000 ± 5.800.mg•h/ml, 1,49 ± 2,13 mg/ml y 88,3 ± 41,4 mg/ml, respectivamente. El cociente de acumulación fue mayor para la Cmín (1,96). Se alcanzó el equilibrio después de la primera administración para la Cmáx y para el ABC respectivamente, y después de 16 semanas para la Cmín. Distribución: En pacientes con artritis reumatoidea, el volumen central de distribución fue de 3,72 litros, el volumen periférico de distribución fue de 3,35 litros, lo que da un volumen de distribución en equilibrio de 7,07 litros. Eliminación: Después de la administración intravenosa, tocilizumab presenta una eliminación bifásica de la circulación. El clearance total de tocilizumab fue dependiente de la concentración y es la suma del clearance lineal y el no lineal. El clearance lineal se estimó como parámetro en el análisis de farmacocinética poblacional y fue de 9,5 ml/hora. El clearance no lineal dependiente de la concentración desempeña una función importante en concentraciones bajas de tocilizumab. Una vez saturada la vía de clearance no lineal, en concentraciones mayores de tocilizumab, la eliminación viene determinada fundamentalmente por el clearance lineal. La vida media (t½) de tocilizumab fue dependiente de la concentración. En equilibrio, después de una dosis de 8 mg/kg cada 4 semanas, la t1/2 eficaz se redujo con concentraciones descendentes dentro de un intervalo posológico de 18 días a 6 días. Linealidad: Los parámetros farmacocinéticos de tocilizumab no cambiaron con el tiempo. Se observó un aumento mayor que el proporcional al incremento de la dosis en el área bajo la curva (ABC) y la concentración mínima (Cmín) con las dosis de 4 y 8 mg/kg, cada 4 semanas. La concentración máxima (Cmáx) aumentó en forma proporcional a la dosis. En equilibrio, el ABC y la Cmín previstos fueron 3,2 y 30 veces mayores con 8 mg/kg que con 4 mg/kg, respectivamente. Uso subcutáneo: La farmacocinética de tocilizumab se determinó usando un análisis farmacocinético poblacional de una base de datos compuesta de 3.552 pacientes con artritis reumatoidea tratados con 162 mg por vía subcutánea cada semana, 162 mg por vía subcutánea cada dos semanas, y/o 4 u 8 mg/kg por vía intravenosa cada 4 semanas durante 24 semanas. Los parámetros farmacocinéticos de tocilizumab no se modificaron con el tiempo. Para la dosis de 162 mg administrada por vía subcutánea, cada semana, los valores medios previstos (±DE) en equilibrio, del área bajo la curva (ABC) semana 1, la Cmín y la Cmáx de tocilizumab fueron 7.970 ± 3.432 mg•h/ml, 43,0 ± 19,8 mg/ml, y 49,8 ± 21,0 mg/ml, respectivamente. El cociente de acumulación del ABC, la Cmín y la Cmáx fue de 6,32; 6,30 y 5,27, respectivamente. El estado estacionario se alcanzó después de 12 semanas para el ABC, la Cmín y la Cmáx. Para la dosis de 162 mg administrada por vía subcutánea, cada dos semanas, los valores medios previstos (±DE) en equilibrio, del ABC semana 2, la Cmín y la Cmáx de tocilizumab fueron 3.430 ± 2.660 mg•h/ml, 5,7 ± 6,8 mg/ml, y 13,2 ± 8,8 mg/ml, respectivamente. El cociente de acumulación para el ABC, la Cmín y la Cmáx fue de 2,67; 6,02 y 2,12, respectivamente. El estado estacionario se alcanzó después de 12 semanas para el ABC y la Cmín, y después de 10 semanas para la Cmáx. Absorción: Después de la dosificación subcutánea en pacientes con artritis reumatoidea, el tiempo para alcanzar las concentraciones séricas de tocilizumab tmáx fue de 2,8 días. La biodisponibilidad para la formulación subcutánea fue de 79%. Eliminación: Para la administración subcutánea, la vida media (t1/2) de tocilizumab dependiente de la concentración aparente, es de hasta 12 días para 162 mg cada semana, y de 5 días para 162 mg cada 2 semanas, en pacientes con artritis reumatoidea en estado estacionario. Poblaciones especiales: Uso intravenoso y Uso subcutáneo: Insuficiencia renal: No se ha realizado ningún estudio formal del efecto de la insuficiencia renal sobre la farmacocinética de tocilizumab. La mayoría de los pacientes del análisis de farmacocinética poblacional tenía una función renal normal o insuficiencia renal leve. Esta (clearance de creatinina basado en Cockcroft-Gault < 80 ml/min y ≥50 ml/min) no tuvo impacto sobre la farmacocinética de tocilizumab. No se requiere ajuste de la dosis en pacientes con insuficiencia renal leve. Insuficiencia hepática: No se ha efectuado ningún estudio formal del efecto de la insuficiencia hepática sobre la farmacocinética de tocilizumab. Edad, sexo y etnia: Los análisis de farmacocinética poblacional en pacientes con artritis reumatoidea demostraron que la edad, el sexo y la raza no afectaron la farmacocinética de tocilizumab. No se requiere ajuste de dosis para estos factores demográficos. Datos preclínicos sobre seguridad: Uso intravenoso y uso subcutáneo: Los datos no clínicos no revelan un riesgo especial para los seres humanos de acuerdo con estudios convencionales sobre farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción y el desarrollo. No se realizaron estudios de carcinogenia, dado que los anticuerpos monoclonales IgGl no se consideran potencialmente carcinogénicos. Los datos no clínicos disponibles demostraron el efecto de IL-6 a la progresión maligna y la resistencia a la apoptosis de diversos tipos de cáncer. Esta información no sugiere un riesgo relevante para la iniciación y la progresión del cáncer bajo tratamiento con tocilizumab. Además, no se observaron lesiones proliferativas en un estudio crónico, de 6 meses, de toxicidad en monos cynomolgus o en ratones con deficiencia de IL-6. Los datos no clínicos disponibles no sugieren un efecto sobre la fertilidad bajo el tratamiento con tocilizumab. No se observaron efectos sobre los órganos endócrinos activos y del aparato reproductor en un estudio de toxicidad crónica en monos cynomolgus y el rendimiento reproductor no se alteró en ratones deficitarios en IL-6. Se comprobó que tocilizumab administrado a monos cynomolgus durante la gestación precoz no tiene efecto lesivo directo o indirecto sobre el embarazo o el desarrollo embrionario-fetal. Sin embargo, se registró un leve aumento de los abortos/las muertes embrionarias-fetales con una alta exposición sistémica ( > 100 x la exposición humana) en el grupo de dosis alta de 50 mg/kg/día en comparación con placebo y otros grupos de dosis bajas. La incidencia de aborto estaba dentro de los antecedentes históricos en monos cynomolgus en cautiverio y los casos individuales de abortos/muerte embriofetal no demostraron ninguna relación constante con la dosificación de tocilizumab o con la duración de la misma. Aunque la IL-6 no parece ser una citocina crítica para el crecimiento fetal o el control inmunológico de la interfaz materno/fetal, no puede excluirse una relación de este hallazgo con tocilizumab. Se ha observado la transferencia de un análogo murino de tocilizumab en la leche de ratones lactantes. El tratamiento con un análogo murino no ha supuesto toxicidad en ratones jóvenes. En particular, no se han registrado alteraciones en el crecimiento esquelético, la función inmune y la maduración sexual. El perfil de seguridad no clínico de tocilizumab en monos cynomolgus no sugiere una diferencia entre las vías de administración intravenosa y subcutánea.
Indicaciones.
Actemra está indicado, en combinación con metotrexato (MTX), para: el tratamiento de artritis reumatoidea (AR) grave, activa y progresiva en adultos no tratados previamente con MTX. El tratamiento de la AR activa de moderada a grave en pacientes adultos con respuesta inadecuada o intolerancia a un tratamiento previo con uno o más fármacos antirreumáticos modificadores de la enfermedad (FAMEs) o con antagonistas del factor de necrosis tumoral (TNF). En estos pacientes Actemra puede ser administrado como monoterapia en caso de intolerancia a MTX o cuando el tratamiento continuado con MTX es inadecuado. Actemra ha demostrado reducir la tasa de progresión del daño articular medido a través de análisis radiológico y mejorar la función física, cuando se administra en combinación con metotrexato.
Dosificación.
El reemplazo de cualquier otro agente biológico requiere el consentimiento del médico prescriptor. La terapia debe ser iniciada por profesionales sanitarios con experiencia en el diagnóstico y el tratamiento de la artritis reumatoidea. Todos los pacientes tratados con Actemra deben recibir la Tarjeta de Alerta del Paciente de Actemra subcutáneo. Debe evaluarse la capacidad del paciente para utilizar la administración subcutánea domiciliaria, y advertirle sobre la importancia de notificar a un profesional de la salud en caso de experimentar síntomas de una reacción alérgica antes de administrar la próxima dosis. Los pacientes deben solicitar atención médica inmediata si desarrollan síntomas de reacciones alérgicas graves (véase Precauciones). Posología: La posología recomendada es de 162 mg, en inyección subcutánea, una vez por semana. Existe información disponible limitada referente al reemplazo del tratamiento con la formulación intravenosa de Actemra, por la formulación subcutánea con dosis fija de Actemra. Una vez iniciado el cambio, el régimen de administración debería ser una vez por semana. Los pacientes que cambian el tratamiento intravenoso por el subcutáneo deben recibir su primera dosis subcutánea en lugar de la siguiente dosis intravenosa programada, bajo la supervisión de un profesional de la salud capacitado. Recomendaciones y ajustes de dosis si los valores de laboratorio están fuera de los parámetros normales (véase Precauciones). Enzimas hepáticas fuera de los valores normales: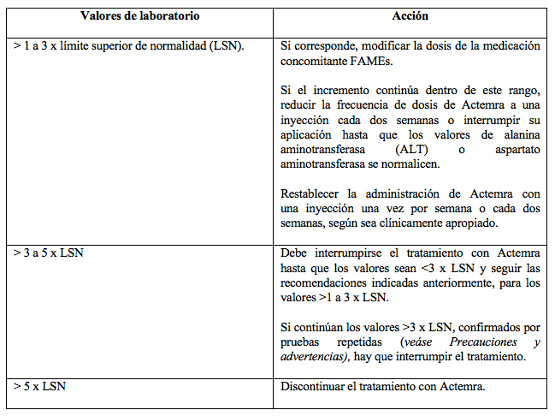
Bajo recuento absoluto de neutrófilos (RAN): No se recomienda iniciar el tratamiento, en pacientes que no han sido tratados previamente con Actemra, si el recuento absoluto de neutrófilos está por debajo de 2 x 109/l.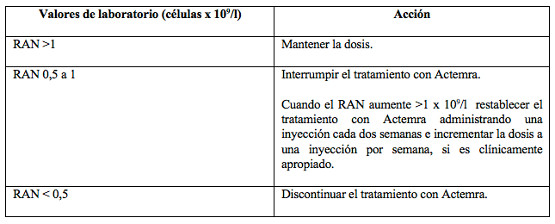
Bajo recuento de plaquetas:
Omisión de dosis: Si un paciente no se administra la inyección semanal de Actemra subcutáneo dentro de los 7 días de la dosis programada, se le deberá indicar que reciba la dosis faltante el día determinado para la próxima dosis. Si el paciente omite una inyección administrada cada dos semanas, dentro de los 7 días de la dosis programada, se le deberá indicar que reciba la dosis omitida de inmediato y la próxima dosis el día señalado en el cronograma. Poblaciones especiales de pacientes: Pacientes pediátricos: No se recomienda administrar la dosis fija de Actemra subcutáneo en niños desde el nacimiento hasta los 18 años de edad, ya que no se dispone de datos suficientes sobre seguridad y eficacia. Pacientes de edad avanzada: No se requiere el ajuste de dosis en pacientes de 65 años de edad y mayores. Pacientes con insuficiencia renal: No se requiere ajuste de la dosis en pacientes con insuficiencia renal leve. Actemra no ha sido estudiado en pacientes con insuficiencia renal de moderada a grave (véase Farmacología - Propiedades, Propiedades farmacocinéticas). La función renal debe ser estrechamente vigilada en esta población. Pacientes con insuficiencia hepática: No se ha estudiado Actemra en pacientes con insuficiencia hepática. Por lo tanto, no pueden hacerse recomendaciones sobre la dosis. Forma de administración: Actemra es para uso subcutáneo. Luego de ser instruidos debidamente acerca de la técnica para la administración de inyecciones, los pacientes pueden autoinyectarse Actemra, si el médico considera que es apropiado. El contenido total (0,9 ml) de la jeringa prellenada debe ser administrado como inyección subcutánea. Los sitios recomendados para la inyección (abdomen, muslos y parte superior del brazo) deberán alternarse, y nunca se debe aplicar la inyección en lunares, cicatrices, o en áreas donde la piel esté sensible, magullada, irritada, dura o no está intacta. La jeringa prellenada no debe agitarse. El Prospecto Información para el paciente contiene instrucciones detalladas para la preparación y administración de Actemra en una jeringa prellenada. Para consultas sobre la preparación, véase el Prospecto Información para el paciente.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de sus excipientes. Infecciones graves y activas (ver Precauciones).
Reacciones adversas.
Resumen del perfil de seguridad: Las reacciones adversas medicamentosas notificadas con más frecuencia (que se produjeron en ≥5% de los pacientes tratados con tocilizumab en monoterapia o en combinación con FAMEs) fueron infecciones en el tracto respiratorio superior, nasofaringitis, cefalea, hipertensión y elevación de la ALT. Las reacciones adversas más graves fueron infecciones graves, complicaciones de la diverticulitis y reacciones de hipersensibilidad. Uso intravenoso: La seguridad de Actemra ha sido evaluada en 4 ensayos controlados con placebo (estudios II, III, IV y V), un estudio con MTX como control (estudio I) y sus fases de extensión (ver Farmacología, Propiedades farmacodinámicas). El período controlado doble-ciego fue de 6 meses en cuatro estudios (I, III, IV y V) y de hasta 2 años en el estudio II. En los estudios controlados, doble-ciego, 774 pacientes recibieron 4 mg/kg de tocilizumab en combinación con MTX, 1.870 8 mg/kg de tocilizumab asociado con MTX/u otros FAMEs y 288 8 mg/kg de tocilizumab en monoterapia. La población expuesta a largo plazo incluye a todos los pacientes que recibieron por lo menos una dosis de tocilizumab en los estudios controlados y doble-ciego o en las fases abiertas de los estudios de extensión. De los 4.009 pacientes expuestos, 3.577 fueron tratados durante por lo menos 6 meses, 3.296 durante por lo menos 1 año, 2.806 durante por lo menos 2 años y 1.222 durante 3 años. Las reacciones adversas medicamentosas enumeradas en la Tabla 5 se presentan según la clasificación por órganos y sistemas y frecuencias. Estas se definen de la siguiente manera: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1.000 a < 1/100), raras ( > 1/10.000 a < 1/1.000) o muy raras ( < 1/10.000). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.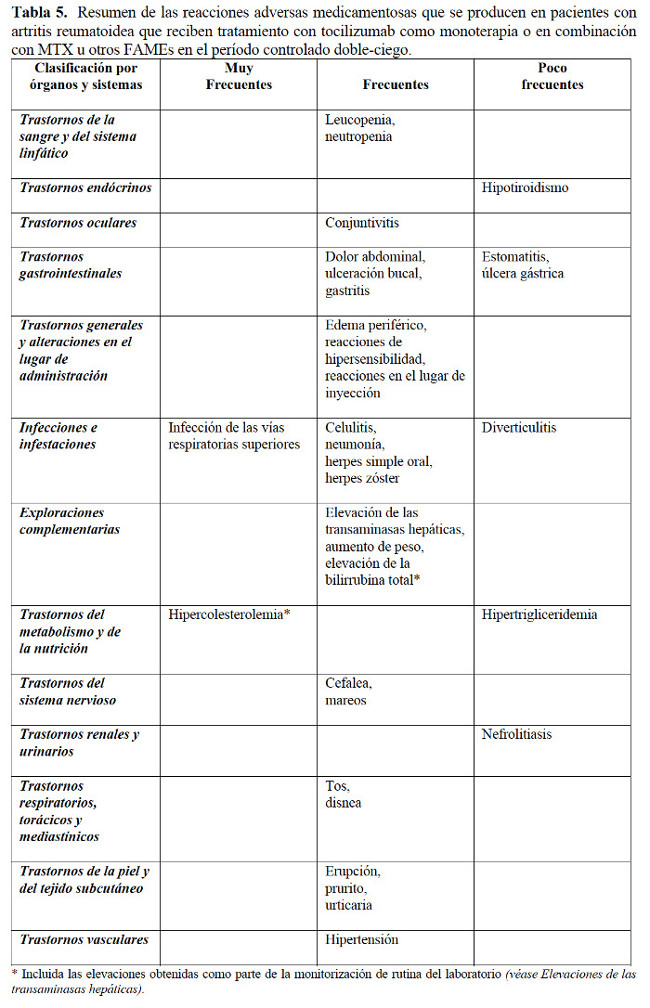
Descripción de