MARVIL® 70
ARISTON
Inhibidor específico no hormonal de la resorción ósea. Antiosteopénico y antiosteoporótico.
Composición.
Cada comprimido recubierto de MARVIL® 70 contiene: Alendronato (como Alendronato monosódico trihidrato 91,37 mg) 70 mg. Excipientes: Cellactosa, Croscaramelosa sódica, Dióxido de silicio coloidal, Estearato de magnesio, Opadry YS-1 blanco, Opaglós AG c.s.
Farmacología.
Mecanismo de Acción. La bibliografía describe para los estudios en animales el siguiente modo de acción: a nivel celular, el alendronato muestra localización preferencial por los sitios de resorción ósea, específicamente debajo de los osteoclastos. Los osteoclastos se adhieren normalmente a la superficie ósea pero carecen del borde plegado que es indicativo de la resorción activa. El alendronato no interfiere con el reclutamiento o acoplamiento de osteoclastos, pero inhibe su actividad. Los estudios en ratones sobre la localización de [3H] alendronato radioactivo en los huesos mostraron que la captación sobre las superficies osteoclásticas es aproximadamente 10 veces mayor que sobre las superficies osteoblásticas. Los huesos examinados 6 y 49 días después de la administración de [3H] alendronato en ratas y ratones, respectivamente, mostraron que el hueso normal se formó en el pico de alendronato, que fue incorporado en la matriz. Mientras se incorpora en la matriz ósea, el alendronato no es farmacológicamente activo. Por lo tanto, el alendronato debe administrarse continuamente para suprimir los osteoclastos en las superficies de resorción recientemente formadas. La histomorfometría en mandriles y ratas demostró que el tratamiento con alendronato reduce el recambio óseo (es decir la cantidad de sitios en los que el hueso es reconstruido). Además, la formación ósea excede la resorción ósea en estos sitios de reconstrucción, originando aumentos progresivos de masa ósea. Farmacodinamia. El alendronato es un bisfosfonato que se une a la hidroxiapatita ósea e inhibe, específicamente, la actividad de los osteoclastos, las células de resorción ósea. El alendronato reduce la resorción ósea sin ningún efecto directo en la formación ósea, aunque este último proceso se reduce, en última instancia, debido a que la resorción y formación ósea se combinan durante el recambio óseo. Osteoporosis en mujeres postmenopáusicas. La osteoporosis se caracteriza por una masa ósea baja que ocasiona un mayor riesgo de fracturas. El diagnóstico puede confirmarse mediante el hallazgo de una masa ósea baja, evidencia de fractura por rayos X, antecedentes de fractura osteoporótica, o pérdida de altura o cifosis, indicativo de fractura vertebral (columna vertebral). La osteoporosis ocurre tanto en varones como en mujeres pero es más común entre las mujeres después de la menopausia, cuando el recambio óseo aumenta y el porcentaje de resorción ósea excede el de la formación ósea. Los estudios de seguridad y eficacia de alendronato fueron realizados con distintos esquemas posológicos. Para proporcionar una información completa aquí se incluye a varios de ellos. Las dosis orales de alendronato (5, 20 y 40 mg durante seis semanas) en mujeres postmenopáusicas produjeron cambios bioquímicos indicativos de una inhibición de la resorción ósea, dependiente de la dosis, que incluyeron disminuciones del calcio urinario y de los marcadores urinarios de la degradación del colágeno óseo (tales como deoxipiridinolina y los N-telopéptidos de enlaces cruzados del colágeno tipo I). Estos cambios bioquímicos volvieron a los valores basales en sólo tres semanas después de la discontinuación de la terapia con alendronato y no difirieron de los observados con el placebo después de 7 meses. El tratamiento a largo plazo de la osteoporosis con alendronato 10 mg/día (hasta cinco años) redujo la excreción urinaria de los marcadores de la resorción ósea, deoxipiridinolina y N-telopéptidos de enlaces cruzados del colágeno tipo I, en aproximadamente un 50% y 70%, respectivamente hasta alcanzar niveles similares a los observados en mujeres postmenopáusicas sanas. Se observaron disminuciones similares en pacientes en estudios de prevención de la osteoporosis que recibieron alendronato 5 mg/día. La disminución en el porcentaje de resorción ósea indicada por estos marcadores fue evidente tan pronto como al mes, y tres a seis meses después alcanzó una meseta que se mantuvo durante todo el tratamiento con alendronato. En los estudios de tratamiento de la osteoporosis, alendronato 10 mg/día disminuyó los marcadores de la formación ósea, la osteocalcina y la fosfatasa alcalina específica del hueso, en aproximadamente un 50% y la fosfatasa alcalina sérica total en aproximadamente un 25 a 30%, hasta alcanzar una meseta después de 6 a 12 meses. En los estudios de prevención de la osteoporosis, alendronato 5 mg/día, disminuyó la osteocalcina y la fosfatasa alcalina sérica total en aproximadamente un 40% y 15%, respectivamente. Se observaron reducciones similares en el porcentaje de recambio óseo en mujeres postmenopáusicas durante estudios de un año de duración con la administración de alendronato 70 mg una vez por semana para el tratamiento de la osteoporosis y alendronato 35 mg una vez por semana para la prevención de la osteoporosis. Estos datos indican que el porcentaje de recambio óseo alcanzó un nuevo estado de equilibrio, a pesar del aumento progresivo en la cantidad total de alendronato depositado en el hueso. En estudios de un año de duración con alendronato 35 y 70 mg administrado una vez por semana, se observaron reducciones similares en los meses 6 y 12. La reducción del fosfato sérico puede reflejar no sólo el equilibrio mineral óseo positivo debido al alendronato sino también una disminución en la reabsorción renal del fosfato. Osteoporosis en el varón. El tratamiento de hombres con osteoporosis con alendronato 10 mg/día durante dos años redujo la excreción urinaria de N-telopéptidos de enlaces cruzados del colágeno de tipo I en aproximadamente un 60% y redujo la fosfatasa alcalina específica del hueso en aproximadamente un 40%.
Farmacocinética.
Absorción. En la bibliografía publicada, se puede encontrar que en relación a una dosis intravenosa (IV) de referencia, la biodisponibilidad oral media del alendronato en mujeres fue del 0,64% con dosis que oscilan de 5 a 70 mg administradas después de un ayuno nocturno y dos horas antes de un desayuno estándar. La biodisponibilidad oral de alendronato 10 mg en varones (0,59%) fue similar a la de las mujeres cuando se administró después de un ayuno nocturno y dos horas antes del desayuno. De acuerdo con un estudio que examina el efecto de sincronización de una comida sobre la biodisponibilidad del alendronato en 49 mujeres postmenopáusicas, la biodisponibilidad disminuye (en aproximadamente un 40%) cuando se administra 10 mg de alendronato ya sea media o una hora antes de un desayuno estándar, en comparación con la dosificación administrada dos horas antes de la ingesta de alimentos. En estudios publicados de tratamiento y prevención de la osteoporosis, el alendronato resultó eficaz cuando se administró por lo menos 30 minutos antes del desayuno. La biodisponibilidad resulta insignificante independientemente de si el alendronato se administra con o hasta dos horas después de un desayuno estándar. La administración concomitante de alendronato con café o jugo de naranja reduce la biodisponibilidad en aproximadamente un 60%. Se ha realizado un estudio de disolución y bioequivalencia con MARVIL® 70 (test) en comparación con los comprimidos de alendronato 70 mg utilizados como referencia. Dicho estudio fue abierto, aleatorizado, con tratamientos cruzados, de dosis única en sujetos sanos. Se evaluó como parámetro, la cantidad total de alendronato eliminado a través de la orina a las 6 y 24 horas luego de su administración (UE6 y UE24h). Se demostró la disolución de más del 85% del componente activo en 15 minutos. El intervalo de confianza para la UE24, se situó entre el 72-122%. Si se realiza una evaluación de aquellos pacientes con metabolismo óseo estable, el intervalo de confianza se encuentra entre el 86 y el 137%, ambos parámetros que se consideran en rango de bioequivalencia. Distribución. Estudios preclínicos (en ratas macho) muestran que el alendronato se distribuye transitoriamente a los tejidos blandos después de la administración IV de 1 mg/kg, pero se redistribuye rápidamente al hueso o se excreta en la orina. El volumen de distribución, a concentraciones plasmáticas estables medias es por lo menos 28 L en los seres humanos. Las concentraciones de la droga en el plasma después de dosis orales terapéuticas son demasiado bajas (menos de 5 ng/mL) para la detección analítica. La unión proteica en el plasma humano es de aproximadamente un 78%. Metabolismo. No existe evidencia de que el alendronato sea metabolizado en los animales o en los seres humanos. Excreción. Después de una dosis IV única de [14C] alendronato, aproximadamente el 50% de la radioactividad se excretó en la orina dentro de las 72 horas y se recuperó poca, o no se recuperó, radioactividad en las heces. Después de una dosis IV única de 10 mg, la depuración renal del alendronato fue de 71 ml/min (64, 78; intervalo de confianza [IC] del 90%) y la depuración sistémica no excedió los 200 ml/min. Las concentraciones plasmáticas disminuyeron en más del 95% dentro de las seis horas posteriores a la administración IV. Se calcula que la vida media terminal en los seres humanos excede los 10 años y probablemente refleje la liberación del alendronato del esqueleto. Sobre la base de lo expuesto anteriormente, se calcula que después de 10 años tratamiento oral con alendronato (10 mg por día) la cantidad de alendronato liberado diariamente del esqueleto es de aproximadamente un 25% de lo absorbido del tracto gastrointestinal. Poblaciones Especiales: Población pediátrica. La biodisponibilidad oral del alendronato en niños es similar a la observada en adultos. Sin embargo, MARVIL® 70, no está indicado en niños (Ver Precauciones). Sexo. La biodisponibilidad y la fracción de una dosis IV excretada en la orina fueron similares en varones y mujeres. Población geriátrica. La biodisponibilidad y la disposición (excreción urinaria) fueron similares en los pacientes de edad avanzada y los más jóvenes. No se requiere ningún ajuste de dosis en este grupo etáreo (ver Posologia y forma de administracion). Raza. No se han realizado estudios farmacocinéticos con diferencias de razas. Insuficiencia renal. Los estudios preclínicos muestran que, en las ratas con falla renal, hay mayores cantidades de droga en el plasma, riñón, bazo y tibia. En los controles sanos, la droga que no se deposita en el hueso se excreta rápidamente en la orina. No se halló evidencia de saturación en la captación ósea después de una dosificación de tres semanas con dosis IV acumulativas de 35 mg/kg en ratas macho jóvenes. Si bien no se dispone de información clínica, es probable que, al igual que en los animales, la eliminación del alendronato por vía renal se reduzca en pacientes con deterioro de la función renal. Por lo tanto, se podría esperar una acumulación un tanto mayor de alendronato en el hueso en pacientes con deterioro de la función renal. No es necesario un ajuste de la dosis en pacientes con insuficiencia renal de leve a moderada (clearance de creatinina de 35 a 60 ml/min). Al igual que con otras formulaciones no se recomienda el uso de alendronato, en pacientes con insuficiencia renal severa (clearance de creatinina < 35 ml/min) debido a la falta de experiencia con alendronato en la falla renal. Insuficiencia hepática. Debido a que existe evidencia de que el alendronato no es metabolizado ni excretado en la bilis, no se llevaron a cabo estudios en pacientes con insuficiencia hepática. No es necesario un ajuste de la dosis.
Indicaciones.
MARVIL® 70 está indicado en: Tratamiento de la osteoporosis en mujeres postmenopáusicas. MARVIL® 70, aumenta la masa ósea y reduce la incidencia de fracturas, inclusive las de cadera y columna (fracturas por compresión vertebral). La osteoporosis se puede confirmar por medio del hallazgo de masa ósea baja (al menos 2 desviaciones) estándares por debajo de la media premenopáusica) o mediante la presencia o antecedentes de fracturas por osteoporosis. (Ver acción farmacológica, Farmacodinamia). Tratamiento para aumentar la masa ósea en varones con osteoporosis.
Dosificación.
La dosis recomendada de MARVIL® 70, es de 1 comprimido, una vez a la semana. MARVIL® 70 debe ingerirse íntegramente y de una sola vez. MARVIL® 70 debe tomarse por lo menos media hora antes de la ingestión de la primer comida, bebida o medicamento del día, acompañado de un vaso con agua corriente potable, solamente (ver Precauciones, Información para Pacientes). Es probable que otras bebidas (incluyendo el agua mineral, té, café, leche, jugo de naranja), comidas y algunos medicamentos reduzcan la absorción del alendronato (ver Precauciones, Interacciones Farmacológicas). Para favorecer la llegada al estómago y por lo tanto reducir el potencial de irritación esofágica, MARVIL® 70, debe ingerirse al levantarse a la mañana, con un vaso lleno de agua corriente (no menos de 200 ml) y los pacientes no deben recostarse durante al menos 30 minutos y hasta después de su primera comida del día. MARVIL® 70, no debe tomarse al acostarse ni antes de levantarse por la mañana. Los comprimidos de MARVIL® 70, no deben masticarse ni triturarse, ni dejar que se disgreguen en la boca por el potencial riesgo de ulceración orofaríngea. No seguir estas instrucciones puede aumentar el riesgo de experiencias adversas esofágicas (ver Advertencias, Precauciones, Información para Pacientes). En el caso de omitir la dosis semanal de MARVIL® 70, se debe tomar el comprimido en la mañana siguiente de haberlo recordado. No se deben tomar 2 dosis (comprimidos) el mismo día, pero se debe mantener el esquema de dosis de un comprimido semanal, en el día de la semana que se ha elegido, originalmente. Los pacientes deben recibir calcio y vitamina D suplementarios, si su ingesta dietaria es inadecuada (ver Precauciones, General). No es necesario un ajuste de dosis para las personas de edad avanzada ni para pacientes con insuficiencia renal leve a moderada (clearance de creatinina de 35 a 60 ml/min). No se recomienda para pacientes con insuficiencia renal severa (clearance de creatinina < 35 ml/min.) debido a la falta de experiencia. No se ha definido aún la duración del tratamiento. En el tratamiento de la osteoporosis, la seguridad y la eficacia del alendronato monosódico han sido establecidas según la información clínica disponible, que abarca un período de cuatro años. Los pacientes que reciben bifosfonatos deberían ser evaluados en forma regular, para verificar la necesidad de continuar con el tratamiento. Aquellos pacientes con un perfil de riesgo bajo de fractura, deben ser considerados para interrumpir la droga luego de 3 a 5 años de tratamiento. En el caso de suspensión del tratamiento, se debe analizar en forma periódica el riesgo de fractura del paciente.
Contraindicaciones.
Anormalidades del esófago que demoran el vaciado esofágico tales como la estenosis o acalasia. Incapacidad para mantenerse de pie o sentado erguido durante por lo menos 30 minutos. Hipersensibilidad a cualquiera de los componentes de este producto.- Hipocalcemia (ver Precauciones, General).
Reacciones adversas.
Estudios clínicos. Tratamiento de la osteoporosis Mujeres postmenopáusicas: En un ensayo clínico realizado en mujeres postmenopáusicas con osteoporosis, de un año de duración, la evaluación de seguridad global de alendronato 70 mg una vez por semana en comprimidos (n= 519) y alendronato 10 mg diarios (n=370), arrojó resultados similares. En dos estudios de tres años de duración, en mujeres postmenopáusicas recibiendo alendronato 10 mg (n=196) o placebo (n=397), la seguridad global de alendronato 10 mg día resultó similar al grupo placebo. Las experiencias adversas consideradas por los investigadores como posible, probable o definitivamente relacionadas con la droga en ≥ 1% de los pacientes en cualquiera de los grupos de tratamiento se presentan en la tabla siguiente. Estudios de tratamiento para osteoporosis en mujeres postmenopáusicas: Experiencias adversas consideradas posible, probable o definitivamente relacionadas con la droga por los investigadores e informadas en > 1% de los pacientes: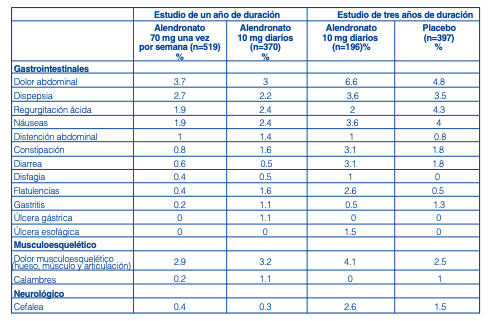
Osteoporosis en Hombres: En dos ensayos clínicos doble ciego, controlados con placebo, multicéntricos en hombres (un estudio de dos años de alendronato 10 mg por día y otro un año de alendronato 70 mg semanal) la tasa de discontinuación por la presencia de eventos adversos fue de 2.7% (alendronato 10mg/d) y de 10.5% para el placebo, y de 6.4% (alendronato 70 mg semanal) y de 8.6% para el placebo. Estudios de tratamiento para osteoporosis en hombres: Experiencias adversas consideradas posible, probable o definitivamente relacionadas con la droga por los investigadores e informadas en una frecuencia igual o > 2% de los pacientes: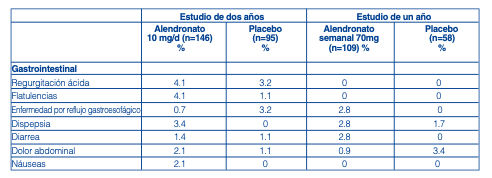
Hallazgos de Pruebas de Laboratorio: En estudios a doble ciego, multicentricos, controlados con placebo, se obervaron disminuciones asintomáticas, leves y transitorias en calcio y fosfatos séricos en aproximadamente el 18 y 10%, respectivamente, de pacientes que tomaron alendronato vs. aproximadamente el 12 y 3% respectivamente de quienes tomaron placebo. Sin embargo, las incidencias en las disminuciones en el calcio sérico a fueron similares en ambos grupos de tratamiento. < 8 mg/dl (2mM) y fosfato séricos a ≤ 2 mg/dl (0.65 mM). Experiencia Fase IV: Se han informado las siguientes reacciones adversas en el uso posterior a la comercialización: A nivel sistémico: reacciones de hipersensibilidad incluyendo urticaria y con poca frecuencia angioedema. Se informaron síntomas transitorios de mialgia, malestar y con poca frecuencia fiebre con alendronato, por lo general en asociación con el inicio del tratamiento. Con poca frecuencia, ocurrió hipocalcemia sintomática, por lo general en asociación con condiciones predisponentes. Edema periférico. Gastrointestinales: esofagitis, erosiones esofágicas, úlceras esofágicas, con poca frecuencia estenosis o perforación esofágica y ulceración orofaríngea. También se informaron úlceras gástricas o duodenales, algunas severas y con complicaciones. (ver Advertencias, Precauciones, Información para Pacientes, Posologia y forma de administracion). Raramente se ha informado osteonecrosis localizada de la mandíbula, generalmente asociada con extracción dental y/o infección local, a menudo con retraso en la curación (ver Advertencias). Músculoesqueléticas: dolor de los huesos, las articulaciones y/o los músculos, ocasionalmente severo y raramente incapacitante, inflamación articular. Fracturas atípicas subtrocantéricas y diafisarias del fémur (ver Advertencias). Sistema Nervioso: mareos y vértigo. Respiratorio: exacerbación de asma bronquial. Piel: rash (ocasionalmente con fotosensibilidad), prurito, alopecia, con poca frecuencia reacciones dérmicas severas, incluyendo el síndrome de Stevens-Johnson y necrólisis epidérmica tóxica. Órganos de los Sentidos: con poca frecuencia uveítis, en raras ocasiones escleritis o epiescleritis. Otros: linfopenia, síntomas pseudo gripales.
Precauciones.
Deben considerarse las causas de la osteoporosis que no sean la deficiencia de estrógenos, edad y uso de glucocorticoides. La hipocalcemia debe corregirse antes de iniciar el tratamiento con alendronato (ver Contraindicaciones). Otros trastornos que afectan el metabolismo mineral (tales como la deficiencia de la vitamina D y/o el hipoparatiroidismo) también deben ser tratados de manera efectiva. En pacientes con estas condiciones, el calcio sérico y los síntomas de hipocalcemia deben monitorearse durante la terapia con alendronato. Presumiblemente debido a los efectos de alendronato sobre el aumento mineral óseo, se pueden presentar pequeñas disminuciones asintomáticas de calcio y fosfato sérico, especialmente en los pacientes con osteítis deformante, en quienes el índice de recambio óseo previo al tratamiento puede ser muy elevado y en pacientes que reciben glucocorticoides en quienes la absorción de calcio puede ser menor. El hecho de asegurar una ingesta adecuada de calcio y vitamina D es especialmente importante en los pacientes con osteítis deformante y en pacientes que reciben glucocorticoides. Insuficiencia Renal: No se recomienda el uso de alendronato en los pacientes con insuficiencia renal severa (clearance de creatinina < 35 ml/min). (Ver Posologia y forma de administracion). Excipientes: Este medicamento contiene lactosa. No deben tomar este medicamento los pacientes con problemas hereditarios raros de intolerancia a la galactosa, intolerancia a la lactasa Lapp o absorción insuficiente de glucosa-galactosa. Interacciones farmacológicas: La ranitidina intravenosa demostró duplicar la biodisponibilidad del alendronato oral. Se desconoce la relevancia clínica de esta mayor biodisponibilidad y si ocurrirán aumentos similares en pacientes a los que se les administran antagonistas orales H2 u otras formulaciones. En pacientes sanos, la prednisona oral (20 mg tres veces por día durante cinco días) no produjo un cambio clínicamente significativo en la biodisponibilídad oral del alendronato (un aumento medio que oscila del 20 al 44%). Los productos que contienen calcio y otros cationes multivalentes probablemente interfieran con la absorción del alendronato. Suplementos de calcio/Antiácidos: Es probable que los suplementos de calcio, los antiácidos y algunos medicamentos orales interfieran con la absorción de alendronato. Por lo tanto, los pacientes deben esperar por lo menos media hora después de tomar alendronato antes de tomar cualquier otro medicamento oral. Aspirina: En los estudios clínicos publicados, la incidencia de los eventos adversos gastrointestinales superiores aumentó en los pacientes que habían recibido tratamiento concomitante con dosis diarias de alendronato superiores a los 10 mg y productos que contenían aspirina. Drogas antiinflamatórias no esteroides (AINEs): El alendronato puede administrarse en pacientes que toman AINEs. En un estudio clínico controlado de 3 años de duración (n=2027) durante el cual la mayoría de los pacientes recibieron AINEs concomitantemente con alendronato, la incidencia de eventos adversos gastrointestinales superiores fue similar en los pacientes que tomaron alendronato 5 o 10 mg/día en comparación con los que tomaron placebo. No obstante, dado que el uso de AINEs se asocia con la irritación gastrointestinal, se debe actuar con precaución durante el uso concomitante con alendronato. En ensayos clínicos, las pacientes que recibieron alendronato y estrógenos (intravaginales, trasdérmicos u orales) no presentaron reacciones atribuidas a su uso concomitante. Carcinogénesis, Mutagénesis, Trastornos de la Fertilidad: Los adenomas de la glándula de Harderian (una glándula retroorbital que no está presente en los seres humanos) aumentaron en ratones hembra con dosis altas (p=0,003) en un estudio sobre carcinogenicidad oral de 92 semanas de duración con dosis de alendronato de 1, 3 y 10 mg/kg/día (machos) o 1, 2 y 5 mg/kg/día (hembras). Estas dosis son equivalentes a 0,1 -1 vez una dosis máxima diaria recomendada de 40 mg (osteítis deformante) sobre la base del área de la superficie mg/m2.Se desconoce la importancia de este hallazgo en humanos. Los adenomas de las células parafoliculares (tiroides) aumentaron en ratas macho con altas dosis (p=0,003) en un estudio de carcinogenicidad oral de 2 años de duración con dosis de 1 y 3,75 mg/kg del peso corporal. Estas dosis son equivalentes a 0,3 y 1 veces la dosis diaria humana de 40 mg basada sobre el área de la superficie mg/m2. Se desconoce la importancia de este hallazgo en humanos. El alendronato no resultó genotóxico en el ensayo in vitro sobre mutagénesis microbiana con y sin activación metabólica, en un ensayo in vitro sobre mutagénesis celular en mamíferos, en un ensayo in vitro de elución alcalina en hepatocitos de ratas y en un ensayo de aberración cromosómica in vivo en ratones. Un ensayo in vitro de aberración cromosómica en células de ovario de hámster chino, sin embargo, arrojó resultados inciertos. El alendronato no tuvo ningún efecto sobre la fertilidad (tanto en machos como en hembras) en ratas tratadas con dosis orales de hasta 5 mg/kg/día (1 vez la dosis diaria humana de 40 mg basada sobre el área de la superficie mg/m2). Embarazo: Los estudios de reproducción llevados a cabo en ratas mostraron una disminución en la sobrevida posterior al implante con 2 mg/kg/día y redujeron el aumento del peso corporal en crías normales con 1 mg/kg/día. Los sitios de osificación fetal incompleta aumentaron en forma significativa desde el punto de vista estadístico en ratas comenzando con 10 mg/kg/día en las vértebras (cervicales, torácicas y lumbares), el cráneo y los huesos del esternón. Las dosis que se mencionaron anteriormente oscilaron entre 0,26 veces (1 mg/kg) y 2,6 veces la dosis máxima diaria recomendada (10 mg/kg) de 40 mg (osteítis deformante) sobre la base del área de la superficie mg/m2. No se observaron efectos fetales similares con conejas gestantes cuando se trataron con dosis de hasta 35 mg/kg/día (10,3 veces una dosis diaria de 40 mg en humanos sobre la base del área de la superficie mg/m2). Tanto el calcio total como el calcio ionizado disminuyeron en las ratas gestantes con 15 mg/kg/día (3,9 veces una dosis diaria de 40 mg en humanos sobre la base del área de la superficie mg/m2) dando como resultado demoras y fallas en el alumbramiento. Se produjo el alumbramiento prolongado debido a la hipocalcemia materna ocurrida en ratas con dosis bajas de 0,5 mg/kg/día (0,13 veces una dosis diaria de 40 mg en humanos sobre la base del área de la superficie mg/m2) cuando las ratas fueron tratadas antes del apareamiento hasta la gestación. La toxicidad materna (muertes por embarazo tardío) se produjo en ratas hembra tratadas con 15 mg/kg/día durante períodos variables de tiempo que oscilaron solamente entre el tratamiento durante el período previo al apareamiento hasta el tratamiento durante la gestación temprana, media o tardía; estas muertes disminuyeron pero no se eliminaron con la interrupción del tratamiento. El suplemento de calcio administrado ya sea en el agua para beber o por medio de una minibomba no pudo mejorar la hipocalcemia ni evitar las muertes maternas y neonatales debido a las demoras en el alumbramiento. El suplemento de calcio administrado por vía intravenosa evitó las muertes maternas pero no las muertes fetales. Los bifosfonatos se incorporan en la matriz ósea, desde la cual se liberan gradualmente durante un período de años. La cantidad de bifosfonatos incorporada en el hueso adulto, y por lo tanto la cantidad disponible para su posterior liberación en la circulación, está directamente relacionada con la dosis y la duración del uso de bifosfonatos. No existen datos sobre el riesgo fetal en humanos, sin embargo, existe un riesgo teórico de daño fetal, predominantemente esquelético, si una mujer queda embarazada después de terminar un ciclo de tratamiento con bifosfonatos. No se ha estudiado el impacto de las variables sobre el riesgo tales como el tiempo entre el cese del tratamiento con bifosfonatos hasta la concepción, el bisfosfonato utilizado en particular y la vía de administración (intravenosa vs. oral). No existen estudios realizados en mujeres embarazadas. No debe ser utilizado durante el embarazo. Lactancia: Uso pediátrico: No se sabe si el alendronato se excreta a través de la leche materna. No debe ser usado durante el amamantamiento. En un pequeño número de pacientes menores de 18 años de edad, con osteogénesis imperfecta, el alendronato ha sido estudiado con resultados insuficientes para recomendar su utilización. No está indicado en pacientes pediátricos. Uso geriátrico: De los pacientes que recibieron alendronato en el Ensayo de Intervención de Fracturas (FIT), el 71% (n=2302) fueron ≥ 65 años de edad y el 17% (n=550) fueron ≥ 75 años de edad. De los pacientes que recibieron alendronato en estudios en los Estados Unidos y multinacional sobre tratamientos de osteoporosis en mujeres, en el estudio sobre osteoporosis en hombres, en los estudios sobre osteoporosis inducida por glucocorticoides y en los estudios de osteítis deformante (Enfermedad de Paget), el 45%, el 50%, el 37% y el 70% respectivamente, se observaron entre pacientes de 65 años de edad o más. No se observaron diferencias generales en la eficacia y seguridad entre estos pacientes y los pacientes más jóvenes, pero no puede descartarse una mayor sensibilidad en algunos pacientes mayores. Habilidad para conducir vehículos: No se han realizado estudios sobre habilidad para conducir vehículos. Sin embargo, algunos efectos adversos que han ocurrido con alendronato podrían afectar la habilidad para conducir máquinas. La respuesta al alendronato puede variar en cada paciente.
Advertencias.
MARVIL® 70, al igual que otros bifosfonatos, puede provocar irritación local de la mucosa gastrointestinal superior. En pacientes que recibieron tratamiento con alendronato se han registrado experiencias adversas, tales como esofagitis, úlceras, erosiones esofágicas, ocasionalmente con hemorragia y raramente seguidas de estenosis esofágica. Dado que en algunos casos estas experiencias han sido severas, los médicos deberán advertir a los pacientes que ante cualquier signo o síntoma que indique una posible reacción adversa esofágica (disfagia, odinofagia, dolor retroesternal o aparición de acidez o empeoramiento de la misma), deben suspender la administración de MARVIL® 70 y solicitar atención médica. Aparentemente, el riesgo de desarrollar efectos adversos esofágicos severos es mayor en aquellos pacientes que se recuestan después de tomar alendronato, que no logran ingerirlo con suficiente agua, o bien que continúan ingiriendo la droga posterior al desarrollo de síntomas indicativos de irritación esofágica. En consecuencia, es muy importante que el paciente cumpla con las instrucciones para la administración del producto. En aquellos pacientes que no puedan cumplir con las mismas debido a una incapacidad mental, se deberá realizar el tratamiento con alendronato bajo estricta supervisión. Debido a los posibles efectos irritantes del alendronato sobre la mucosa del aparato gastrointestinal superior y a un posible empeoramiento de la enfermedad subyacente, se debe tener precaución al administrar MARVIL® 70 a pacientes con trastornos activos del aparato gastrointestinal superior tales como: disfagia, alteraciones esofágicas, gastritis, duodenitis o úlceras, o con antecedentes en el último año, de enfermedad gastrointestinal mayor como sangrado gastrointestinal activo, úlcera péptica, cirugía del tracto gastrointestinal superior u otras como piloroplastia. En pacientes con esófago de Barrett, debe realizarse un cuidadoso análisis, tras valorar beneficios y riesgos potenciales en forma individual antes de su indicación. Cáncer de Esófago: a la fecha existe información controvertida con respecto a la posibilidad de desarrollar cáncer de esófago en pacientes bajo tratamiento con bifosfonatos orales, las anormalidades persistentes de la mucosa esofágica en pacientes con esofagitis erosiva usualmente secundario a la administración inapropiada de bifosfonatos orales podría potencialmente conducir al desarrollo de cáncer. Han ocurrido algunas notificaciones luego de la comercialización del producto, úlceras gástricas y duodenales, en algunos casos severas y en otros con complicaciones. No se ha observado un incremento del riesgo en los estudios clínicos extensos. Dolor Musculo-esquelético: Luego de la comercialización del producto, ha sido notificado dolor severo y ocasionalmente incapacitante a nivel articular, óseo y/o muscular, en pacientes recibiendo bifosfonatos, incluyendo alendronato. Los síntomas varían en su aparición desde un día a varios meses luego del inicio del tratamiento. La mayoría de las afectadas fueron mujeres postmenopáusicas. Se debe interrumpir la administración si se presentan síntomas graves. En general, en la mayor parte de los pacientes los síntomas calman al discontinuar el tratamiento. Puede ocurrir recurrencia sintomática ante la reexposición con la misma droga u otro bifosfonato. En ensayos clínicos controlados con placebo, la proporción de pacientes sintomáticos en ambos grupos fue similar. Osteonecrosis de la Mandíbula: Ha sido reportada osteonecrosis de mandíbula, generalmente asociada con extracción y/o infección local dental, frecuentemente con demora en la curación en pacientes que toman bifosfonatos. La mayoría de los casos reportados de bifosfonatos asociados a osteonecrosis han sido de pacientes con cáncer tratados con bifosfonatos intravenoso, aunque algunos han ocurrido en pacientes con osteoporosis postmenopáusica. Debe tenerse en cuenta realizar antes de comenzar el tratamiento con bifosfonatos, un examen odontológico y otras conductas preventivas adecuadas, en pacientes con factores de riesgo. Los factores de riesgo conocidos para la osteonecrosis incluyen: procedimientos odontológicos invasivos (extracción dentaria, implantes, cirugía), diagnóstico de cáncer, terapias concomitantes (Ej. quimioterapia, radioterapia, corticoesteroides), mala higiene bucal y trastornos comórbidos (Ej. Enfermedad dental pre-existente, anemia, coagulopatía, infección). El riesgo podría incrementarse con el tiempo de exposición a los bifosfonatos. Los pacientes que desarrollen osteonecrosis de mandíbula mientras se encuentren bajo una terapia con bifosfonatos deberían recibir atención de un Cirujano Dental. La cirugía dental puede exacerbar la situación. La interrupción del tratamiento con bifosfonatos debe ser considerada según la valoración de riesgos y beneficios en forma individual. En aquellos casos donde se requiera realizar procedimientos dentales invasivos, la interrupción del tratamiento con bifosfonatos podría reducir en algunos casos, el riesgo de osteonecrosis de la mandíbula. La opinión clínica del médico tratante y/o del cirujano dental debería guiar el plan a seguir en cada paciente basado en la valoración de los riesgos y beneficios individuales. Fracturas atípicas subtrocantéricas y diafisarias del femur: En algunos pacientes tratados con bifosfonatos se han informado fracturas atípicas, de baja energía o por traumatismo de bajo impacto, de la diáfisis femoral. Estas fracturas pueden ocurrir en cualquier porción de la diáfisis del fémur, desde justo por debajo del trocánter menor hasta el área supracondílea; tienen orientación transversal o son cortas y oblicuas y no hay evidencia de pulverización. No se ha establecido una relación causal, ya que estas fracturas también ocurren en pacientes osteoporóticos que no han sido tratados con bifosfonatos. Las fracturas atípicas del fémur pueden ser bilaterales y comúnmente ocurren sin que haya impacto o luego de un impacto mínimo en el área afectada. Muchos pacientes refieren dolor prodrómico en el área afectada, que generalmente se presenta como un dolor sordo en el muslo semanas a meses antes de que ocurra una fractura completa. Algunos reportes notan que los pacientes afectados también estaban bajo tratamiento con glucocorticoides al momento de producirse de la fractura. En todo paciente con antecedente de exposición a bifosfonatos que presente dolor en el muslo o en la ingle, debe sospecharse una fractura atípica y debe realizarse una evaluación para descartar fractura de fémur. Los pacientes con fracturas atípicas también deben ser examinados para verificar si hay signos o síntomas de fractura en el miembro contralateral. Se debe considerar la interrupción del tratamiento con bifosfonatos hasta que los riesgos y beneficios puedan ser evaluados en forma individual.
Conservación.
Conservar a temperatura Ambiente entre 15°C y 30°C. Protegido de la Luz.
Sobredosificación.
Se observó una letalidad significativa después de dosis orales únicas en ratas y ratones hembra con dosis de 552 mg/kg (3256 mg/m2) y 966 mg/kg (2898 mg/m2), respectivamente. En machos, estos valores fueron ligeramente mayores, 626 y 1280 mg/kg, respectivamente. No hubo letalidad en perros con dosis orales de hasta 200 mg/kg (4000 mg/m2). No se encuentra disponible información específica para el tratamiento de la sobredosis. Puede ocurrir hipocalcemia, hipofosfatemia y eventos adversos gastrointestinales superiores, tales como un malestar estomacal, acidez, esofagitis, gastritis o úlcera por una sobredosis por vía oral. Se debe administrar leche o antiácidos para unirse al alendronato. Debido al riesgo de irritación gastroesofágica, no se debe inducir el vómito y el paciente debe permanecer erguido. No sería beneficiosa la diálisis. "Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse con los centros de Toxicología: Hospital de Pediatria Ricardo Gutierrez: (011) 4962-6666/2247. Hospital Alejandro Posadas: (011) 4654-6648/4658-7777. Optativamente otros Centros de Toxicología". Tratamiento orientativo inicial de la sobredosis: luego de la cuidadosa evaluación clínica del paciente, de la valorización del tiempo transcurrido desde la ingesta o administración, de la cantidad de tóxicos ingeridos y descartando la contraindicación de ciertos procedimientos, el profesional decidirá la realización o no del tratamiento general de rescate.
Presentación.
Envases conteniendo 4 comprimidos recubiertos.
Revisión.
09/2015.