ESPEROCT®
NOVO NORDISK
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de coagulación sanguínea.
Composición.
Polvo: Principio activo: turoctocog alfa pegol (factor VIII de coagulación humana (ADNr), pegilado). Excipientes: Polvo: Cloruro de sodio, L-histidina, sacarosa, polisorbato 80, L-metionina, cloruro de calcio dihidratado, hidróxido de sodio (para ajuste del pH), ácido clorhídrico (para ajuste del pH). Solvente: cloruro de sodio, agua para inyectables. Turoctocog alfa pegol (factor VIII recombinante humano) producido mediante tecnología de ADN recombinante en una línea de células de ovario de Hámster Chino (CHO), conjugados de manera covalente con un polietilenglicol (PEG) de 40 kDa. Excipiente con efecto conocido Más de 1 mmol de sodio (23 mg) por vial. Forma farmacéutica: Polvo liofilizado y solvente para solución inyectable. Cada vial contiene 500 UI, 1000 UI o 1500 UI de turoctocog alfa pegol, según lo declarado. Después de la reconstitución de la solución, 1 ml de Esperoct® contiene aproximadamente 125 UI, 250 UI, 375 UI, 500 UI o 750 UI de turoctocog alfa pegol. El polvo es blanco a blanquecino. El solvente es una solución transparente e incolora. La actividad específica de Esperoct® es de aproximadamente 9500 UI/mg de proteína.
Farmacología.
Propiedades farmacodinámicas: Mecanismo de acción: Esperoct® es un producto purificado del factor VIII recombinante humano (FVIIIr) que contiene un polietilenglicol (PEG) de 40 kDa conjugado con la proteína. El PEG está unido al O-glicano en el dominio B truncado del FVIIIr (turoctocog alfa). El mecanismo de acción de Esperoct® se basa en la sustitución del factor VIII deficiente o ausente en pacientes con hemofilia A. Cuando Esperoct® es activado por la trombina en el sitio de la lesión, el dominio B que contiene la fracción de PEG y la región a3 se escinden, de modo que el factor VIII recombinante activo (FVIIIra) tiene una estructura similar a la del factor VIIIa nativo. La terapia de sustitución aumenta los niveles plasmáticos del factor VIII, corrigiendo así temporalmente la deficiencia de factor VIII y la tendencia al sangrado. Datos de eficacia clínica y seguridad: Eficacia clínica en la profilaxis y el tratamiento de los episodios hemorrágicos: La eficacia clínica de Esperoct® en la profilaxis y el tratamiento de hemorragias se evaluó en cinco estudios clínicos multicéntricos prospectivos en pacientes tratados previamente (PTP) que padecían hemofilia A grave. El efecto hemostático se confirmó en adultos/adolescentes y niños. Profilaxis de rutina en adultos/adolescentes: El efecto profiláctico de Esperoct® ha sido demostrado en adultos/adolescentes (a partir de 12 años de edad) a razón de 50 UI por kg de peso corporal cada 3 o 4 días en 175 pacientes. La mediana de la tasa anual de hemorragia (ABR, por sus siglas en ingles) en adultos y adolescentes que recibieron Esperoct® cada 3-4 días fue de 1,18 (rango intercuartílico (IQR)): 0,00; 4,25), mientras que la tasa anual de hemorragias espontáneas fue de 0,00 (IQR: 0,00; 1,82), la tasa anual de hemorragias traumáticas fue de 0,00 (IQR: 0,00; 1,74) y la tasa anual de hemorragias articulares fue de 0,85 (IQR: 0,00; 2,84). De los 175 adultos/adolescentes en profilaxis, 70 (40%) no tuvieron ninguna hemorragia. Los adultos/adolescentes que tuvieron una tasa de hemorragia baja, de 0 a 2 hemorragias en los últimos 6 meses, y que recibieron al menos 50 dosis de Esperoct® tuvieron la opción de ser aleatorizados a un tratamiento profiláctico cada 7 días (75 UI/kg cada 7 días) o cada 4 días (50 UI/kg cada 4 días). Un total de 55 pacientes aptos decidieron ser aleatorizados (17 a la dosis cada 4 días y 38 a la dosis de 75 UI cada 7 días). En general, los participantes del estudio que fueron tratados cada 7 días pudieron mantener una tasa de hemorragia baja con una dosis más elevada de tratamiento profiláctico a intervalos más prolongados. La mediana de la tasa anual de hemorragia fue de 0,00 para los pacientes tratados cada 4 días (IQR: 0,00; 2,23) y 0,00 para los pacientes tratados cada 7 días (IQR: 0,00; 2,36). No se observaron diferencias obvias en el ABR entre los grupos de edad Profilaxis de rutina en niños (menores de 12 años): Un total de 68 niños menores de 12 años recibieron tratamiento profiláctico con Esperoct® con una dosis de 65 UI/kg de peso corporal (50 a 75 UI/kg) dos veces por semana. El efecto profiláctico de Esperoct® se demostró en todos los niños menores de 12 años con una tasa anual de hemorragia de 1,95 (IQR: 0,00; 2,79), mientras que la mediana de la tasa anual de hemorragia espontánea fue de 0,00 (IQR: 0,00; 0,00), la tasa anual de hemorragia traumática fue de 0,00 (IQR: 0,00; 2,03) y la tasa anual de hemorragia articular fue de 0,00 (IQR: 0,00; 1,95). En 29 (42,6%) de los 68 niños no se produjo ninguna hemorragia durante el tratamiento profiláctico con Esperoct® a una dosis de 65 UI/kg (50 a 75 UI/kg). 10 de 13 pacientes con 17 articulaciones blanco (target) como base de referencia, no presentaron hemorragias en 14 de sus articulaciones blanco (target) durante la fase de tratamiento de 12 meses. Si se incluyen los datos de la fase de extensión del estudio con una exposición media de 3,4 años, la mediana de la tasa anual de hemorragia fue de 0,98 (IQR: 0,27; 1,44). Eficacia clínica de Esperoct® en el tratamiento de episodios hemorrágicos y durante el tratamiento a demanda La eficacia del Esperoct® en el tratamiento de los episodios hemorrágicos se ha demostrado en todos los grupos de edad. La gran mayoría de las hemorragias tratadas con Esperoct® fueron leves/moderadas. En el grupo de pacientes que recibieron tratamiento a demanda, se trataron 1.126 hemorragias en 12 pacientes mayores de 18 años de edad, siendo 38,1 UI/kg la dosis media para hemorragias leves a moderadas. En total, el 86,9% de las 1.126 hemorragias fueron tratadas eficazmente después de 1 inyección de Esperoct®. En total, el 96,8% de las 1.126 hemorragias fueron tratadas eficazmente después de 1 a 2 inyecciones de Esperoct®. La tasa de éxito general para el tratamiento de las hemorragias fue del 87,7%, con un 94,4% de hemorragias tratadas con una o dos inyecciones. Eficacia clínica de Esperoct® durante la cirugía mayor: Esperoct® ha demostrado ser eficaz en el mantenimiento de la hemostasia durante la cirugía mayor (43 de 45 fueron calificadas como "excelentes" o "buenas"), con una tasa de éxito del 95,6% en todas las cirugías mayores realizadas. Seguridad clínica: véase "Contraindicaciones", "Reacciones adversas", "Advertencias y precauciones especiales de uso", "Interacción con otros medicamentos y otras formas de interacción" y "Sobredosificación". Propiedades farmacocinéticas: En total, se evaluaron 129 perfiles farmacocinéticos (PK) para una dosis única de Esperoct® en 86 pacientes (incluidos 24 pacientes pediátricos (de 0 a < 12 años de edad)). Todos los estudios farmacocinéticos con Esperoct® se realizaron en pacientes con hemofilia A grave (factor VIII < 1%), previamente tratados. Los pacientes recibieron una dosis única de 50 UI/kg. Se tomaron muestras de sangre antes de la administración y en varios momentos hasta 96 horas después de la administración. La farmacocinética de Esperoct® se comparó con la de medicamentos del factor VIII no modificados, es decir, productos recombinantes y productos derivados del plasma. La vida media de Esperoct® fue 1,6 veces más larga que la de los productos de factor VIII no modificados. Se examinaron muestras de plasma para determinar la actividad del factor VIII mediante ensayos cromogénicos y de coagulación de una fase. Los parámetros farmacocinéticos resultantes de las dos pruebas fueron comparables. Parámetros farmacocinéticos: En total, se evaluaron 108 perfiles farmacocinéticos de dosis única de 50 UI/kg de Esperoct® en 69 pacientes. Los parámetros farmacocinéticos de dosis única son comparables entre los niños pequeños (0 a < 6 años de edad) y los niños mayores (6 a < 12 años de edad), así como en los adolescentes (12 a 17 años de edad) y los adultos (≥18 años de edad). Como era de esperar, en los niños, la recuperación progresiva fue menor que en adolescentes y adultos, mientras que el clearance ajustado al peso corporal fue mayor. La tendencia general fue que la recuperación incremental aumentaba con la edad, mientras que el clearance (mL/h/kg) disminuía. Esto ya se ha descrito para otros medicamentos de factor VIII y corresponde a un mayor volumen de distribución por kg de peso corporal en los niños en comparación con los adultos (Tabla 1). Los parámetros farmacocinéticos de dosis única determinados después de 28 semanas de tratamiento profiláctico con Esperoct® fueron congruentes con los parámetros farmacocinéticos iniciales.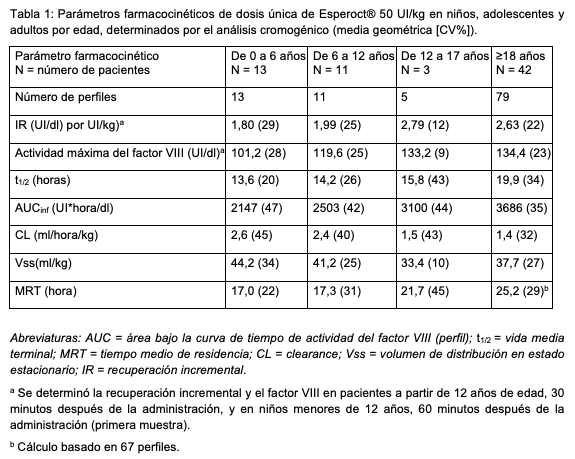
El nivel plasmático medio del factor VIII en estado estacionario antes de su administración para la profilaxis con Esperoct®, que se administra a razón de 50 UI/kg cada 4 días, es de 3,0 UI/dl (IC del 95%: 2,6; 3,4) en pacientes de 12 años de edad y mayores. En los pacientes menores de 12 años que recibieron 60 UI/kg (50 a 75 UI/kg) dos veces por semana, el nivel plasmático medio del factor VIII en estado estacionario antes de su administración durante la profilaxis fue de 1,5 UI/dl (IC del 95%: 1,2; 1,9). Duración prevista de la actividad del factor VIII superior al 5%. Los perfiles de actividad del factor VIII en estado estacionario se simularon utilizando un modelo de compartimiento único con cinética de eliminación de primer orden y con parámetros de clearance farmacocinético (CL) y volumen de distribución (Vss) en estado estacionario. Las predicciones farmacocinéticas mostraron que los pacientes tratados generalmente cada 3 a 4 días (72 a 95% de las veces) tenían una actividad del factor VIII superior al 5% (es decir, una hemofilia leve). Los pacientes tratados con 75 UI/kg cada 7 días deberían tener una actividad de factor VIII superior al 5% en el 57% de los casos. Datos preclínicos de seguridad: Esperoct® fue administrado como parte de un estudio de toxicidad a dosis repetidas en ratas inmunodeficientes (50 a 1200 UI/kg/4 días durante 52 semanas). No se produjeron cambios histopatológicos relacionados con el tratamiento ni resultados no deseados. El PEG no pudo detectarse en el tejido cerebral (incluyendo el plexo coroideo) por medio de una tinción inmunohistoquímica específica del PEG. No se observaron efectos sobre los parámetros de seguridad farmacológica (funciones cardiovascular, renal, respiratoria y central) en monos javaneses machos que recibieron Esperoct® hasta 2500 UI/kg/3 días. No se han realizado estudios en animales a largo plazo para evaluar el potencial carcinogénico de Esperoct® ni estudios para determinar los efectos de Esperoct® en la genotoxicidad, la fertilidad y las funciones de reproducción y de desarrollo. Se realizó una revisión del potencial carcinogénico de Esperoct® y no se detectó ningún riesgo carcinogénico.
Indicaciones.
Tratamiento y profilaxis de hemorragias en pacientes con Hemofilia A (deficiencia congénita del factor VIII) tratados previamente. Esperoct® no contiene ninguna cantidad farmacológicamente eficaz del factor von Willebrand y, por lo tanto, no es adecuado para el tratamiento del síndrome de von Willebrand Jürgens.
Dosificación.
El tratamiento debe ser supervisado por un médico con experiencia en el tratamiento de la hemofilia. Pacientes no tratados previamente: No se ha establecido aún la seguridad y eficacia de Esperoct® en pacientes no tratados previamente. Control del tratamiento: Durante el transcurso del tratamiento, se recomienda controlar adecuadamente los niveles del factor VIII para ajustar la dosis a administrar y la frecuencia de administración de Esperoct®. La respuesta al factor VIII varía con cada paciente, demostrando diferentes vidas medias y recuperaciones incrementales. Especialmente en el caso de las intervenciones de cirugía mayor, es necesario monitorear la terapia de sustitución del factor VIII midiendo la actividad del factor VIII en plasma. La actividad del factor VIII de Esperoct® puede medirse de manera más confiable mediante análisis convencionales del factor VIII, tales como el ensayo de coagulación de una fase o el análisis cromogénico. Ciertos reactivos a base de sílice (por ejemplo, APTT-SP, STA-PTT, TriniCLOT) no deben usarse en el ensayo de coagulación de una sola fase para determinar la actividad del factor VIII, ya que pueden causar una subestimación. Esto es particularmente importante en caso de cambio de laboratorio y/o los reactivos utilizados en el análisis. Posología: La dosis, el intervalo de administración y la duración de la terapia de sustitución dependerán de la gravedad, de la deficiencia de factor VIII, de la ubicación y extensión de la hemorragia, del nivel de actividad del factor VIII en cuestión y del estado clínico del paciente. La cantidad de factor VIII se expresa en Unidades Internacionales (UI), de acuerdo con el estándar de concentrado actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa en porcentaje (en relación con el plasma humano normal) o en Unidades Internacionales por dl (en relación con el Estándar Internacional actual para factor VIII en plasma). Tratamiento a demanda y tratamiento de episodios hemorrágicos: El cálculo de la dosis necesaria de factor VIII se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad del factor VIII en plasma en 2 UI/dl. La dosis requerida se determina utilizando la siguiente fórmula: Unidades requeridas (UI) = peso corporal (kg) × aumento deseado de factor VIII (%) (UI/dl) × 0,5 (UI/kg por UI/dl). La respuesta farmacocinética (por ejemplo, vida media, recuperación in vivo) y la respuesta clínica de los pacientes puede variar de un paciente a otro. La dosis y la frecuencia de administración de Esperoct® deberán basarse en la respuesta clínica individual. En la Tabla 2 se incluyen las guías para la dosificación de Esperoct® para el tratamiento a demanda y el tratamiento de los episodios hemorrágicos. Se deben mantener los niveles de actividad del factor VIII en los niveles plasmáticos indicados o por encima de ellos (en UI/dl o % del estandar). La frecuencia de administración y la duración del tratamiento deben ajustarse caso por caso para lograr una eficacia clínica óptima.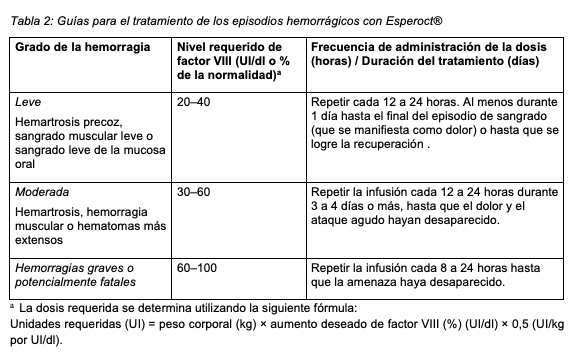
Tratamiento perioperatorio: La dosis y los intervalos de administración durante la cirugía dependen de la intervención y de la práctica local. La frecuencia de la administración y la duración del tratamiento se deben ajustar a la respuesta clínica individual. En la tabla 3 se incluyen recomendaciones generales para la dosificación de Esperoct® para el tratamiento perioperatorio. Se debe considerar la posibilidad de mantener la actividad del factor VIII dentro o por encima de la zona objetivo.
Profilaxis de rutina con Esperoct®: Adultos y adolescentes (a partir de los 12 años de edad): la dosis inicial recomendada es de 50 UI de Esperoct® por kg de peso corporal cada 4 días. Posteriormente, el régimen de dosificación puede ser ajustado a 50 UI/kg cada 3-4 días o a 75 UI/kg cada 7 días, en función de la respuesta del paciente (tasa de hemorragia baja de 0-2 episodios en los últimos 6 meses) y a criterio del médico tratante. Niños (menores de 12 años): deberá administrarse una dosis de 65 UI (50 a 75 UI) de Esperoct® por kg de peso corporal dos veces por semana. Modo de administración: Vía intravenosa. Esperoct® se debe administrar mediante inyección intravenosa (durante aproximadamente 2 minutos) luego de la reconstitución del polvo liofilizado con 4 ml de solvente de cloruro de sodio 0,9% (incluido). Esperoct® no debe mezclarse ni reconstituirse con otras soluciones inyectables que no sean el solvente de cloruro de sodio suministrado. No administrar Esperoct® reconstituido en los mismos tubos o recipientes que hubieran contenido otros medicamentos. Para las instrucciones correspondientes a la reconstitución del medicamento antes de su administración, ver "Precauciones de manipulación". Se requiere una capacitación adecuada si la administración debe ser realizada por el paciente o un cuidador.
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes mencionados en la sección "Composición". Reacción alérgica conocida a las proteínas del hámster.
Reacciones adversas.
Resumen del perfil de seguridad: Se han observado reacciones de hipersensibilidad y/o reacciones alérgicas (que pueden incluir hipersensibilidad, erupción cutánea, eritema y prurito) y, en algunos casos, pueden producir una anafilaxia grave (incluido shock anafiláctico). En muy raras ocasiones se ha observado la aparición de anticuerpos dirigidos contra las proteínas de hámster, asociadas a reacciones de hipersensibilidad. En los pacientes con hemofilia A tratados con factor VIII, incluido Esperoct®, se puede producir el desarrollo de anticuerpos neutralizantes (inhibidores). Si aparecen inhibidores de este tipo, la situación se manifestará como una respuesta clínica inadecuada. En estos casos, se recomienda contactar con un centro especializado en hemofilia. Lista de reacciones adversas: En la tabla 4 se presenta la frecuencia de las reacciones adversas que se producen en 270 pacientes con hemofilia A grave ( < 1% de actividad del factor VIII endógeno), tratados previamente (PTP) y sin antecedentes de inhibidores, en cinco estudios clínicos multicéntricos, prospectivos. La tabla que figura a continuación corresponde a la clasificación de órganos del sistema MedDRA (clasificación de órganos del sistema y nivel de términos preferente). Las categorías de frecuencia se definen conforme a las siguientes convenciones: muy frecuentes (≥1/10); frecuentes (≥1/100 a < 1/10); poco frecuentes (≥1/1.000 a < 1/100); raras (≥1/10.000 a < 1/1.000); muy raras ( < 1/10.000) y de frecuencia desconocida (no puede estimarse sobre la base de los datos disponibles).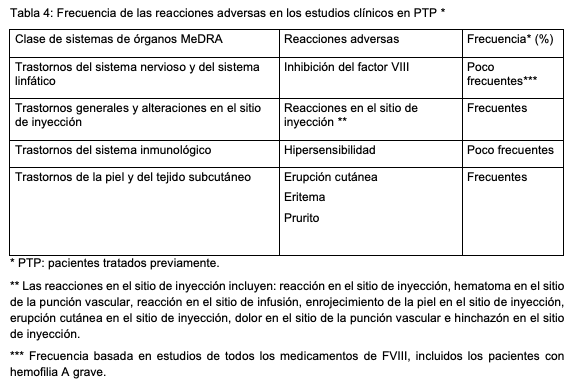
Niños y adolescentes: Pacientes tratados previamente: el perfil de seguridad de Esperoct® no mostró diferencias entre niños, adolescentes y adultos tratados previamente con Esperoct®. Pacientes no tratados previamente: La seguridad de Esperoct® aún no ha sido demostrada en pacientes no tratados previamente.
Advertencias.
Hipersensibilidad: El tratamiento con Esperoct® puede causar reacciones de hipersensibilidad del tipo alérgico. El medicamento contiene trazas de proteínas de hámster, que pueden provocar reacciones alérgicas en algunos pacientes. Si aparecen síntomas de hipersensibilidad, se debe informar a los pacientes que deben interrumpir inmediatamente el tratamiento Esperoct® y consultar a su médico. Se debe informar a los pacientes sobre los primeros signos de las reacciones de hipersensibilidad (urticaria localizada, urticaria generalizada, opresión en el pecho, respiración sibilante, hipotensión y anafilaxia). En caso de shock anafiláctico, se seguirán las pautas médicas habituales para su tratamiento. Inhibidores: La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores suelen ser inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en Unidades Bethesda (UB) por ml de plasma usando el ensayo Bethesda modificado. El riesgo de desarrollar inhibidores se correlaciona con la gravedad de la enfermedad, y la exposición al factor VIII, siendo mayor durante los primeros 20 días de exposición. Los inhibidores a veces pueden formarse incluso después de los primeros 100 días de exposición. Se han observado casos de inhibidores recurrentes (de título bajo) en pacientes previamente tratados, con una duración de exposición superior a 100 días y que tenían antecedentes de formación de inhibidores después de pasar de un producto de factor VIII a otro. Por consiguiente, se recomienda que todos los pacientes sean monitoreados cuidadosamente para detectar la presencia de inhibidores después de un cambio de formulación. La relevancia clínica del desarrollo del inhibidor dependerá del título del inhibidor. Los inhibidores de título bajo, que solo están presentes temporalmente o que permanecen de bajo título, tienen menor riesgo de respuesta clínica insuficiente que aquellos con título alto. En general, todos los pacientes tratados con medicamentos con factor VIII deben ser monitoreados de cerca a fin de que sea posible detectar la presencia de inhibidores mediante observaciones clínicas y pruebas de laboratorio adecuadas. Si no se alcanzan la actividad esperada del factor VIII en plasma, o si la hemorragia no puede controlarse con una dosis adecuada, se deben verificar la presencia de inhibidores del factor VIII. En pacientes con niveles altos de inhibidores, es posible que el tratamiento con factor VIII no sea efectivo. En ese caso se deben considerar otras opciones de tratamiento. El tratamiento de estos pacientes debe ser realizado por un médico con experiencia en el tratamiento de la hemofilia y los inhibidores del factor VIII. Episodios cardiovasculares: En pacientes con factores de riesgo cardiovascular, la terapia de sustitución con factor VIII puede aumentar el riesgo cardiovascular. Complicaciones relacionadas con el catéter: Si se requiere un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, tales como, infecciones locales, bacteriemia y trombosis en el sitio de inserción del catéter. Consideraciones relacionadas con los excipientes Este medicamento contiene, luego de la reconstitución, 30,5 mg de sodio por vial. Esto corresponde al 1,5% de la dosis diaria de 2,0 g de sodio recomendada por la OMS para adultos. Niños y adolescentes: Las advertencias y las precauciones especiales de uso mencionadas aplican tanto para los adultos como para los niños. Efectos sobre la capacidad de conducir y utilizar máquinas: Esperoct® no afecta la capacidad de conducir o utilizar máquinas. Interacción con otros medicamentos y otras formas de interacción: No se han notificado interacciones entre los productos del factor VIII de coagulación humana (ADNr) y otros medicamentos. Embarazo y lactancia: No se han realizado estudios de reproducción en animales con Esperoct®. Debido a la rareza de la hemofilia A en las mujeres, no se dispone de datos clínicos sobre el uso de Esperoct® durante el embarazo o la lactancia. Por lo tanto, Esperoct® no debe usarse durante el embarazo o la lactancia a menos que sea estrictamente necesario.
Incompatibilidades.
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
Conservación.
Vial sin abrir (antes de la reconstitución): Periodo de conservación: 30 meses si se almacena en la heladera (2°C-8°C). Durante la vida útil, el producto puede ser almacenado: a temperatura ambiente (≤30°C) hasta 12 meses o por encima de la temperatura ambiente (entre > 30°C y 40°C) hasta 3 meses. Una vez que el producto ha sido almacenado fuera de la heladera, no debe volver a colocarse en la heladera para su almacenamiento. Anote el inicio del período de almacenamiento y la temperatura de almacenamiento fuera de la heladera en el espacio previsto para ello en el envase. Mantener el vial en su envase para protegerlo de la luz. Después de la reconstitución: Desde el punto de vista microbiológico, el medicamento debe utilizarse inmediatamente después de la reconstitución. Se ha demostrado la estabilidad química y física "en uso" durante: 24 horas cuando se almacena en la heladera (2°C - 8°C) o 4 horas cuando se almacena a ≤30 °C o 2 horas si se almacena entre > 30°C y 40°C para productos que, antes de su reconstitución, no hayan sido almacenados más de 3 meses por encima de la temperatura ambiente (entre > 30°C y 40°C). Si el medicamento no se utiliza inmediatamente, el usuario es responsable de los tiempos de almacenamiento durante el uso y de las condiciones de almacenamiento antes del uso. Se recomienda no almacenar o utilizar durante períodos más largos que los mencionados anteriormente, a menos que la reconstitución se haya realizado en condiciones estériles controladas y validadas. La preparación reconstituida debe ser almacenada en el vial. Precauciones de almacenamiento: Almacenar en la heladera (entre 2°C y 8°C). No congelar. Conservar en el envase original para protegerlo de la luz. Para obtener más información sobre las condiciones de almacenamiento a temperatura ambiente (≤30°C) o hasta 40°C y después de la reconstitución, ver "Vial sin abrir (antes de la reconstitución): Período de conservación" y "Después de la reconstitución". Precauciones de manipulación: Esperoct® debe administrarse por vía intravenosa después de la reconstitución del polvo liofilizado con el solvente proporcionado en la jeringa. Después de la reconstitución, la solución es un líquido transparente e incoloro sin partículas visibles. El medicamento reconstituido se debe inspeccionar visualmente para detectar partículas o decoloración antes de su administración. La solución debe ser transparente e incolora. No utilizar las soluciones turbias o que presentan depósitos. Para consultar las instrucciones de reconstitución del medicamento antes de su administración, ver el prospecto del estuche. La velocidad de administración de aproximadamente 2 minutos debe ajustarse de acuerdo con la condición del paciente. También necesitará un set de infusión (sonda y aguja mariposa), toallitas estériles con alcohol, gasas y apósitos. Estos elementos no están incluidos en el envase de Esperoct®. Siempre utilice una técnica aséptica. Eliminación: Después de la inyección, desechar de forma segura la jeringa con el set de infusión y el vial con el adaptador de vial. Todo medicamento no utilizado o material de descarte debe eliminarse de acuerdo con la normativa local vigente.
Sobredosificación.
En estudios clínicos con Esperoct®, no se ha notificado sobredosificación de Esperoct® a dosis de hasta 114 UI/kg. No se ha informado de ningún síntoma clínico asociado con la sobredosificación de Esperoct®. Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los centros de toxicología: Hospital de Pediatría Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital A. Posadas: (011) 4654-6648/4658-7777. Optativamente, otros centros de intoxicaciones.
Presentación.
Cada envase de Esperoct® de 500 UI a 1500 UI de polvo liofilizado y solvente para solución inyectable contiene: 1 vial de vidrio (tipo I) conteniendo el polvo liofilizado, con un tapón de liofilización de goma de clorobutilo y una tapa snap-off de aluminio y plástico. 1 jeringa prellenada conteniendo 4 ml de solvente, con un émbolo de goma (bromobutilo), un tope (polipropileno) y un capuchón de goma (bromobutilo). 1 varilla del émbolo (polipropileno). 1 adaptador de vial estéril para la reconstitución. Los tapones de goma, el capuchón de plástico con tapón y el émbolo de goma no contienen látex natural.